

Abstract
Facile cycloisomerization of (2-ethynylphenyl)alkynes is proposed to be promoted synergistically by two molecules of BrettPhosAuNTf2, affording tricyclic indenes in mostly good yields. A gold vinylidene is most likely generated as one of the reaction intermediates based on both mechanistic studies and theoretical calculations. Different from the well-known Rh, Ru and W counterparts, this novel gold species is highly reactive and undergoes facile intramolecular C(sp3)-H insertions as well as O-H and N-H insertions. The formation step for the gold vinylidene is predicted theoretically to be complex with a bifurcated reaction pathway. The pyridine N-oxide acts as a weak base to facilitate the formation of an alkynylgold intermediate, and the bulky BrettPhos ligand in the gold catalyst likely plays a role in sterically steering the reaction toward the gold vinylidene formation.
Due to intense efforts by many research groups in the past decade, various initially novel aspects of homogenous gold catalysis1 have now been well accepted and increasingly applied in synthesis of complex structures.2 Further development of gold chemistry likely hinges on the discovery of novel gold intermediates and the application of their new reactivities.
Vinylidenes complexes3 of various metals such as Ru, Rh and W are versatile intermediates that promote the formation of various carbon-heteroatom and carbon-carbon bonds in a range of powerful catalytic reactions. Little is known, however, about gold vinylidenes. Several studies have invoked their intermediacy,4 but later calculations suggested alternative pathways.5 The only exception is the formation of gold β-halovinylidene intermediates via AuCl-promoted [1,2]-halo migration of terminally halogenated alkynes (i.e., haloalkynes),4d which was later supported by DFT calculations.6 For synthetic usefulness, terminal alkynes are ideal, simple starting materials to access metal vinylidenes. So far, no proven case of gold vinylidene formation from terminal alkynes has been reported. Herein we report a gold-catalyzed cycloisomerization of (2-ethynylphenyl)alkynes, where a gold vinylidene is the most likely intermediate based on both mechanistic studies and theoretical calculations; importantly, this novel gold intermediate, in contrast to its Ru, Rh or W counterparts, which often can be isolated,3 is highly reactive and undergoes facile C(sp3)-H insertion to form tricyclic indenes.
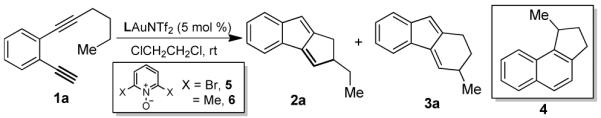
During our continuing study on gold-catalyzed intermolecular alkyne oxidation,7 we subjected diyne 1a to various combinations of gold catalysts and pyridine/quinoline N-oxides. While complex mixtures often resulted, a clean transformation was noticed when BrettPhosAuNTf27a,8 (5 mol %) and 2,6-dibromopyridine N-oxide (5, 1.1 equiv) was used. To our surprise, the oxidant 5 was not consumed at all and the major product (92% NMR yield) was a hydrocarbon whose structure was assigned as 1,2-dihydrocyclopenta[a]indene 2a (Table 1, entry 1).9 Notably, this reaction involved an apparent C(sp3)-H insertion that leads to a tricyclic structural motif found in natural products such as pallidol10 and sporolides11 and patented compounds of medical interest.12 A minor product, also involving C-H insertion, was assigned as the 6-membered structural isomer 3a.
Table 1.
