

Abstract
Vigabatrin, a GABA aminotransferase (GABA-AT) inactivator, is used to treat infantile spasms and refractory complex partial seizures and is in clinical trials to treat addiction. We evaluated a novel GABA-AT inactivator (CPP-115) and observed that it does not exhibit other GABAergic or off-target activities and is rapidly and completely orally absorbed and eliminated. Using in vivo microdialysis techniques in freely moving rats and micro-PET imaging techniques, CPP-115 produced similar inhibition of cocaine-induced increases in extracellular dopamine and in synaptic dopamine in the nucleus accumbens at 1/300–1/600th the dose of vigabatrin. It also blocks expression of cocaine-induced conditioned place preference at a dose 1/300th that of vigabatrin. Electroretinographic (ERG) responses in rats treated with CPP-115, at doses 20–40 times higher than those needed to treat addiction in rats, exhibited reductions in ERG responses, which were less than the reductions observed in rats treated with vigabatrin at the same dose needed to treat addiction in rats. In conclusion, CPP-115 can be administered at significantly lower doses than vigabatrin, which suggests a potential new treatment for addiction with a significantly reduced risk of visual field defects.
Keywords: GABA aminotransferase, Enzyme inactivator, Addiction, Cocaine, Visual field defect, Pharmacokinetics, Micro-PET imaging, Conditioned place preference
The neurochemical response to cocaine and other drugs of abuse is well characterized by a rapid elevation in the release of dopamine in the nucleus accumbens (NAc).1 This increase in dopamine, and associated behaviors, can be antagonized by an increase in the concentration of γ-aminobutyric acid (GABA), which has been shown to occur with use of the FDA-approved epilepsy drug vigabatrin (also known as CPP-1091), a known mechanism-based inactivator2,3 of γ-aminobutyric acid aminotransferase (GABA-AT).4 Vigabatrin is currently marketed for the treatment of infantile spasms (West’s syndrome) and refractory complex partial seizures.
Vigabatrin has been used in the treatment of stimulant addiction,5,6 specifically in animal models for cocaine,1 nicotine,7 methamphetamine, heroin, ethanol,8 and combination addictions.9 In human studies, vigabatrin treatment is effective for stimulant addiction,10,11 including a recently reported randomized, double-blind, placebo-controlled, trial of 103 subjects,12 in which 28% of subjects treated with vigabatrin achieved abstinence compared to 7.5% of subjects treated with a placebo.
The acceptance of vigabatrin for the treatment of both epilepsy and as a potential treatment for stimulant addiction has been hampered primarily by concerns about abnormalities of the peripheral visual field (visual field defects or VFDs) in 25–50% of patients following chronic administration of vigabatrin.13,14 As a result, an unmet medical need remains for a GABA-AT inhibitor with improved potency and reduced, or eliminated, visual field defects for the treatment of both addiction and epilepsy.
The cellular mechanisms leading to the visual field defects are not known; however, it remains an active area of research. Visual field defects could be from (1) a direct toxic effect of vigabatrin, (2) elevated GABA levels, (3) inactivation of GABA-AT, (4) the consequence of an enzymatically produced byproduct from one of the enzyme inactivation mechanisms, (5) off-target activities, or (6) a combination of these potential modes of action. In albino rats acute vigabatrin exposure damages the outer retina by a GABA-independent and vigabatrin-specific mechanism, resulting in sensitization of photoreceptors to light-induced damage.15 In this case, it is probable that reactive oxygen species are involved since they participate in light-mediated retinal toxicity.16 Recently, the visual field defects in an animal model were reported to result from drug related toxic effects that lead to a reduction in taurine, an essential amino acid related to retinal function.17
A new synthetic compound, (1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-1151), was designed as a mechanism-based inactivator of GABA-AT, which could
