

Abstract
Targeting altered cancer cell metabolism with the glycolysis inhibitor, 2-deoxyglucose (2DG), is a viable therapeutic strategy, but the effects of 2DG on lymphoma cells and the mechanism of action are unknown. Five T cell lymphoma lines (TCLs) and two B cell lymphoma lines (BCLs) were shown to be highly sensitive to 2DG. Examination of the cell death pathway demonstrated proapoptotic protein Bax “activation” and caspase cleavage in 2DG-treated cells. However, Q-VD-OPh, a potent inhibitor of caspase activity, provided minimal protection from death. In contrast, overexpressing the anti-apoptotic protein Bcl-2 dramatically enhanced survival of 2DG-treated cells that was negated by a Bcl-2 antagonist. BH3-only members, Bim and Bmf, were upregulated by 2DG, and shRNAs targeting Bim protected from 2DG toxicity demonstrating that Bim is a critical mediator of 2DG toxicity. 2DG also induced GADD153/CHOP expression, a marker of ER stress and a known activator of Bim. Mannose, a reagent known to alleviate ER stress, transiently protected from 2DG-induced cell death. Examination of the effects of 2DG on energy metabolism showed a drop in ATP levels by 30 min that was not affected by either Bcl-2 or mannose. These results demonstrate that ER stress appears to be rate limiting in 2DG-induced cell death in lymphoma cells and this cell killing is regulated by the Bcl-2 family of proteins. Bcl-2 inhibition combined with 2DG may be an effective therapeutic strategy for lymphoma.
Keywords: 2-deoxyglucose (2DG), Apoptosis, Bcl-2, Bax, BH3 mimetics, Oncogenesis
Introduction
Cancer cells do not regulate glycolysis and oxidative phosphorylation in the same fashion as non-transformed cells and this phenomenon has been termed the Warburg effect. In an attempt to exploit metabolic differences between cancer and normal cells, drugs like the glucose analog, 2-deoxyglucose (2DG), have been used to inhibit hexokinase, the rate limiting enzyme in glycolysis (Crane and Sols, 1954; Tower, 1958). The inhibition of the hexokinase and glycolysis eventually leads to metabolic stress and cell death that occurs more rapidly in cancer vs. normal cells (Aykin-Burns et al., 2009). Since tumor cells have a greater tendency to depend on glycolysis than normal cells, 2DG has been studied as a promising therapeutic agent that targets metabolic alterations of cancer cells (Ben Sahra et al., 2010; Simons et al., 2007; Zhang et al., 2006).
In addition to blocking glycolysis, 2DG has also been shown to inhibit N-linked glycosylation in the endoplasmic reticulum (ER) by interfering with mannose addition to the oligosaccharides (Kurtoglu et al., 2007). This latter activity can be overcome by the addition of excess mannose. Research indicates that 2DG inhibition of N-linked glycosylation is a significant contributor to toxicity in multiple tumor cell lines (Kurtoglu et al., 2007). Interference with N-linked glycosylation results in improper protein folding, leading to ER stress and the activation of the unfolded protein response (UPR), and, if the stress is sufficiently severe, – initiation of cell death pathways. The UPR-induced transcription factor GADD153/CHOP is particularly important in ER stress-induced cell death, as both cells and animals lacking CHOP are protected against a variety of insults that cause ER stress (Oyadomari and Mori, 2004). ER stress can be triggered by a number of factors, including aberrations in calcium regulation (Ma and Hendershot, 2004), hypoxic interference of proper protein folding (Koumenis et al., 2002), increased synthesis of secretory proteins (Raden et al., 2005), and inhibition of glycosylation (Kurtoglu et al., 2007). With prolonged or pronounced ER stress, cells activate the mitochondrial apoptotic pathway. While the complete pathway that results in cell death following ER stress is still under investigation, both Bim and Puma deficiency have been shown to protect from ER stress induced apoptosis both in vivo and in vitro (Puthalakath et al., 2007; Reimertz et al., 2003).
It is well known that Bcl-2 family members are important regulators of apoptotic cell death and consist of proapoptotic and antiapoptotic members. The proapoptotic members are divided into multidomain and BH3-only subgroups. Activating the multidomain members, such as Bax and Bak, is considered a gateway to apoptotic cell death. Cell death is believed to be initiated by the BH3-only subgroup members, among which are Bim and Puma (Adams and Cory, 1998; Gross et al., 1999). The regulation of BH3-only proteins is complex and involves both transcriptional as well as post-translational events that affect their activity. The current view is that BH3-only proteins promote apoptosis by interacting with prosurvival Bcl-2 family members (indirect activation) and/or activating multi-domain proapoptotic members Bax or Bak (direct activation) (Kuwana et al., 2005; Letai et al., 2002). Chemical compounds (BH3 mimetics) that trigger apoptotic signaling by antagonizing anti-apoptotic Bcl-2 proteins have been developed and explored therapeutically, since Bcl-2 activity is frequently elevated and causes drug resistance in cancer cells (Letai et al., 2004).
One of these Bcl-2 antagonists, ABT-737, has high affinity to the anti-apoptotic proteins Bcl-2, Bcl-XL and Bcl-W, although not to Mcl-1 and A1 (Oltersdorf et al., 2005). Applying ABT-737 with chemotherapeutic agents demonstrated a marked increase in response to treatment in vitro and exhibited good activity as a single agent in small cell lung cancer xenograft models (Oltersdorf et al., 2005). Multiple studies have shown that ABT-737 is effective as a single agent or in combination with cytotoxic agents targeting lymphoma (van Delft et al., 2006), lymphoblastic leukemia (Kang et al., 2007), and small cell lung cancer (Shoemaker et al., 2006). Considering deriviatives of ABT-737 are in early Phase clinical trials (www.clinicaltrials.gov), exploring mechanisms and determining optimum combined modality therapies involving these drugs is potentially of great clinical significance.
In the current study, we sought to determine the sensitivity of lymphoma cell lines to 2DG and examine the involvement of the Bcl-2 family members in lymphoma resistance to 2DG. In addition, we sought to determine whether combination therapy with ABT-737 would further increase the efficacy of 2DG treatment in lymphoma. Understanding Bcl-2 family proteins' involvement in 2DG-induced toxicity may lead to the development of combination treatments that optimize the clinical response in patients with lymphoma.
Results
TCLs are uniformly sensitive to 2DG treatment
Murine T cell lymphoma cell lines (TCLs) were obtained from five independent thymic lymphomas from Lck-Bax transgenic mice. These cell lines were treated with 20 mM 2-deoxyglucose (2DG), and cell viability was monitored. All five cell lines demonstrated a rapid decrease in viability following 2DG treatment (Fig. 1A). Importantly, freshly extracted murine thymocytes were not sensitive to 2DG treatment compared to TCLs when monitored for up to 24 hours following the treatment, suggesting selective sensitivity to 2DG treatment by lymphomas and not normal cells (Fig. 1B). To determine whether caspases were activated following 2DG treatment, caspase 3 and PARP processing were examined in two of the TCLs. Both cell lines showed cleavage of caspase 3 and near complete cleavage of its downstream target PARP. Q-VD-OPh, a broad spectrum pan-caspase inhibitor, effectively prevented the cleavage of both caspase 3 and PARP (Fig. 1C). Surprisingly, Q-VD-OPh had little effect on cell death following 2-DG treatment (Fig. 1D), suggesting that 2DG induces cell death via caspase-independent pathway.