Optimization of reaction conditions.a
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | additive | time | yieldb | 2a/3a |
| 1 | BrettPhosAuNTf2 | 5 (1.1 equiv) | 2 h | 92% c | 21/1 |
| 2 | BrettPhosAuNTf2 | 5 (0.5 equiv) | 4 h | 82% | 21/1 |
| 3 | BrettPhosAuNTf2 | 5 (0.2 equiv) | 10 h | 66% | 21/1 |
| 4 | BrettPhosAuNTf2 | - | 24 h | 64% | 21/1 |
| 5 | BrettPhosAuNTf2 | 6 (1.1 equiv) | 4 h | 91% | 21/1 |
| 6 | BrettPhosAuNTf2 | 6 (0.5 equiv) | 6 h | 88% | 21/1 |
| 7 | BrettPhosAuNTf2 | 6 (0.2 equiv) | 24 h | 88% | 21/1 |
| 8 | BrettPhosAuNTf2 | 2,6-Br2Py (0.5 equiv) | 24 h | 70% | 20/1 |
| 9 | BrettPhosAuNTf2 | lutidine (0.5 equiv) | 48 h | 15% (75% 1a) | 20/1 |
| 10 | BrettPhosAuNTf2 | TsNa (0.5 equiv) | 12 h | 79% | 21/1 |
| 11 | Cy-JohnPhos | 6 (0.5 equiv) | 48 h | 55% (20% 1a) | 10/1 |
| 12d | IPrAuNTf2 | 6 (0.5 equiv) | 24 h | 35% (40% 1a) | 3/1 |
| 13 | Ph3PAuNTf2 | 6 (0.5 equiv) | 48 h | 3% (92% 1a) | - |
| 14 | Et3PAuNTf2 | 6 (0.5 equiv) | 20 h | 9% (84% 1a) | - |
| 15 | PtCl2 | toluene, 100 °C | 10 h | 45% of 4 | - |
Reaction run in vials using regular DCE as the solvent; [1a] = 0.1 M.
Measured by 1H NMR using diethyl phthalate as the internal standard.
90% Isolated yield.
Reaction temperature: 60 °C.
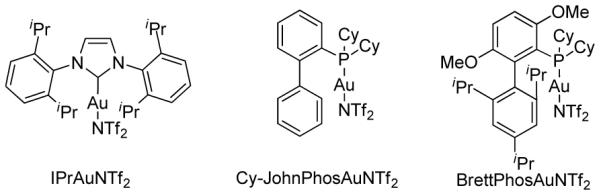
We were rather curious about the role of 5 and first probed the impact of its quantity on the reaction outcome. As shown in entries 1-4, Table 1, the reaction became slower and less efficient as the amount of the N-oxide was decreased. Lutidine N-oxide (6) also worked well as an additive, though the reactions were a bit slower (entries 5-7). We suspected that the N-oxides might simply function as bases. Indeed, 2,6-dibromopyridine offered a small improvement to the reaction (comparing entries 8 and 4) albeit lutidine was deleterious (entry 9), perhaps due to its strong basicity (pKa of its conjugated acid: 6.77). Sodium toluenesulfinate, with a moderate basicity (pKa of its conjugate acid: 1.9913), led to an acceptable reaction rate and 79% yield (entry 10). Subsequent catalyst screening (entries 11-14) revealed that bulky BrettPhos was uniquely effective as the Au(I) ligand, rendering both a high yield and excellent chemoselectivity (i.e., 2a/3a). Contrary to BrettPhosAuNTf2, PtCl2 promoted selective formation of 4 (entry 15), consistent with Liu’s previously published results.14
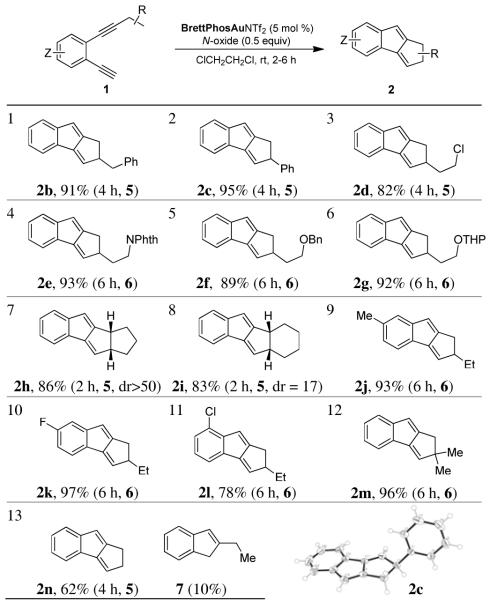
The reaction scope for this rapid construction of 1,2-dihydrocyclopenta[a]indenes was then studied. As shown in Table 2, the aliphatic substituent at the internal alkyne tolerated a range of functional groups including phenyl (entries 1 and 2), chloro (entry 3), protected amino (entry 4) and hydroxy (entries 5 and 6). Moreover, cycloalkyl groups such as cyclopentyl (entry 7) and cyclohexyl (entry 8) could be the alkyne substituent, leading to tetracyclic products. Substitutions on the benzene ring (entries 9-11) at different positions were readily allowed. In addition to efficient insertion into methylene C-H bonds, the insertion into a methine C-H proceeded equally well (entry 12). Insertion into a methyl C-H was less efficient (entry 12) and a small amount of 2-ethyl-1H-indene (i.e., 7) was isolated in addition to 2n. X-ray diffraction studies of product 2c confirmed our structural assignments.