generate a more reactive intermediate along the pathway to attachment to the active site of GABA-AT via a Michael addition.18 In contrast to the high KI value reported for vigabatrin as an inactivator of GABA-AT (3.2 mM18 or 10 mM4), CPP-115 has a KI value (at suboptimal pH) of 31 μM.18 A comparison of the kinact/KI values, a measure of the efficiency of the inactivator, indicated that CPP-115 is 187 times more effective as an inactivator of GABA-AT than vigabatrin under suboptimal conditions (at optimal conditions for substrate turnover the rate of inactivation is too rapid to measure; these values were obtained at a pH and temperature well below the optimum). Despite irreversibility of the inhibition, the low potency and poor blood-brain barrier penetration of vigabatrin translates into treatment doses of 1–3 g/day.19 Because CPP-115 displayed superior enzyme inactivation properties compared to vigabatrin, we have carried out pharmacological and preclinical studies with CPP-115. Because of the preponderance of data indicating vigabatrin is effective for the treatment of addiction, the effect of CPP-115 was investigated on cocaine-induced conditioned place preference in rats, an established animal model for effectiveness of addiction treatments. We also used microPET imaging to measure the ability of CPP-115 to antagonize cocaine-induced increases in synaptic nucleus accumbens dopamine. Because of the higher potency of CPP-115 relative to vigabatrin, we hypothesized that CPP-115 would exhibit less retinal toxicity by either reducing the formation of toxic enzyme inactivation by-products or by a reduction of direct toxic effects of the drug as a result of the lower dose needed to inactivate GABA-AT. Therefore, we used electroretinography (ERG) in an animal model to evaluate the retinal toxicity potential of CPP-115 compared to vigabatrin.17,20
Results
Effects of (1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) at GABA transporters and receptors
Interaction of CPP-115 with GABA transporters
CPP-115 displayed no inhibitory activity at 1 mM concentration at each of the four human or mouse GABA transporter subtypes, in neurons, astrocytes, or mammalian cells recombinantly expressing human or mouse transporter subtypes (see Supporting Information Table 1).21
Interaction of CPP-115 with GABA receptors
To investigate a possible interaction of CPP-115 with GABA receptors, we tested the compound for its ability to displace [3H]GABA binding to ionotropic GABAA receptors or metabotropic GABAB receptors in rat brain cortical homogenates.22 At a concentration of 100 μM, no inhibition of binding was observed at either receptor tested, whereas 1 μM cold GABA inhibited radioligand binding as expected (see Supporting Information Table 2). Additionally, we tested CPP-115 for activity at recombinant human ρ1 GABAC receptors expressed in oocytes and was found to exhibit no effect as an agonist or antagonist at a concentration of 100 μM (see Supporting Information Figure 1).
Effects of (1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) at non-GABAergic off targets
Off-target tests of 111 different biological targets were carried out at Cerep (Poitiers, France). There was no significant effect of CPP-115 on any of these off targets. See Supporting Information Tables 3 and 4 for the summary of testing. There also was no in vitro effect on aspartate aminotransferase, alanine aminotransferase, or succinic semialdehyde dehydrogenase, unlike vigabatrin, which is known to reduce activities of alanine aminotransferase23,24 and succinic semialdehyde dehydrogenase25 in vivo. CPP-115 also did not inhibit or induce CYP1A2, CYP2B6 and CYP3A4/5 or inhibit CYP2C8, CYP2C9, CYP2D6, which are the more common human liver microsomal cytochrome P450 enzymes (100 μM). The in vitro effects of CPP-115 (300 μM) on the human ether-à-go-go-related gene (hERG) channel current at near-physiological temperature was determined. CPP-115 inhibited hERG current by (mean ± SEM) 1.1 ± 0.2% at 10 μM (n = 3) and 1.5 ± 0.2% at 300 μM (n = 3) versus 0.8 ± 0.3% (n = 3) in control. At both test concentrations hERG inhibition was not statistically significant (P < 0.05) when compared to vehicle control values, indicating a minimal risk for CPP-115 induced cardiac arrhythmias.
Pharmacokinetics and metabolic stability of CPP-115
CPP-115 is rapidly (1.7 h−1 for rats and 2.3 h−1 for beagle dogs) and completely (79% for rats and >100% for dogs) orally absorbed, and rapidly eliminated (T1/2 is 1 h for rats and 2.3 h for dogs). Rats received a 10 mg/kg i.v. dose or a 30 mg/kg oral dose. Dogs received a 5 mg/kg i.v. dose or a 10 mg/kg oral dose. The greater than 100% oral absorption observed for dogs may be due to random variability (only three animals were employed) or due to non-linear dose-response relationships in the rates of absorption and/or elimination for the i.v. and oral doses. The metabolic stability of CPP-115 was evaluated in cryopreseved human hepatocytes, and 16% loss was observed after 4 hours (T1/2 is 20 h), which is consistent with a drug that would survive first pass metabolism and through several half lives in the blood stream. CPP-115 was incubated, in triplicate, in human hepatocytes (1,000,000 cells/mL) for 4 hours. The calculated T1/2 is well past the end of the incubation period and is only approximate.