Figure 1. TCLs are uniformly sensitive to 2DG treatment.
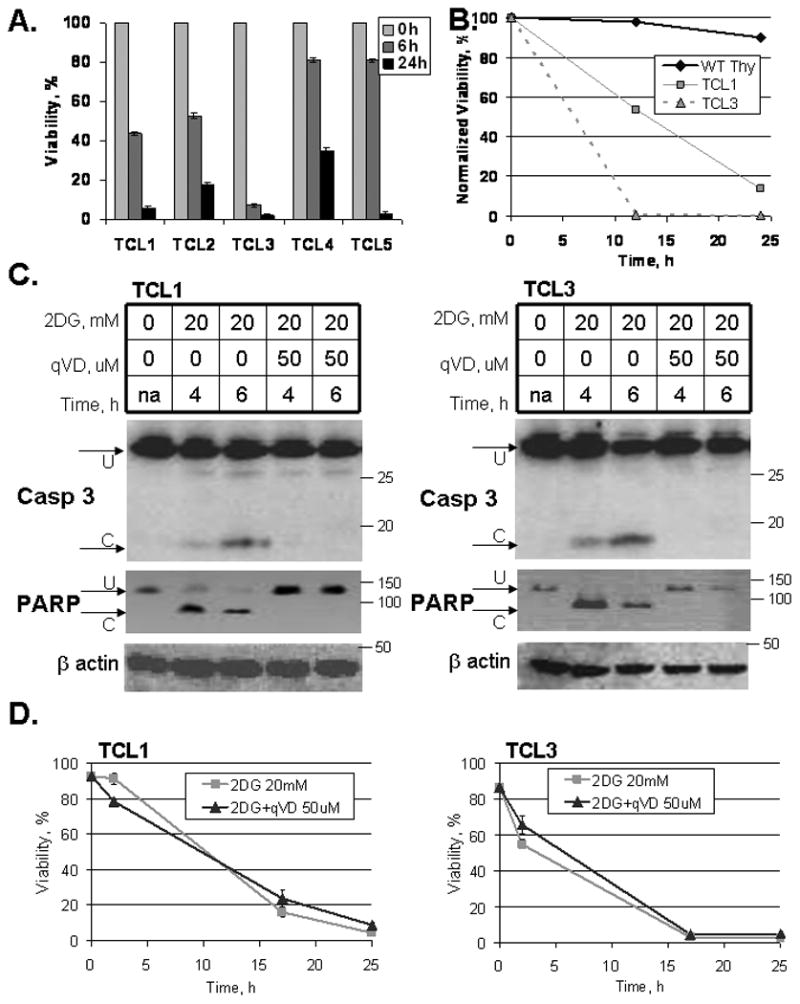
(A) Mouse T cell lymphoma cells (TCLs) were plated at 200,000/mL in Adv.RPMI medium with 20 mM 2DG. Viability was assessed using Guava ViaCount dye exclusion assay (0-24 h). (B) Murine thymocytes (WT Thy) and TCLs were plated at 200,000/mL in Adv.RPMI medium with 20 mM 2DG for the time indicated. Viability was assessed by Guava ViaCount dye exclusion assay. Data represent average viabilities ± S.D. (C) TCL1 and TCL3 were treated with 20 mM 2DG and with 50 μM Q-VD-OPh (qVD) for the times indicated. caspase 3 and PARP cleavage were assessed by Western blot. U = uncleaved, C = cleaved. (D) Cells were treated with 20 mM 2DG and with 50 μM Q-VD-OPh (qVD) as indicated. Viability was assessed using Guava ViaCount dye exclusion assay at the times indicated.
To examine whether the Bcl-2 family-regulated mitochondrial apoptotic pathway was involved in 2DG killing, we examined whether 2DG induced Bax activation. Bax is known to undergo conformational change and oligomerization, when the mitochondrial pathway is activated for cell death (Smaili et al., 2003). We detected Bax conformational change by FACS using the conformation specific antibody (Hsu and Youle, 1998). While 2DG robustly induced Bax activation, caspase inhibition by Q-VD-OPh had only a modest effect on Bax activation (Fig. 2). Taken together, these results suggest that the mitochondrial apoptotic pathway is activated following 2DG treatment but that cell death is not dependent on the efficient activation of caspases.
Figure 2. Bax is activated following 2DG treatment.
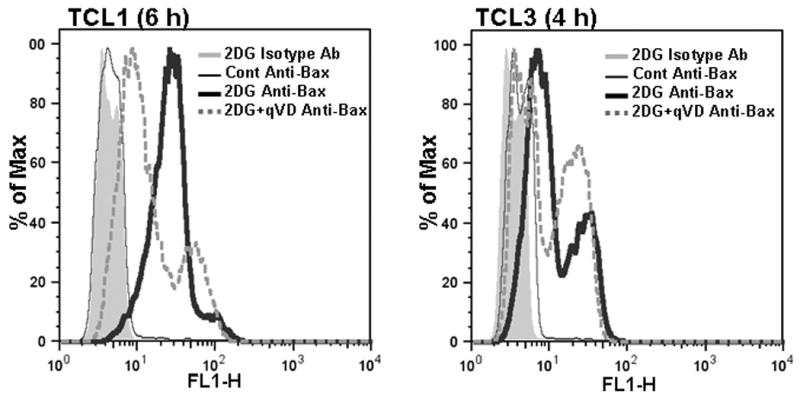
TCLs were treated with 2DG (20 mM) and with Q-VD-OPh (50 μM, qVD) as indicated. After 4 h (TCL3) or 6 h (TCL1) cells were labeled for the active conformation of Bax and analyzed by flow cytometry as described in methods. Histograms of the following samples are shown: 2DG treated cells stained with isotype control antibody (shaded gray), untreated cells stained with Anti-Bax (thin black line), 2DG treated cells stained with Anti-Bax (thick black line), 2DG+qVD treated cells stained with Anti-Bax (thick dotted gray line).
Inhibition of 2DG toxicity by Bcl-2
It is known that Bcl-2 inhibits Bax activation, thus we reasoned that overexpressing Bcl-2 would protect 2DG-treated lymphomas from death. TCL1 and TCL3 were therefore generated to overexpress Bcl-2 (Fig. 3A). Elevated Bcl-2 completely protected from cell death following 24 hours of 2DG treatment (Fig. 3A). Moreover, Bcl-2 overexpression in TCLs treated with 2DG for 24 hours fully preserved their clonogenic proliferative ability (Fig. 3B), suggesting that pro-apoptotic Bcl-2 proteins mediate 2DG-induced cell death. As predicted, Bax activation was effectively blocked by Bcl-2 in 2DG-treated TCLs (Fig. 3C). To examine 2DG effects on energy metabolism, ATP levels were measured in TCLs and TCL-Bcl-2 cells following 2DG treatment. TCL1 and TCL3 showed a 60-70% drop in ATP levels as early as 30 min following 2DG treatment (Fig. 3D). This drop was not altered when co-treated with Q-VD-OPh (not shown). Bcl-2 overexpression did not change 2DG-caused drop in ATP levels within the first two hours demonstrating that the effects on ATP are not due to cell death (Fig. 3D).
Figure 3. Bcl-2 expression protects from 2DG toxicity.