Table 2.
|
Reactions run in vials; isolated yields are reported.
The 6-membered ring isomer 3 is <5%.
To probe the reaction mechanism, we performed deuterium labeling studies. The results shown in Eq. 1 indicated that the hydrogen isotope at the C-8 of 2a came mostly from the added
 |
(1) |
 |
(2) |
H2O or D2O and little from the alkyne terminus in the substrate. By labeling the aliphatic side chain with deuteriums (Eq. 2), the origin of the C-3 hydrogen/deuterium in the products was firmly established and is consistent with the formation of the C-2 and C-3 bond via an intramolecular carbene C-H insertion.
 |
(3) |
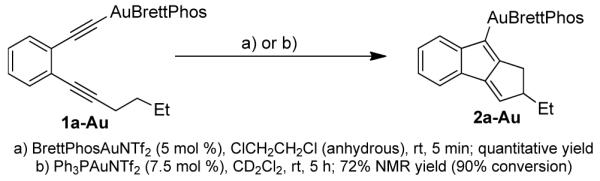
Equation 1 further suggested that the C-8 hydrogen in 2a might be installed via protodeauration, and the alkyne terminal hydrogen in 1a might become labile in the course of the reaction. Using phenylacetylene as the model alkyne, we indeed observed facile exchange of its alkyne terminal hydrogen with added D2O (10 equiv) in CDCl3 in the presence of BrettPhosAuNTf2 and N-oxide 6 (50% conversion in 5 min); an alkynylgold is the most likely intermediate.15 Importantly, without 6, the exchange was much slower (50% conversion after 1.5 h). This result suggests that 5 or 6 acts as a base to facilitate the removal of the alkyne terminal hydrogen to form alkynylgold 1a-Au (see Eq. 3). Fortunately, this intermediate could be prepared in its pure form. When we treated 1a-Au with 5 mol % of BrettPhosAuNTf2, the reaction proceeded to completion in 5 min, and 8-aurated-1,2-dihydrocyclopenta[a]indene 2a-Au was formed in a quantitative yield (Eq. 3). To confirm the uniuqely effective nature of BrettPhosAuNTf2, Ph3PAuNTf2 was used as the catalyst instead, and the reaction was much slower and less efficient.16 The use of Ph3P to replace the BrettPhos ligand in 1a-Au with either BrettPhosAuNTf2 or Ph3PAuNTf2 as the catalyst led to even slower reactions and little desired products.16
A mechanism consistent with the above results is proposed in Scheme 1. Firstly, the N-oxide acts as a base to abstract the proton from the terminal alkyne, which is activated by BrettPhosAu+. This affords the alkynylgold complex 1-Au. The reverse process, i.e., protodeauration, may occur, but the presence of a base should help shift the equilibrium toward 1-Au. A strong base such as lutidine, however, would bind to the gold catalyst and therefore hinder the overall reaction. Another cationic gold(I) complex can then activate the other C-C triple bond of 1-Au via the intermediacy of 1-Au2, which undergoes a 5-endo-dig cyclization that we believe leads to the gold vinylidene intermediate I.17 This novel species is apparently highly reactive and may undergo a facile C-H insertion,18 leading to 2-Au, followed by protodeauration to form the observed product. The formation of 6-membered structural isomer (e.g., 3a, Table 1) could be readily explained by a competing 1,6-C-H insertion by the gold vinylidene. A likely competing 6-endo-dig cyclization by 1-Au2 could lead to the intermediate II, which is somewhat analogous to that leading to 4 with platinum catalysis;14 however, no naphthalenic products were observed.