Toxicity of CPP-115
CPP-115 was tested in the in vitro mammalian chromosome aberration test using human peripheral blood lymphocytes (HPBL) in both the absence and presence of an Aroclor-induced rat liver S9 metabolic activation system. At a concentration of 10 mM, the percentage of cells with structural or numerical aberrations in the test article-treated groups was not significantly increased relative to solvent control (p > 0.05, Fisher’s Exact test). Results of the Bacterial Reverse Mutation Assay (Salmonella typhimurium tester strains TA98, TA100, TA1535, and TA1537 and Escherichia coli tester strain WP2 uvrA) indicate that CPP-115 (234 mM, 5000 μg/plate) did not cause a positive response with any of the tester strains in either the presence or absence of Aroclor-induced rat liver S9.
Vigabatrin also has been shown to cause microvacuolization in the white matter of the CNS, which has been characterized as intramyelinic edema.26 This histopathological finding resolves in a few weeks after discontinuation of therapy. There is currently no histological data for CPP-115 to confirm whether or not this effect occurs with CPP-115 and to what degree.
Effects of CPP-115 on cocaine induced CNS neurochemical changes
MicroPET imaging of the effects of CPP-115 on cocaine-induced lowering of [11C]-raclopride
To compare the pharmacological effects of CPP-115 to that previously reported for vigabatrin, we determined the effect of cocaine administration on NAc-released dopamine (Figure 1). In these microPET imaging studies, cocaine reduced [11C]-raclopride binding by an average of 22%, consistent with an increase in synaptic dopamine.1 However, when treated with CPP-115, cocaine had no effect on [11C]-raclopride binding. That is, [11C]-raclopride binding was similar to the control data, consistent with CPP-115 (at 0.5 mg/kg) producing a blockade of cocaine-induced increases in synaptic dopamine at a dose 600 times lower (0.5 mg/kg) than the required 300 mg/kg dose of vigabatrin that was effective previously.27 In two microPET imaging studies, CPP-115 administered 2.5 hours prior to radiotracer injection did not produce a change in [11C]-raclopride binding.
Figure 1.
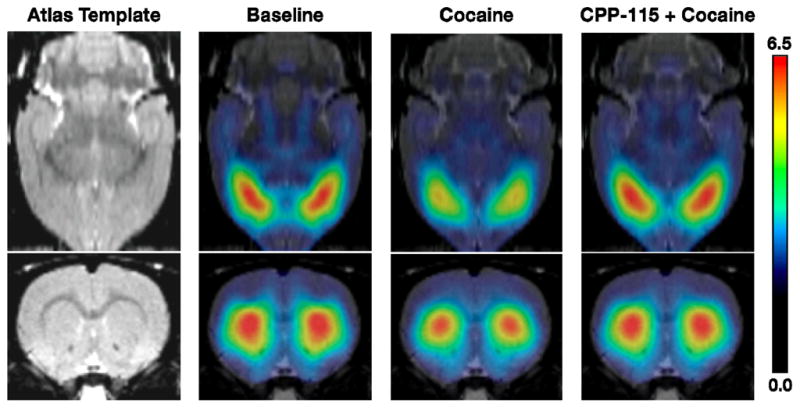
Stimulatory effects of cocaine by pretreatment with CPP-115. In vivo microPET imaging shows the stimulatory effects of cocaine are reduced by pretreatment with CPP-115. [11C]-Raclopride is sensitive to competition with synaptic dopamine, so its binding (higher levels in red) is inversely related to dopamine levels. Increases in dopamine produced by a cocaine challenge (20 mg/kg; cocaine) displace [11C]-raclopride binding. This effect is blocked by pretreatment with CPP-115 (0.5 mg/kg; CPP-115 + cocaine) 2.5 hours prior to cocaine.