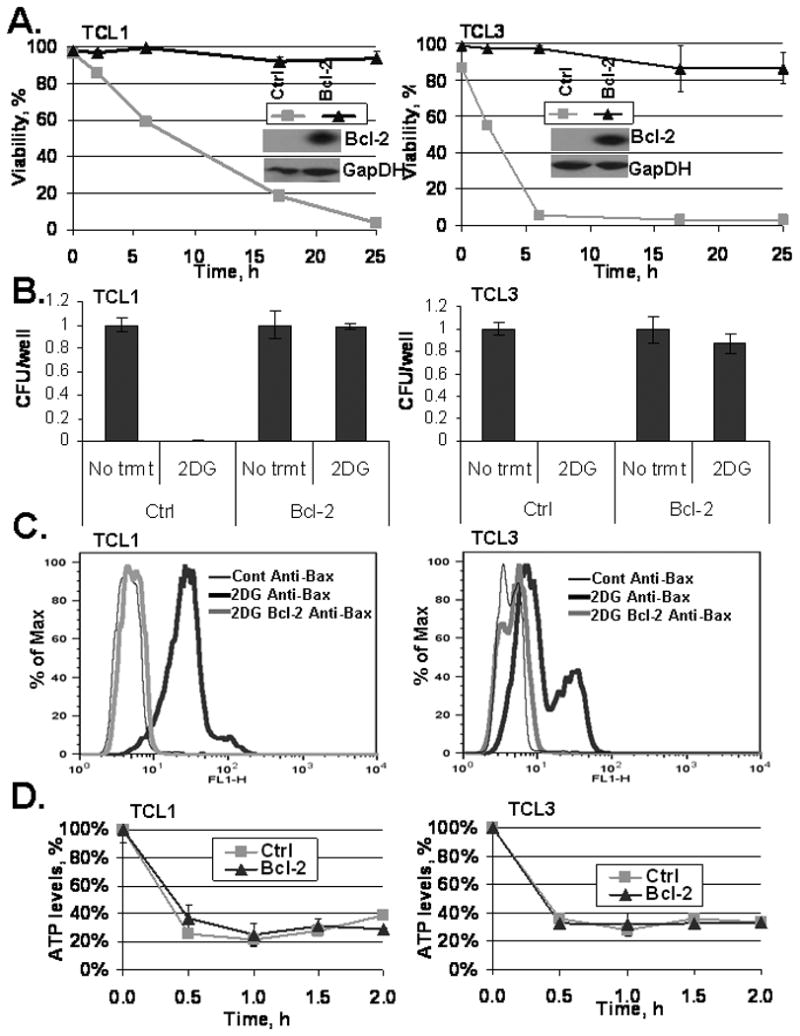
(A) Western blot of TCL1 and TCL3 following lentiviral transfection with either Bcl-2 Luciferase plasmid or Luciferase control plasmid (as described in methods). Control (ctrl) and TCL-Bcl-2 cells (Bcl-2) were treated with 20 mM 2DG and viability was assessed by Guava ViaCount dye exclusion assay at the times indicated. Data represent average viabilities ± S.D. (B) Clonogenic survival of control (ctrl) and TCL-Bcl-2 (Bcl-2) lines was measured following 24 h 2DG (20 mM) treatment as described in methods. Data represent average calculations of CFU/well from three 96-well plates ± S.D. (C) TCLs were treated with 20 mM 2DG (TCL1, TCL1-Bcl-2 for 6 h; TCL3, TCL3-Bcl-2 for 4 h), harvested, fixed, and stained to examine Bax activation as described in Fig. 2 and the methods. Histograms of the following samples are shown: Untreated TCLs (thin black line), 20 mM 2DG treated TCLs (thick black line), 20 mM 2DG treated TCL-Bcl-2 cells (thick gray line). (D) TCLs and TCL-Bcl-2 cells were treated with 20 mM 2DG for the time indicated and ATP levels were measured using a bioluminescent cell assay kit as described in methods. Data represent average ATP levels ± S.D.
In order to confirm the role of Bcl-2 in preventing cell death, we antagonized Bcl-2 by a BH3 mimetic, ABT-737. As expected, ABT-737 sensitized both TCL1-Bcl-2 and TCL3-Bcl-2 to 2DG (5 mM) treatment (Fig. 4A). Of note, ABT-737 alone was not toxic to these cells. Importantly, TCL1 and TCL3 were also sensitized by ABT-737, suggesting that endogenous levels of anti-apoptotic proteins in TCLs also limit the toxicity of 2DG (Fig. 4B). To determine whether Bax and/or Bak are required for 2DG-induced toxicity, IL-3 dependent hematopoietic lines deficient in both Bax and Bak were examined (Lum et al., 2005). Double knockout (DKO) cells were resistant to 2DG, ABT-737, and their combination (Fig. S1). Control cells expressing either Bax or Bak were sensitized to all of these treatments. Of note, treatment with both 2DG and ABT-737 showed no toxicity in the DKO cells while the Bax and Bak expressing cells had less than 10% viability. Taken together, these results demonstrate 2DG-induced toxicity is dependent on Bax or Bak and in cancer cells the toxicity can be enhanced by a BH3 mimetic.
Figure 4. ABT-737, a Bcl-2 antagonist, restores and induces sensitivity to 2DG.
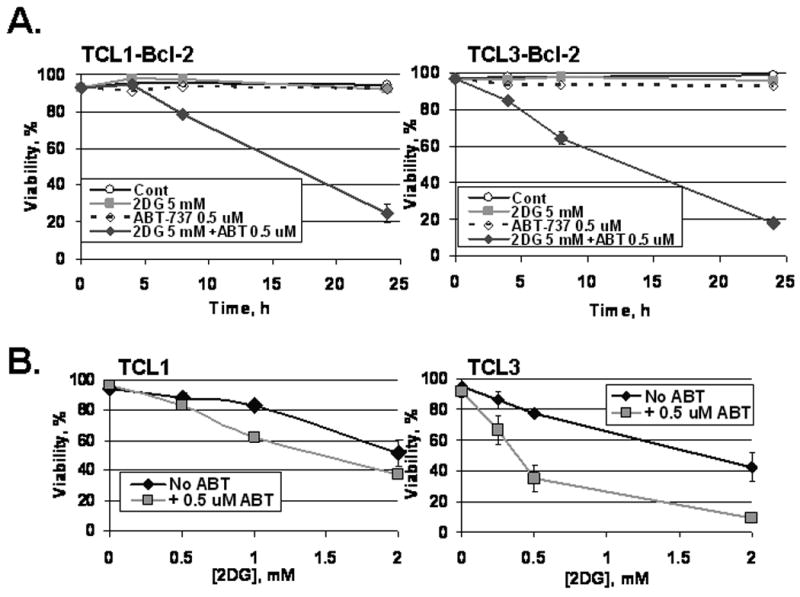
(A) TCLs with Bcl-2 overexpression were treated as indicated with 2DG and ABT-737 (0.5 μM). Viability was assessed by Guava ViaCount at the times indicated. Data represent average viabilities ± S.D. (B) Non-transduced TCLs were treated as indicated with 2DG and ABT-737 (0.5 μM) for 24 hours. Viability was assessed by Guava ViaCount. Data represent average viabilities ± S.D.