Scheme 1.
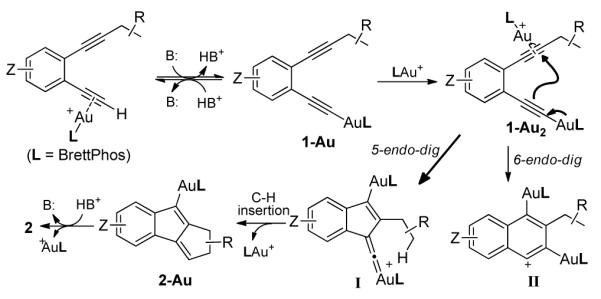
Proposed reaction mechanism
The preference for the 5-endo-dig cyclization over the 6-endo-dig cyclization might be attributed on one hand to the bulky19 BrettPhosAu moiety at the alkyne end as its bulk might help steer the approach of the other C-C triple bond to the less hindered distal alkyne end, and on the other hand to back bonding of the Au center to the adjacent carbocation in the form of a gold vinylidene (i.e., I).
Density functional theory (DFT) calculations of the reaction energy surface of a model reaction (Scheme 2) at the M06/6-31+G(d,p)/LANL2DZ(Au)(SMD) in CHCl3 level have shown that this is a reasonable mechanism, supporting the hypothesis that a gold vinylidene intermediate would be stable enough to form, yet reactive enough to readily carry out the required C-H insertion reaction. The energetics for the expected proton abstraction by pyridine N-oxides is favorable (uphill by only 2.2 kcal/mol in chloroform, see Supporting Information) and the 5-endo-dig ring closure was found to occur with a free-energy barrier in chloroform of only 14.7 kcal/mol in single-point calculations at the M06/aug-cc-pVTZ/aug-cc-pVTZ-PP(Au)(SMD) level. This reaction step forms the novel gold vinylidene intermediate B, with a predicted ΔG°CHCl3 of 6.9 kcal/mol from A. The calculated bond lengths and charges (see Supporting Information) clearly support the vinylidene character of B. This intermediate is highly reactive and can rapidly undergo an intramolecular C-H insertion reaction with a calculated free-energy barrier of 9.1 kcal/mol for primary C-H bonds to form the tricyclic intermediate D. The corresponding energy barrier for insertion into the secondary C-H bonds of a pendent n-propyl group (see SI) is calculated to be 4.9 kcal/mol. The observed product is formed from D following sequential decomplexation and protodeauration steps.
Scheme 2.
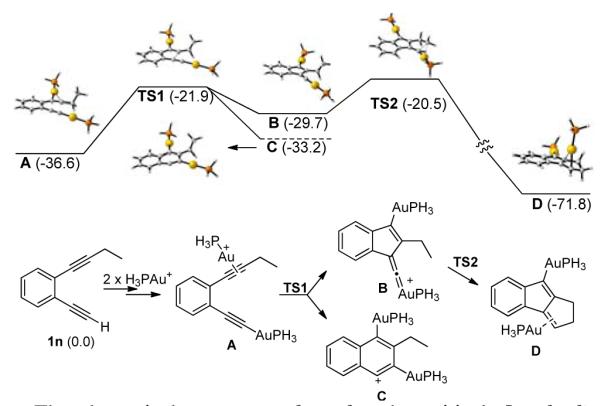
Free energy diagram calculated in chloroform at the M06/aug-cc-pVTZ-PP(SMD)//M06/6-31+G(d,p)/LANL2DZ(Au) level. All energies relative to 1n in kcal/mol at 298 K.