Effects of CPP-115 on cocaine-induced increases in extracellular dopamine
To compare the pharmacological effects of vigabatrin and CPP-115, we measured the effect of cocaine stimulation on extracellular dopamine (Figure 2). Control rats pretreated with saline prior to cocaine showed an increase in dopamine concentration of 550 ± 21% relative to untreated rats. Sixty minutes prior to the cocaine challenge, rats were pretreated with either 300 mg/kg of vigabatrin or 1 mg/kg of CPP-115. Pretreatment with vigabatrin attenuated this response by 40%, or an increase in dopamine of 331 ± 32%. In contrast, pretreatment with CPP-115 attenuated the response to cocaine by 54%, a cocaine-induced increase in dopamine of only 256 ± 26%. Thus, in freely moving, unrestrained animals, CPP-115 at 1/300th the dose used for vigabatrin, produced a greater inhibition of the stimulatory effects of cocaine than vigabatrin, without altering baseline dopamine levels. The 10–20 minute delay in response in Figure 2 occurs because of a 10-minute sampling interval. In vivo microdialysis techniques, unlike microPET, require that extracellular samples are collected over some period of time and injected into an HPLC. This period of time is directly related to the sensitivity of the electrochemical detector, in this case 10 minutes.
Figure 2.

Effect of CPP-115 on cocaine-induced increases in nucleus accumbens dopamine in freely moving rats. CPP-115 preferentially attenuates cocaine-induced increases in nucleus accumbens dopamine in freely moving rats. All drugs (saline, vigabatrin (also known as CPP-109), and CPP-115) were given by intraperitoneal injection 60 minutes prior to cocaine (20 mg/kg). Significant differences in peak effect were determined using a Student’s t-test, ***p<0.001 from saline controls, §p<0.05 from CPP-109.
Effect of CPP-115 on the expression of conditioned place preference, a model for cocaine addiction
Increases in NAc dopamine following administration of cocaine produces a dose-dependent and profound effect on the expression of conditioned place preference in rats. Conditioned place preference is a well-documented model that assesses the saliency of drugs of abuse in a drug-free state.1 The ability to pharmacologically block the expression of a cocaine-induced conditioned place preference suggests that these compounds might have an indication for treating cocaine addiction.
Cocaine produced a dose-dependent conditioned place preference response, with the most reliable and robust response occurring at 20 mg/kg. Therefore, a 20 mg/kg cocaine dose was chosen to examine the effect of the administration of CPP-115 on the expression of a cocaine-induced conditioned place preference (Table 1). The results clearly indicate that 1.0 mg/kg of CPP-115 blocked the expression of cocaine-induced conditioned place preference. By itself, CPP-115 produced neither a conditioned place preference nor a conditioned aversive response, indicating that CPP-115 exhibits no abuse potential. These data are particularly exciting in that similar findings with vigabatrin required a dose of 300 mg/kg, while the effects of CPP-115 were obtained using a dose of 1.0 mg/kg. Specifically, in the saline/saline pairings, animals spent an equal amount of time in both chambers (7.3 ± 0.5 versus 7.7 ± 0.6 min). However, in the saline/cocaine pairings, animals spent a significantly greater amount of time in the cocaine-paired chamber 11.8 ± 0.5 versus 3.2 ± 0.4 min (p< 0.01, Student’s two-tailed t-test). In the saline/compound CPP-115 pairings, animals spent an equal amount of time in both chambers (8.7 ± 0.2 versus 6.9 ± 0.9 min), suggesting that CPP-115 did not produce a conditioned place preference on its own (i.e., is not addictive). In the cocaine/saline + compound CPP-115 pairings, animals again spent an equal amount of time in both chambers (7.8 ± 0.5 versus 7.2 ± 0.9 min), demonstrating that at a dose of 1.0 mg/kg, CPP-115 completely blocked the expression of a cocaine-induced conditioned place preference with 300 times greater potency than that of vigabatrin.
Table 1.