Bim and Bmf upregulation following 2DG treatment
In order to determine which BH3-only proteins are involved in 2DG toxicity, we examined the levels of multiple BH3-only members following 2DG treatment in TCLs and found that both Bim and Bmf levels were elevated in TCLs with and without Bcl-2 overexpression at 6 and 24 hours after 2DG treatment, respectively (Fig. 5A, B). Other BH3-only members, Bid, Bad and Puma, remained unchanged following 2DG treatment (Fig. 5B). Additionally, examining the antiapoptotic protein levels of Bcl-xL and Mcl-1 did not show apparent changes following 2DG treatment (Fig. S2). To directly examine which BH3-only proteins interact with Bcl-2 following 2DG treatment, Bcl-2 was immunoprecipitated from Bcl-2 overexpressing TCLs that were either untreated or treated with 2DG for 24 hours. The Bcl-2 antibodies effectively pulled down Bcl-2 protein as no Bcl-2 protein was detected in the supernatant after immunoprecipitation (not shown). Of the BH3-only proteins examined, only Bim and Bmf were bound to Bcl-2 under basal conditions (Fig. 5C, not shown). With 2DG treatment, Bcl-2-bound Bim and Bmf were greatly increased while Bid, Bad and Puma remained unbound (not shown). Significantly, when cells were treated with ABT-737 for the final 3 hours of the 24 hour 2DG treatment, the levels of Bim and Bmf that were bound by Bcl-2 were markedly reduced (Fig. 5C). These results clearly demonstrate that 2DG toxicity involves Bim and Bmf upregulation and binding to Bcl-2 and suggest they are involved in the activation of Bax leading to cell death (Chen et al., 2005; Kuwana et al., 2005).
Figure 5. ABT-737 inhibits 2DG-caused binding of Bim and Bmf BH3-only proteins to Bcl-2.
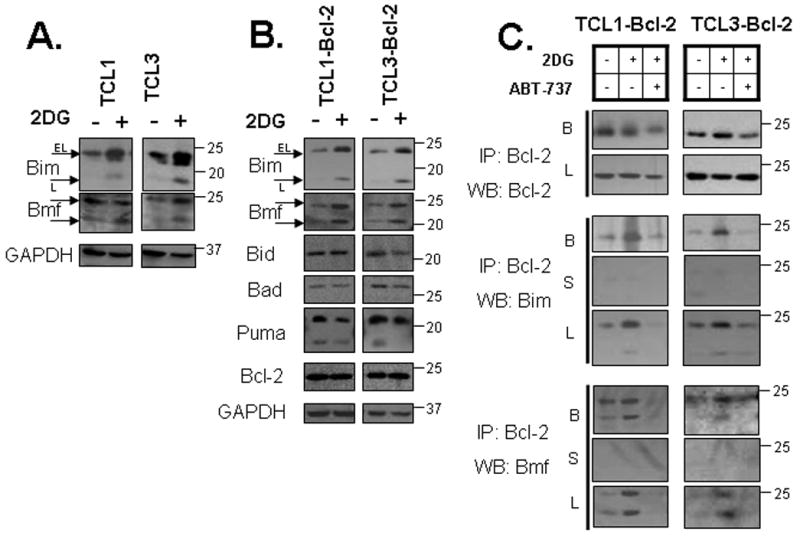
(A) TCL cells were treated with 2DG (20 mM) for 6 hours as indicated. Protein levels were assessed by Western blot. (B) TCL-Bcl-2 cells were treated with 2DG (20 mM) for 24 hours as indicated. Protein levels were verified by Western blot. (C) TCL-Bcl-2 cells were treated with 2DG (20 mM) for 24 hours and with 0.5 μM ABT-737 for the final 3h as indicated. Immunoprecipitation was done with Bcl-2 antibody as described in methods. Protein levels were verified by Western blot. B = beads, L = lysates, S = supernatant.
Downregulation of Bim protein inhibits 2DG toxicity
To determine if Bim is important in causing 2DG toxicity, shRNAs were developed to stably downregulate Bim levels in TCLs. TCL1, TCL3, and TCL4 were stably transduced with retroviral shBIM-containing plasmids. Of the four hairpins designed to downregulate Bim, cell lines with shBIM279 and shBIM280 were studied due to their effective reduction of Bim levels (> 50%) compared to the non-targeting vector and the empty vector control in all three cell lines examined (Fig. 6A). As predicted, all three TCL lines expressing shBIM279 and shBIM280 were significantly protected from cell death at all concentrations of 2DG examined relative to vector control (Fig. 6B). The shBIM277 vector, which failed to downregulate Bim levels, had no inhibitory effect on cell death in each of the cell lines examined. We conclude that Bim is a critical mediator of 2DG toxicity in TCLs.
Figure 6. Downregulation of Bim protein inhibits 2DG toxicity.
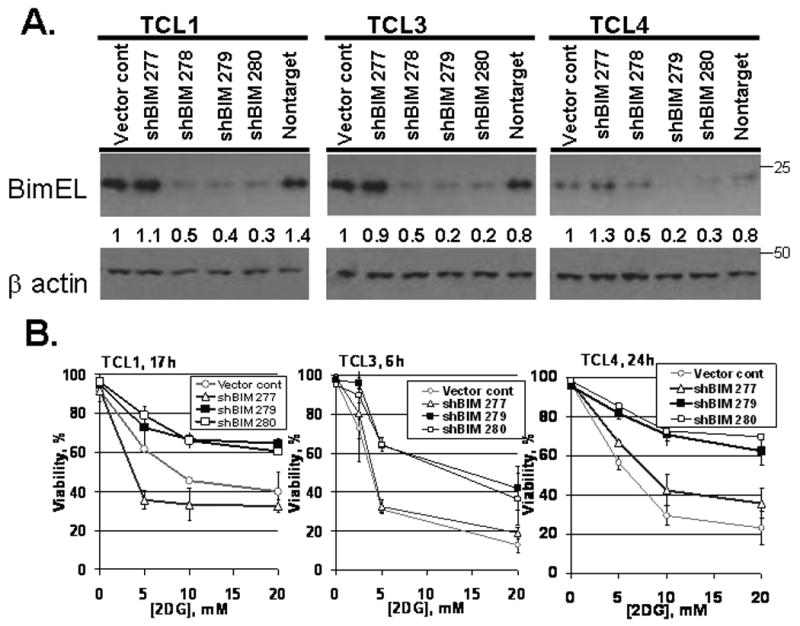
(A) Cells were transduced with shBIM retroviral plasmids. Bim protein levels were assessed by Western blot and actin was used as a loading control. The ratios of Bim to actin levels were determined by UVP (methods) and the untreated vector control cells were assigned a normalized ratio of 1.0. (B) Cells were treated with various concentrations of 2DG and viability was monitored by Guava ViaCount at the time indicated. The duration of 2DG treatment was individually selected for each line such that control cells would be expected to be between 30 and 70% viable at that time point. Data represent average viabilities ± S.D.