The theoretical energy surface for the critical 5-endo-dig cyclization step to B shows a most interesting situation in which the alternative 6-endo-dig cyclization to form C actually forms from the same transition state TS1. This constitutes a bifurcated reaction pathway with a ridge-valley inflection point on a relatively flat energy surface, as shown in Figure 1. A second transition state structure TS1a, 0.89 kcal/mol lower in energy than the first, was found for interconversion of the 5- and 6-membered ring intermediates B and C. In situations of this sort, the choice between B and C is made on the basis of dynamical factors along the ridge (between TS1 and TS1a) separating the valleys for B and C, and product ratios must be predicted by dynamical effects from trajectory calculations, rather than classical transition state theory.20,21 Preliminary atom-centered density matrix propagation (ADMP) molecular dynamics calculations for a simplified model reaction with PH3 ligands, a methyl substituent, and no benzo fusion give reaction trajectories starting from the first transition state that go to both the 5-membered and 6-membered ring products within 150-300 fs with no intermediate formation. The trajectories predict that the 5-membered ring product will prevail, as observed, but not overwhelmingly. It was expected that the stronger selectivity toward the 5-endo-dig cyclization observed experimentally might be attributed to the steric bulk of one of the BrettPhos ligands. The bulky BrettPhos ligand on the gold vinylidene moiety in B is held a C=C bond length further away from the ring and pendant ethyl group than in C. This steric preference for B with a BrettPhos ligand was confirmed with calculations on structures optimized at the B3LYP/6-31G(d)/LANL2DZ(Au) level that showed a steric shift in free energy differences toward the five-membered ring intermediate of 5.1 kcal/mol.22 The calculation was performed on a model with the full BrettPhos structure for the relavent ligand and PMe3 for the other ligand. The conformation chosen for the BrettPhos ligand was the same as that found in the crystal structure of BrettPhosAuNTf2.7a Interestingly, such bifurcation pathways are still very rare in organometallic chemistry, but have recently been seen for gold-catalyzed reactions.23
Figure 1.
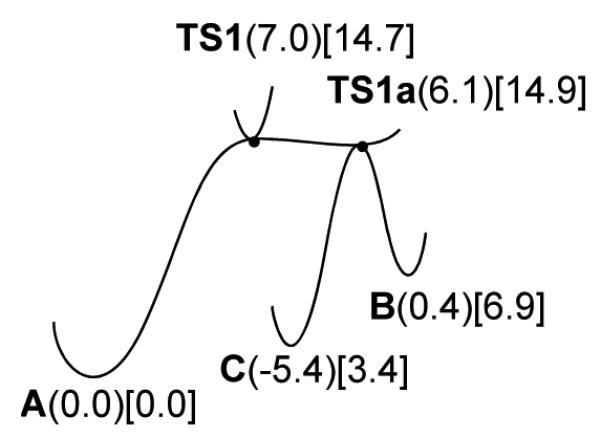
Energy diagram showing a bifurcation at a ridge-valley inflection, calculated at the M06/aug-cc-pVTZ-PP(SMD)//M06/6-31+G(d,p)/LANL2DZ(Au) level. Relative electronic energies in parentheses, relative free energies in chloroform in brackets, all in kcal/mol at 298 K. For pictures of typical 3D surfaces see Reference 20.
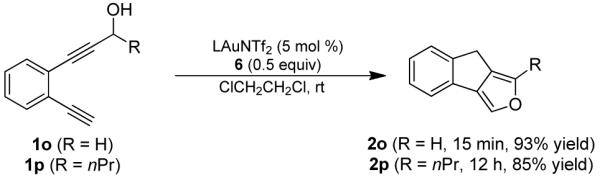
The gold vinylidene intermediate of type I was also found to readily insert into O-H (Eq. 4) and N-H bonds (Eq. 5), that led to highly efficient formation of useful furan- and pyrrole-fused tricyclic structures, respectively. Interestingly, when there was a competition between O-H and C-H bonds (i.e, in the case of 1p in Eq. 4), the gold vinylidene preferred the O-H bond, and no C-H insertion product was found.
 |
(4) |
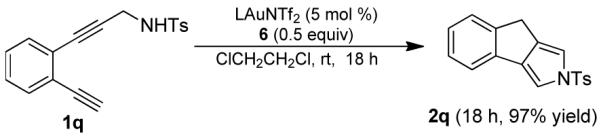 |
(5) |
In summary, we report a gold-catalyzed cycloisomerization of benzene-1,2-dialkynes, where two molecules of gold catalysts are proposed to work synergistically. A gold vinylidene intermediate is most likely generated based on both mechanistic studies and theoretical calculations. Importantly, this novel gold species, different from the well-known Rh, Ru and W counterparts, is highly reactive and undergoes facile intramolecular C(sp3)-H insertion to form tricyclic indenes. Insertions into O-H and N-H bonds are also highly efficient. The formation step for the gold vinylidene is predicted theoretically to be complex with a bifurcated reaction pathway. The pyridine N-oxides act as weak bases to facilitate the formation of alkynylgold intermediates, and the bulky BrettPhosAu is believed to play a role in sterically steering the reaction toward the gold vinylidene formation. This strategy for the generation of novel gold vinylidenes will be pursued further in our laboratory.
Supplementary Material
Acknowledgment
We are grateful for the generous financial support by NIH (R01 GM084254), the National Center for Supercomputing Applications, NSF (CHE100123) utilizing the NCSA Ember system, and UCSB and the kind donation of BrettPhos by Sigma-Aldrich.
Footnotes
Supporting Information Available: Experimental procedures, compound characterization data and computational details are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).For selected reviews, see: Corma A, Leyva-Pérez A, Sabater MJ. Chem. Rev. 2011;111:1657–1712. doi: 10.1021/cr100414u. Wang S, Zhang G, Zhang L. Synlett. 2010;2010:692–706. Abu Sohel SM, Liu R-S. Chem. Soc. Rev. 2009;38:2269–2281. doi: 10.1039/b807499m. Patil NT, Yamamoto Y. Chem. Rev. 2008;108:3395–3442. doi: 10.1021/cr050041j. Li Z, Brouwer C, He C. Chem. Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l. Gorin DJ, Sherry BD, Toste FD. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. Arcadi A. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d. Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. Fürstner A, Davis PW. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335.