Role of ER stress in Bim upregulation
Our data demonstrating that overexpression of Bcl-2 protects against 2DG without restoring cellular ATP levels suggests an alternate pathway for 2DG-induced cell death. It has been reported that 2DG causes ER stress through its interference with lipid-linked oligosaccharide formation, and that this effect can be blocked by the addition of mannose (Kurtoglu et al., 2007). ER stress induces cell death at least in part by upregulation of CHOP/GADD153, which has been shown to upregulate Bim (Puthalakath et al., 2007). We therefore hypothesized that 2DG induces apoptosis via ER stress-induced upregulation of GADD153, which then leads to Bim upregulation. This hypothesis predicts that (1) mannose will inhibit 2DG-induced cell death; (2) 2DG will activate the UPR and mannose will block this activation; and (3) 2DG will result in rapid upregulation of CHOP, which will correlate with Bim upregulation and be blocked by mannose. The first of these predictions was confirmed by demonstrating that mannose greatly delayed 2DG-induced cell killing (Fig. 7A). 2DG treatment led to rapid upregulation (6 h) of UPR target genes, including Gadd153/Chop; and Gadd34, which is downstream of Gadd153 (Marciniak et al., 2004); as well as the ER-associated degradation factor Edem and the ER chaperone Bip (not shown). Mannose cotreatment effectively blocked upregulation of each of these genes. 2DG also caused splicing of Xbp1 mRNA, which is induced by activation of the IRE1 pathway of the UPR which was also effectively blocked by mannose (Fig. 7C). These results demonstrate that 2DG activates a canonical UPR that can be blocked by mannose. Importantly, mannose also blocked upregulation of Bim by 2DG, while having no effect on ATP depletion (Fig. 7D). Similarly, the upregulation of Bmf levels was inhibited by mannose indicating its upregulation at least in part is due to the ER stress pathway (Fig. S3).
Figure 7. Bim upregulation and ER stress following 2DG treatment.
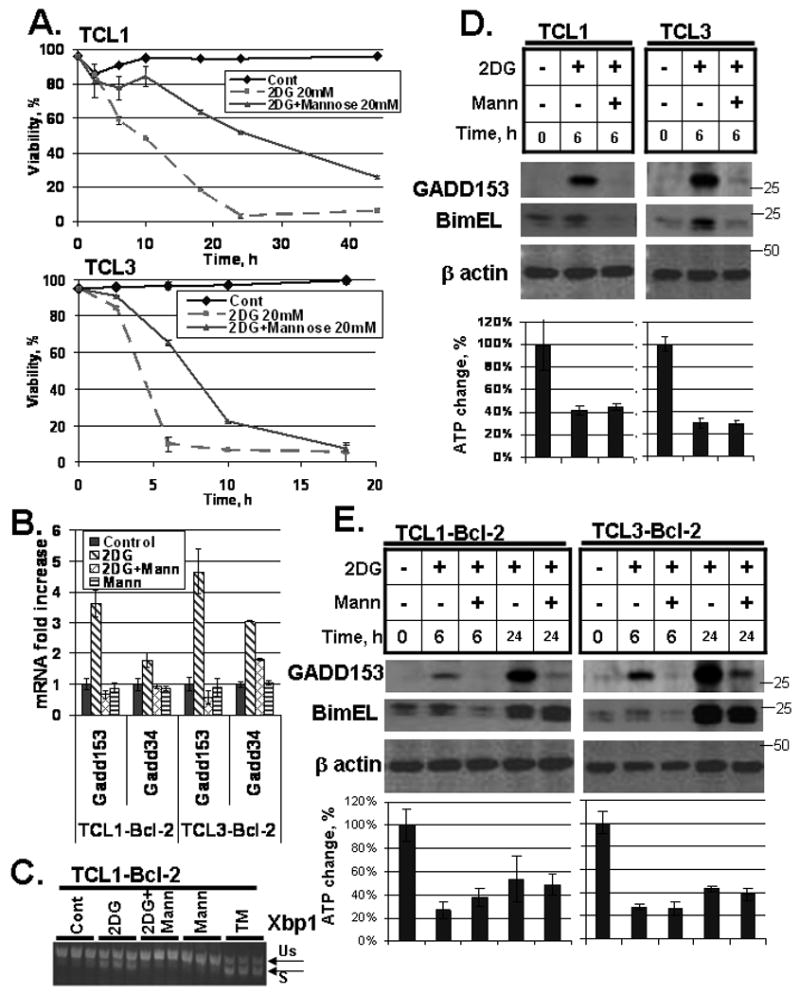
(A) Cells were treated with 20 mM 2DG and 20 mM mannose as indicated. Viability was monitored by Guava ViaCount. Data represent average viabilities ± S.D. (B) Cells were treated with 20 mM 2DG and/or 20 mM mannose for 6 h as indicated. mRNA levels of Gadd153 and Gadd34 are expressed relative to the levels in untreated samples. Data represent average mRNA levels ± S.D. from three independent samples. (C) Cells were treated with 20 mM 2DG, 20 mM mannose or 5 μM tunicamycin (TM; a positive control for ER stress) for 6 h as indicated. Splicing of Xbp1 mRNA was assessed by conventional RT-PCR, with spliced (S) and unspliced (Us) forms indicated. Replicate samples are shown. (D) Cells were treated with 20 mM 2DG and 20 mM mannose for 6 h as indicated. Protein levels were determined by Western blot as described in the methods section. ATP levels were measured as described in methods. Data represent average ATP levels ± S.D. (E) Cells were treated with 20 mM 2DG and 20 mM mannose for 6 h and 24 h as indicated. Protein levels were assessed by Western blot and ATP levels were measured as in (D).
We noticed that mannose conferred little protection against 2DG at longer time points (Fig. 7A). Consistent with this observation, while mannose strongly blocked GADD153 upregulation both early and late during 2DG exposure, it failed to suppress Bim upregulation at later time points in Bcl-2-overexpressing cells (which were used because of their lower sensitivity to longer 2DG treatment) (Fig. 7E). Mannose also had no effect on cellular ATP levels at either time point. These results suggest that 2DG affects Bim in a complex regulatory manner.
2DG induces toxicity in cell lines from B cell lymphomas that is enhanced by ABT-737 and inhibited by mannose
To determine whether other hematopoietic cell lines show similar responses to 2DG and ABT-737, two cell lines from Myc-induced B cell neoplasms were examined: iMyc Eμ-1 represents an immature lymphoblastic B cell lymphoma while iMycEμ-2 is a more mature plasmacytoma (Han et al., 2006). Both cell lines were sensitive to 2DG, and the sensitivity was enhanced by co-treatment with ABT-737. Furthermore, co-treatment with mannose protected from 2DG-induced killing (Fig. 8). WEHI-231 cells, a well characterized lymphoblastic B cell lymphoma line, were similarly sensitive to 2DG and protected by co-treatment with mannose (not shown). These findings demonstrate that B cell neoplasms are sensitive to 2DG and suggest that the cell death pathways involved in 2DG toxicity are similar to those in TCLs.
Figure 8. B cell lymphoma cell lines are sensitive to 2DG treatment.
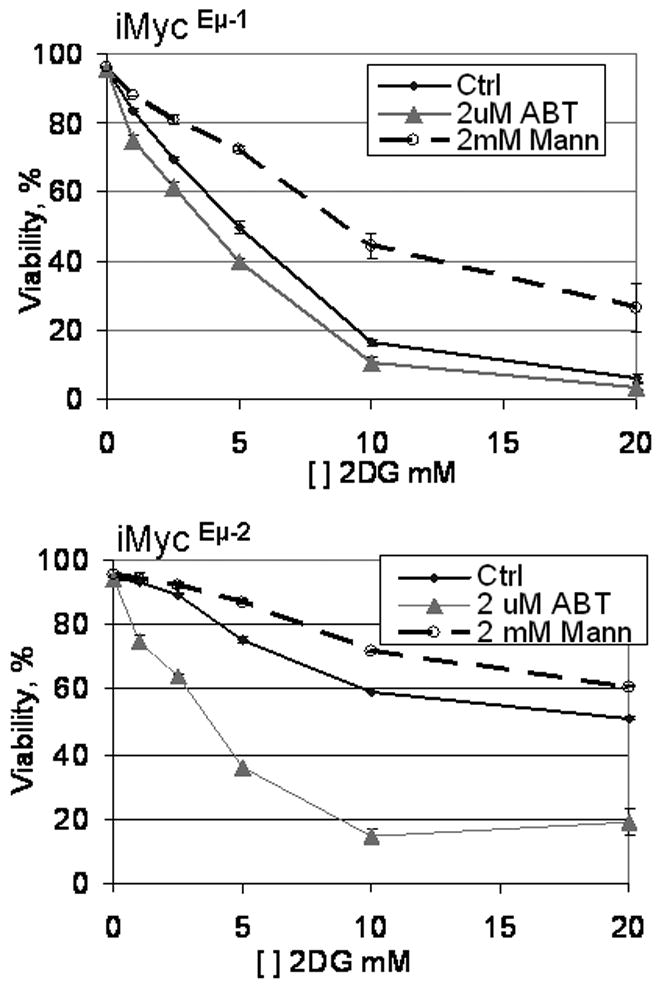
Mouse B cell lymphoma cell lines were treated with various concentrations of 2DG either alone (Ctrl) or in combination with 2 μM ABT-737 (ABT) or 2 mM mannose (Mann) as indicated. Viability was assessed by PI exclusion 18 h after the treatment.