- (2).(a) Hashmi ASK, Rudolph M. Chem. Soc. Rev. 2008;37:1766–1775. doi: 10.1039/b615629k. [DOI] [PubMed] [Google Scholar]; (b) Furstner A. Chem. Soc. Rev. 2009;38:3208–3221. doi: 10.1039/b816696j. [DOI] [PubMed] [Google Scholar]
- (3).(a) Bruneau C, Dixneuf PH, editors. Metal Vinylidenes and Allenylidenes in Catalysis from Reactivity to Applications in Synthesis. Weinheim; Wiley-VCH: 2008. [Google Scholar]; (b) Lynam JM. Chem. Eur. J. 2010;16:8238–8247. doi: 10.1002/chem.201000695. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, McClory A. Chem. Asian J. 2008;3:164–194. doi: 10.1002/asia.200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bruneau C, Dixneuf PH. Angew. Chem., Int. Ed. 2006;45:2176–2203. doi: 10.1002/anie.200501391. [DOI] [PubMed] [Google Scholar]; (e) Varela JA, Saá C. Chem. Eur. J. 2006;12:6450–6456. doi: 10.1002/chem.200600388. [DOI] [PubMed] [Google Scholar]; (f) McDonald FE. Chem. Eur. J. 1999;5:3103–3106. [Google Scholar]; (g) Bruneau C, Dixneuf PH. Acc. Chem. Res. 1998;32:311–323. [Google Scholar]; (h) Bruce MI. Chem. Rev. 1991;91:197–257. [Google Scholar]
- (4).(a) Seregin IV, Schammel AW, Gevorgyan V. Tetrahedron. 2008;64:6876–6883. doi: 10.1016/j.tet.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lavallo V, Frey GD, Kousar S, Donnadieu B, Bertrand G. Proc. Nat. Acad. Sci. 2007;104:13569–13573. doi: 10.1073/pnas.0705809104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seregin IV, Gevorgyan V. J. Am. Chem. Soc. 2006;128:12050–12051. doi: 10.1021/ja063278l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mamane V, Hannen P, Fürstner A. Chem. Eur. J. 2004;10:4556–4575. doi: 10.1002/chem.200400220. [DOI] [PubMed] [Google Scholar]
- (5).(a) Xia Y, Dudnik AS, Li Y, Gevorgyan V. Org. Lett. 2010;12:5538–5541. doi: 10.1021/ol1024794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rabaâ H, Engels B, Hupp T, Hashmi ASK. Int. J. Quantum Chem. 2007;107:359–365. [Google Scholar]
- (6).Soriano E, Marco-Contelles J. Organometallics. 2006;25:4542–4553. [Google Scholar]
- (7).(a) Ye L, He W, Zhang L. Angew. Chem., Int. Ed. 2011;50:3236–3239. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He W, Li C, Zhang L. J. Am. Chem. Soc. 2011:8482–8485. doi: 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]; (c) Ye L, He W, Zhang L. J. Am. Chem. Soc. 2010;132:8550–8551. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lu B, Li C, Zhang L. J. Am. Chem. Soc. 2010;132:14070–14072. doi: 10.1021/ja1072614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fors BP, Watson DA, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2008;130:13552–13554. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Lian JJ, Chen PC, Lin YP, Ting HC, Liu RS. J. Am. Chem. Soc. 2006;128:11372–11373. doi: 10.1021/ja0643826. [DOI] [PubMed] [Google Scholar]; (b) Lin GY, Yang CY, Liu RS. J. Org. Chem. 2007;72:6753–6757. doi: 10.1021/jo0707939. [DOI] [PubMed] [Google Scholar]
- (10).Bavaresco L, Vezzulli S. Rec. Prog. Med. Plants. 2006;11:389–410. [Google Scholar]
- (11).Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2005;7:2731–2734. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]
- (12).(a) Jakubowski JA, Utterback BG, Mais DE, Hardinger SA, Braish TF, Richard Nevill C, Fuchs PL. Prostaglandins. 1994;47:189–201. doi: 10.1016/0090-6980(94)90060-4. [DOI] [PubMed] [Google Scholar]; (b) Tomyama T, Ikegami S, Hashimoto S, Imamaki T. Jpn. Kokai Tokkyo Koho. 1995 JP 07076550 A 19950320. [Google Scholar]; (c) Ratilainen J, Huhtala P, Karjalainen A, Karjalainen A, Haapalinna A, Virtanen R, Lehtimaeki J. PCT Int. Appl. 2001 WO2001085698 A1 20011115. [Google Scholar]
- (13).Burkhard RK, Sellers DE, DeCou F, Lambert JL. J. Org. Chem. 1959;24:767–769. [Google Scholar]
- (14).Taduri BP, Ran Y-F, Huang C-W, Liu R-S. Org. Lett. 2006;8:883–886. doi: 10.1021/ol052962m. [DOI] [PubMed] [Google Scholar]
- (15).Cheong PHY, Morganelli P, Luzung MR, Houk KN, Toste FD. J. Am. Chem. Soc. 2008;130:4517–4526. doi: 10.1021/ja711058f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).For details, please see the Supporting Information.
- (17).Chen K-H, Feng YJ, Ma H-W, Lin Y-C, Liu Y-H, Kuo T-S. Organometallics. 2010;29:6829–6836. [Google Scholar]
- (18).(a) Bhunia S, Liu R-S. J. Am. Chem. Soc. 2008;130:16488–16489. doi: 10.1021/ja807384a. [DOI] [PubMed] [Google Scholar]; (b) Horino Y, Yamamoto T, Ueda K, Kuroda S, Toste FD. J. Am. Chem. Soc. 2009;131:2809–2811. doi: 10.1021/ja808780r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lemiere G, Gandon V, Cariou K, Hours A, Fukuyama T, Dhimane AL, Fensterbank L, Malacria M. J. Am. Chem. Soc. 2009;131:2993–3006. doi: 10.1021/ja808872u. [DOI] [PubMed] [Google Scholar]
- (19).Benitez D, Tkatchouk E, Gonzalez AZ, Goddard WA, Toste FD. Org. Lett. 2009;11:4798–4801. doi: 10.1021/ol9018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ess DH, Wheeler SE, Iafe RG, Xu L, Çelebi-Ölçüm N, Houk KN. Angew. Chem., Int. Ed. 2008;47:7592–7601. doi: 10.1002/anie.200800918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).While Scheme 2 suggests a possibility that B and C might equilibrate through TS1a, this equilbration is calculated to be somewhat slower than the competing C-H insertion reaction when the C-H bond is secondary. The possible fate of any C formed in the reaction was predicted computationally. It was found to undergo an exothermic gold migration and C-H insertion with a barrier as low as that for B and faster than equilibration of B and C. This insertion product would lead to 2,3-cyclopentano-fused naphthalenes, rather than the 1,2-fusion seen in 4 from platinum. See Supplementary Information for details.
- (22).To see whether steric effects in the products B and C with BrettPhos ligands would be felt along the reaction surface in the vicinity of the bifurcation points, calculations at the B3LYP/SDD level were carried out. Geometry optimizations with these lower-level calculations show separate transition states for formation of the 5- and 6-membred ring products instead of the ridge-valley inflection and bifurcation surface for higher-level calculations, e. g. M06/6-31G(d)/LANLDZ(Au) and M06/6-31+G(d,p)/LANLDZ(Au). Using these two transition-state geometries for the core structures, partial geometry optimizations with the trimethylphosphine and BrettPhos ligands showed about a 2-3 kcal/mol estimated free energy difference favoring the five-membered ring product, again using a comparable pair of conformations from the crystal structure of BrettPhosAuNTf2. While it seems reasonable that the steric effects evident in the B and C intermediates would be felt along the reaction trajectories and that the product ratio would shift further toward B as a result, such substituent effects may not be readily predictable in mechanisms involving bifurcation pathways, and we are working on this general problem currently. See Supporting Information for details
- (23).(a) Noey EL, Wang X, Houk KN. J. Org. Chem. 2011;76:3477–3483. doi: 10.1021/jo200556f. [DOI] [PubMed] [Google Scholar]; (b) Garayalde D, Gómez-Bengoa E, Huang X, Goeke A, Nevado C. J. Am. Chem. Soc. 2010;132:4720–4730. doi: 10.1021/ja909013j. [DOI] [PubMed] [Google Scholar]; (c) Wang ZJ, Benitez D, Tkatchouk E, Goddard WA, III, Toste FD. J. Am. Chem. Soc. 2010;132:13064–13071. doi: 10.1021/ja105530q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.