Discussion
The property of cancer cells to utilize glycolysis more than normal cells was initially observed by Otto Warburg. This acquisition by cancer cells occurred probably as an adaptation to alter existing cell metabolism in a way that supports cell growth (Vander Heiden et al., 2009). 2DG is a well known synthetic glucose analog that inhibits glycolysis and interferes with protein folding. Despite numerous studies reporting the toxicity of 2DG in cancer cells, its molecular mechanisms remain obscure (El Mjiyad et al., 2011). In particular, the involvement of the Bcl-2 family proteins in 2DG-induced cell death has been underexplored. Understanding the involvement of Bcl-2 family in 2DG-caused toxicity could contribute to deciphering when 2DG could be applied clinically, since antiapoptotic Bcl-2 family genes are known to be elevated in cancers and affect treatment outcomes by increasing cell resistance to drugs (Letai et al., 2004). The need to understand the pathway of 2DG toxicity in cells is urgent, since 2DG is in Phase I clinical trials (www.clinicaltrials.gov).
Our findings indicate that 2DG toxicity in TCLs involves proapoptotic Bax activation, suggesting that the mitochondrial apoptotic pathway is involved in 2DG-induced killing. However, efficient inhibition of caspases, as demonstrated by lack of cleavage of caspase 3 and its target PARP, did not prevent or even delay cell death following 2DG treatment. This demonstrates that cell death is not dependent on the activation of caspases. Other studies have shown that death does not always depend on efficient caspase activation, and under these conditions death may be due to energy loss involving mitochondrial dysfunction (Cheng et al., 2001; Chipuk and Green, 2005). Our results demonstrating the dramatic decrease of ATP levels following 2DG treatment suggest a similar pathway may be involved in cell death in lymphoma cells; but the fact that manipulations capable of rescuing cells from 2DG-induced cell death (i.e., Bcl-2 overexpression and mannose) did not reverse the changes in ATP, argues that ATP depletion may regulate the toxicity of 2DG through alterations in the mitochondrial apoptotic pathway involving the Bcl-2 family.
Bcl-2, on the other hand, did show effective protection from 2DG treatment both by membrane integrity assays as well as clonogenic survival while having no immediate effect on preventing the depletion of ATP levels. Both Bax activation and Bcl-2 protection support the involvement of the Bcl-2 family proteins in 2DG toxicity in these cells. Since BH3-only family members act upstream of the multidomain Bcl-2 family proteins (Cheng et al., 2001; Huang and Strasser, 2000), we asked if ABT-737, a BH3 mimetic, would sensitize TCLs to 2DG treatment. Indeed, while not toxic when used alone, ABT-737 sensitized TCLs and Bcl-2 overexpressing TCLs to low doses of 2DG treatment, suggesting the sequestration of critical BH3-only members by anti-apoptotic Bcl-2 proteins. Interestingly, caspase inhibition did not significantly protect from cell death of TCLs despite effective inhibition of caspase 3 cleavage. This prompted us to examine whether the cell death required Bax/Bak or perhaps occurred by necrosis or autophagy, pathways that have previously been shown to be blocked by Bcl-2 (Kane et al., 1995; Liang et al., 1999; Saeki et al., 2000). 2DG-induced toxicity was shown to require Bax or Bak as Bax/Bak DKO cells were highly resistant to this treatment, wherein isogenic control cells expressing either Bax or Bak were sensitive to 2DG, ABT-737, and their combination. These results demonstrate that Bax and/or Bak are required for 2DG induced toxicity.
2DG treatment increased both Bim and Bmf levels, while Bid, Bad, Puma were unchanged. We also found that the levels of both Bim and Bmf that were associated with Bcl-2 were increased following 2DG treatment. Significantly, the interaction of Bcl-2 with either Bim or Bmf was disrupted by a BH3 mimetic, ABT-737, which sensitized the cells to 2DG toxicity. Most likely, the liberated Bim and Bmf acted on other anti-apoptotics and/or Bax/Bak to induce cell death. Given a more pronounced Bim upregulation in TCLs and previous studies showing that Bim deficiency protects lymphoid cells from cell death (Egle et al., 2004), we focused on studying the role of Bim in 2DG-caused toxicity. Indeed, downregulation of Bim promoted cell survival during 2DG treatment, indicating that Bim is an essential contributor to 2DG-induced cell death. Cells remained somewhat sensitive to high concentrations of 2DG, suggesting other BH3-only members may also participate. Since Bmf was upregulated and bound to Bcl-2 following 2DG treatment, we favor this as a likely contributor of the observed cell death.
Since both Bim and Bmf are activated following ER stress, we examined whether 2DG altered GADD153 levels in TCLs. GADD153 is a transcription factor known to be upregulated following ER stress and can directly activate transcription of Bim. Consistent with this model, we found that GADD153 protein levels were markedly induced following 2DG treatment and this induction was independent of Bcl-2 expression. Upregulation of GADD153 is most likely the consequence of ER stress and UPR activation by 2DG, as other measures of UPR activation were also detected, including induction of other target genes (Gadd34, Bip, Edem) and splicing of Xbp1 mRNA. That mannose blocked UPR activation, suppressed Bim upregulation, and inhibited 2DG-induced cell death suggests that 2DG-induced death occurs at least partly through the UPR-CHOP-Bim axis, particularly during early time points. However, when the ER stress response was alleviated by the addition of mannose, Bim upregulation was not prevented in TCL-Bcl-2 cells. This prompted us to speculate that at early time points, 2DG-induced Bim upregulation is ER stress/GADD153 dependent while prolonged 2DG treatment leads to marked upregulation of Bim that is induced differently. Since mannose effectively blocks GADD153 upregulation without altering the drop in ATP levels, we conclude that mannose selectively blocks 2DG-induced ER stress without blocking its effect on energy metabolism. Based on this, we propose the ER stress response is a rate limiting factor in 2DG-induced death and suggest that 2DG may act synergistically with other agents that may lead to ER stress. One such treatment reagent is bortezomib (Velcade), an FDA approved proteasome inhibitor known to induce ER stress (Dasmahapatra et al., 2009).
Our findings are important for several reasons. First, both 2DG and ABT-737 (or analogs) are in clinical trials, and additional information about how these reagents work may allow the development of a biochemical rationale for improving the efficacy of these agents in clinical applications. Second, combining 2DG and ABT-737 to induce cellular toxicity is a novel finding that could be utilized to sensitize otherwise drug resistant tumor cells. This effect could be particularly applicable for tumors with high levels of the anti-apoptotic Bcl-2 family members. Third, understanding the Bcl-2 member family involvement in 2DG-induced toxicity is an important contribution to deciphering the mechanism and improving the understanding of this long-studied reagent in cancer biology.
Materials and Methods
Cell culture
Five independently derived murine Lck-Bax38/1 thymic tumor cell samples (van de Wetering et al., 2007) were cultured in Advanced RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 g/mL streptomycin, and 250 μg/L Fungizone Amphotericin (Invitrogen) (complete medium). B cell lymphomas and hematopoietic lines doubly deficient in Bax and Bak were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 g/mL streptomycin, 50 μM 2-mercaptoethanol. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were cultured for up to 4 months and then another aliquot was thawed and utilized. No significant changes in the properties of the cells were observed over this time. Thymocytes were isolated from C57/BL6 mice and minced into a single cell suspension between two frosted glass slides. The erythrocytes were removed by 5-10 minute incubation at room temperature in hypotonic lysis buffer (10 mM Tris, 0.83% NH4Cl, pH 7.2). The cells were then pelleted and resuspended in complete medium as described above, and used for viability experiments.
Viability assays
Cell viability and cell count were determined utilizing a Guava PCA cell analyzer (Guava Technologies Inc.). Guava assay distinguishes between viable and non-viable cells based on the differential permeability of DNA-binding dyes in the ViaCount Reagent (Millipore). In brief, 10 μL of cells were added to 50 μL of Guava ViaCount reagent and allowed to stain for 5–20 min. Just prior to placement in the instrument, the cells were diluted with 40 μL of PBS and mixed, and the data were acquired. Experiments were performed in duplicate or triplicate, and the data represent the mean ± S.D. for each sample. For Propidium Iodide (PI) staining, cells were stained with 1 μg/mL PI in media and analyzed on a FACSCalibur flow cytometer.
Clonogenic Survival Assay
Clonogenic survival of TCLs was performed by plating cells by limiting dilution into 96-well plates. Briefly, cells were treated as indicated, pelleted and resuspended in culture medium. Total cells were counted with the Guava PCA system and then plated out at limiting dilution of 1-5 cells/well in 96-well round bottom plates. The number of positive wells was recorded after 9-10 days of growth. For each experiment samples were performed in triplicate and the data were normalized to the clonogenic survival of the untreated cells.
Activated Bax Staining by Flow Cytometry
Cells were pelleted and fixed in 0.25% paraformaldehyde in PBS for 5 min at room temperature. Following fixation, cells were washed with PBS and resuspended in 100 μL of blocking buffer (PBS with 0.01% digitonin, 5 μg/mL anti-FC, and 10% normal rat serum). Each sample was split in half and either stained with anti-Bax antibody (clone 6A7; eBioscience) or isotype control (clone P3; eBioscience) at 2.5 μg/sample. Cells were stained for 30 min at room temperature, washed, and stained with 1 μg/100 μL fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG antibody for 30 min. The cells were then washed with PBS, 0.01% digitonin prior to staining for DNA content with PI and RNase as previously described (Rathmell et al., 2003). Cells were acquired and analyzed on a BD FACSCalibur flow cytometer. Doublets and subdiploid cells were excluded in analysis. Gates were adjusted so that less than 1% of the cells were positive with the isotype control antibody.
Immunoprecipitation experiment
Cells were treated as indicated in each experiment and then collected and washed with cold washing solution (1 mM CaCl2, 1 mM MgCl2 in 1X PBS), resuspended in IP Lysis Buffer (10 mM HEPES, pH 7.4, 10 mM KCl, 0.1 mM EGTA) containing Complete Protease Inhibitor (Roche, 11873580001) and 1 mM PMSF. Cells were incubated 30 min on ice, after which NaCl (200 mM final) and CHAPS (1% final) were added. Samples were vortexed well, and the insoluble material was removed by centrifugation (12,000g × 15 min). The protein concentrations of the original lysates were determined using the DC Bradford assay. Protein A/G beads (50 μL/sample, Santa Cruz, sc-2003) were washed with IP Wash buffer (200 mM NaCl, 1% CHAPS in IP lysis buffer) and 120 μg of lysates were added to the beads followed by the addition of the Bcl-2 antibody (6C8, Pharmingen) (1 μg/sample) in a total volume of 100 μL. Samples were incubated overnight at 4°C on the 3D mixer. Original lysates were stored at -20°C for controls (1.2 μg/μL). The cleared supernatants were collected after pelleting the beads (2,000g × 5 min) and used to confirm complete immunoprecipitation of Bcl-2 in each sample. Beads were washed (5×) in IP Wash Buffer and resuspended with 40 μL of sample buffer containing SDS. Samples were boiled for 5 min and loaded onto Western blot gels with equivalent amounts of lysates and supernatant and the beads loaded at 1.5 to 2 times the equivalent volume.
Western Analysis
Cell pellets were lysed in RIPA buffer (1% NP40, 0.5% DOC, 0.1% SDS in PBS containing Complete Protease Inhibitor Cocktail (Sigma, P8340)), incubated on ice for 30 min, pelleted at 13,000 rpm at 4°C for 30 min. The protein measurements were determined by DC Bradford Protein Assay. Samples were brought to equal concentrations, mixed with the loading buffer containing 2-mercaptoethanol, boiled for 5min, loaded and separated on a gradient (4-20%) Tris–Glycine gel (Invitrogen) and transferred to a nitrocellulose membrane. The membrane was blocked for 1 h in a 5% milk/PBS Tween solution and incubated overnight at 4°C with a polyclonal antibody against the desired protein. The membrane was washed three times over 15 min in PBS Tween and then incubated for 1 h at room temperature with a secondary antibody. The membrane was again washed three times over 15 min in PBS Tween and then analyzed with ECL or ECL+ kit. The following antibodies were used: Bad and CHOP/GADD153 (Santa Cruz, sc-943, sc-575); Bcl-2 (Pharmingen, 15131S); β actin, Bim (Sigma, A4700, B7929); Bmf (Lifespan, LS-c4126); Bid (R&D, AF860); caspase 3, PARP, Puma (Cell Signaling, 9664, 9542, 4976); and GAPDH (AbCam Inc, ab9484). The quantitation of protein ratios was assessed by the Bioimaging Systems, Ultra-Violet Products (UVP).
Cell line transduction
TCL-Bcl-2 cells were generated using a HIV based lentiviral system with the pLA-CMV plasmid containing the cDNA for human Bcl-2 and puromycin resistance as previously described (Budanov et al., 2004). In brief, HEK 293T cells were transfected with both a 5 plasmid packaging mix (pTRE-GAG-PRO-PRE-polyA, pCMV-VpR-RT-IN-polyA, pCMV-VSV-G-polyA, pCMV-TEToff-polyA, and pCMV-TAT-REV) and the target plasmid pLACMV-Bcl-2. Briefly, the lentiviral packaging plasmids and the target plasmid were resuspended in EC buffer (Qiagen) and Enhacer (Qiagen), vortexed and incubated for 5 min at room temperature. Next, Effectene (Qiagen) was added to the plasmid mix, vortexed and incubated at room temperature for 20 min. This new plasmid mix was used to transfect 293T cells in fresh complete media. Every 12 to 18 h for 2.5 days following the initial transfection, virus particles were harvested from the 293T cells and the medium was filtered with a 0.22 μm syringe filter (Millipore), polybrene was added (4 μg/mL, Sigma, 107-689) and then added to the TCLs. Transduced TCLs were then selected with 1 μg/mL puromycin (Invitrogen) for 7-10 days in culture.
TCL shBIM cell lines were generated using microRNA-adapted retroviral system (Thermo Scientific) with the MSCV-LMP plasmid containing shRNAmir inserts and puromycin gene. The inserts were designed on RNAi-Central- RNAi Oligo Retriever (http://katahdin.mssm.edu/homepage/siRNA/RNAi.cgi?type=shRNA). Each of the four designed inserts (shBIM277-280) contained a ‘sense’ sequence of 22 nucleotides in length from the coding sequence of Bim (accession NM_207680.2): 5′-AGTTCTGAGTGTGACAGAGAAG-3′ (start position 252), 5′-CACCCTCAAATGGTTATCTTAC-3′ (start position 753), 5′-CACAAGGAGGGTGTTTGCAAAT-3′ (start position 710), 5′-CTCAGTGCAATGGCTTCCATAC-3′ (start position 597). The insertions were confirmed by sequencing, and positive clones were further used to generate shBIM cell lines. Briefly, Ψ-2 packaging plasmid and target plasmid were resuspended in calcium phosphate containing BES buffer as described previously (Chen and Okayama, 1987). The mix was incubated at room temperature for 15 min and used to transfect tsA201 cells. Viral particles were collected at 48 h and 72 h following transfection, pooled and polybrene (4 μg/mL) was added to enhance transduction. TCLs were transduced by spinfection at 700g for 2 h at room temperature. Following centrifugation, cells were pelleted and resuspended in fresh media. Cells were selected with puromycin as described above.
ATP measurements
Cells were plated in 96-well plates and treated under varying conditions. At indicated time points, cells were mixed and an aliquot was taken for both Guava Viability and ATP measurements. ATP levels were measured in triplicate utilizing ATP bioluminescent cell assay kit (Sigma-Aldrich, FLASC) as described by the manufacturer. ATP levels were normalized per viable cell and then normalized to untreated control sample being 100%. Bioluminescence was measured in white plates using a SpectraMax M5 microplate reader.
RNA analysis
Conventional RT-PCR for Xbp1, and quantitative RT-PCR for Chop, Bip, and Gadd34 was performed as described previously (Rutkowski et al., 2006) with the levels normalized to both Gapdh and Hprt. Briefly, total RNA was isolated by Trizol (Invitrogen). For qRT-PCR, 0.5 μg of RNA was programmed into a 10 ml cDNA synthesis reaction (iScript, BioRad), which was then diluted and used for subsequent qPCR analysis (iQ Superscript, BioRad). The specificity and efficiency of all primer pairs were exhaustively validated before use. Spliced and unspliced Xbp1 mRNA were detected using the Superscript III/Platinum Taq RT kit (Invitrogen) and primers flanking the 26 base intron. Data represent average mRNA levels from three independent samples.
Supplementary Material
Acknowledgments
This work was supported by RO1 #CA104695, RO1#CA133114, and R01 #DK084058. The authors would like to thank Dr. Chris van de Wetering for assistance with generating lymphoma cell lines. Dr. Siegfried Janz and Dr. SS Han for B cell neoplasms. Dr. Agshin Taghiyev and Dr. Van Tompkins for advice and assistance with the TCL transduction experiments. Dr. Craig Kuder and Dr. Ray Hohl for antibodies and assistance with ER stress markers. Heather Tyra for assistance with RT-PCR. Han Du for advice with the Bcl-2 immunoprecipitation experiments. Rebecca Glover and Jacob Wolf for technical advice on Western blotting.
Footnotes
Conflict of Interest: None of the authors have any financial interest or other conflicts of interest related to this work.
Supplementary information: Supplementary figures are available at Oncogene's website.
References
- Adams JM, Cory S. The bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol. 2005;6:268–275. doi: 10.1038/nrm1573. [DOI] [PubMed] [Google Scholar]
- Crane RK, Sols A. The non-competitive inhibition of brain hexokinase by glucose-6-phosphate and related compounds. J Biol Chem. 1954;210:597–606. [PubMed] [Google Scholar]
- Dasmahapatra G, Lembersky D, Rahmani M, Kramer L, Friedberg J, Fisher RI, et al. Bcl-2 antagonists interact synergistically with bortezomib in DLBCL cells in association with JNK activation and induction of ER stress. Cancer Biol Ther. 2009;8:808–819. doi: 10.4161/cbt.8.9.8131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Mjiyad N, Caro-Maldonado A, Ramirez-Peinado S, Munoz-Pinedo C. Sugar-free approaches to cancer cell killing. Oncogene. 2011;30:253–264. doi: 10.1038/onc.2010.466. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Han SS, Peng L, Chung ST, DuBois W, Maeng SH, Shaffer AL, et al. CDDO-imidazolide inhibits growth and survival of c-myc-induced mouse B cell and plasma cell neoplasms. Mol Cancer. 2006;5:22. doi: 10.1186/1476-4598-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Kane DJ, Ord T, Anton R, Bredesen DE. Expression of bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- Kang MH, Kang YH, Szymanska B, Wilczynska-Kalak U, Sheard MA, Harned TM, et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood. 2007;110:2057–2066. doi: 10.1182/blood-2007-03-080325. [DOI] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, et al. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007;6:3049–3058. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. J Chem Neuroanat. 2004;28:51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Raden D, Hildebrandt S, Xu P, Bell E, Doyle FJ, 3rd, Robinson AS. Analysis of cellular response to protein overexpression. Syst Biol (Stevenage) 2005;152:285–289. doi: 10.1049/ip-syb:20050048. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski, et al. PLoS Biology. 2006;4(11):e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki K, Yuo A, Okuma E, Yazaki Y, Susin SA, Kroemer G, et al. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000;7:1263–1269. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- Shoemaker AR, Oleksijew A, Bauch J, Belli BA, Borre T, Bruncko M, et al. A small-molecule inhibitor of bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006;66:8731–8739. doi: 10.1158/0008-5472.CAN-06-0367. [DOI] [PubMed] [Google Scholar]
- Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67:3364–3370. doi: 10.1158/0008-5472.CAN-06-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaili SS, Hsu YT, Carvalho AC, Rosenstock TR, Sharpe JC, Youle RJ. Mitochondria, calcium and pro-apoptotic proteins as mediators in cell death signaling. Braz J Med Biol Res. 2003;36:183–190. doi: 10.1590/s0100-879x2003000200004. [DOI] [PubMed] [Google Scholar]
- Tower DB. The effects of 2-deoxy-D-glucose on metabolism of slices of cerebral cortex incubated in vitro. J Neurochem. 1958;3:185–205. doi: 10.1111/j.1471-4159.1958.tb12625.x. [DOI] [PubMed] [Google Scholar]
- van de Wetering CI, Horne MC, Knudson CM. Chromosomal instability and supernumerary centrosomes represent precursor defects in a mouse model of T-cell lymphoma. Cancer Res. 2007;67:8081–8088. doi: 10.1158/0008-5472.CAN-07-1666. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Deslandes E, Villedieu M, Poulain L, Duval M, Gauduchon P, et al. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res. 2006;26:3561–3566. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.