CPP-115alone is not rewarding and blocks the expression of preference to cocaine. CPP-115 (1 mg/kg) and vigabatrin (300 mg/kg) produced comparable inhibition of cocaineinduced place preference, p<0.001, Student’s t-test
| Treatment Group | number of animals | Time in chamber (min)
|
|
|---|---|---|---|
| paired | unpaired | ||
| Saline/Salinea | 9 | 7.3 ± 0.5 | 7.7 ± 0.6 |
| Saline/Cocainea | 9 | 11.8 ± 0.4 | 3.2 ± 0.4 |
| Saline/CPP-115 | 8 | 8.1 ± 0.2 | 6.9 ± 0.9 |
| CPP-115/Cocaine | 8 | 7.8 ± 0.5 | 7.2 ± 0.9 |
| Vigabatrin/Cocainea | 10 | 7.9 ± 0.5 | 7.1 ± 0.6 |
Dewey et al.7
Retinal Toxicity of CPP-115
We collected electroretinograms (ERG) for scotopic (rod), mesopic (standard combined), photopic (cone), and 10 and 15 Hz flicker ERGs for both eyes from each animal in the study. One of the animals in the vigabatrin group at the 45-day time point exhibited a larger difference between the right and left eyes for all ERGs. Histological examination of both eyes confirmed that the eye producing the lower ERG results showed inflammation of both the vitreous and retina and was indicative of a major infection. Therefore, all results from this animal (both eyes) were omitted from further consideration. During the course of the study, due to the use of a high maximum tolerated dose (MTD) for both drugs (20 mg/kg for CPP-115 and 200 mg/kg for vigabatrin), four animals were euthanized or found dead (two female and two male) in the CPP-115 group and two deaths (one of each sex) in the vigabatrin group, out of 15 animals in each group. The need for the euthanizations resulted from the sedative effects of both drugs in the first three weeks of treatment, which led to malnourishment, dehydration, and general distress. The MTD of 20 mg/kg for CPP-115 was determined directly by a dose ranging study (unpublished results). The MTD for vigabatrin used in this study was determined empirically from previously published results of vigabatrin toxicity,26,28 the 250 mg/kg/day dose that was used in the previously reported retinal toxicity study upon which this study is based,17 and in consideration of the longer duration of this study. Doses several times larger of both CPP-115 and vigabatrin are needed for a given pharmacological effect when given repeatedly over extended times compared to when the drug is administered as a single dosage.28,29 As a result, the single dosages previously described for cocaine addiction animal models used a larger dosage than the retinal toxicity study.
The implicit times of the ERGs were generally unremarkable and showed little or no drug effect for either drug, which is consistent with past reports for vigabatrin in this model.17,20 However, in females the rod, mesopic, and cone b-wave implicit times were significantly increased with vigabatrin compared to controls or CPP-115 (data not shown). The a-wave amplitudes, when present, generally showed the same trends described below, but with less statistical power than the b-wave amplitudes as a result of the smaller size of these signals. We also collected flicker ERGs at both 10 Hz and 15 Hz. The 10 Hz flicker ERGs were significantly lower with vigabatrin compared to both CPP-115 and controls in both sexes at both time points (data not shown). Only the 15 Hz flicker ERG is discussed because the 15 Hz ERG responses exhibit smaller rod contributions because of the higher stimulus frequency and, therefore, would be more indicative of cone photoreceptor recovery time. Vigabatrin showed a larger decrease in the scotopic, mesopic, and photopic b-wave ERG and flicker ERG average amplitude responses when compared to CPP-115. Vigabatrin and CPP-115 treatment in females resulted in a greater reduction in all ERG measurements compared to males (see Table 2 and Figure 3) with vigabatrin generally resulting in more pronounced reductions in amplitude.
Table 2.
Number of eyes examined, ERG means and standard errors, and p-valuesa of comparisons to the matching control values (see text for p-value discussion).
| Scotopic (Rod) ERG | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Females | Males | ||||||||
| n | Mean | SEM | p-value | n | Mean | SEM | p-value | ||
| Control | 45 d | 10 | 290.3 | 39.0 | 10 | 274.6 | 41.7 | ||
| 90 d | 20 | 294.4 | 31.5 | 20 | 302.1 | 46.0 | |||
| CPP-115 | 45 d | 8 | 260.1 | 51.7 | ns | 8 | 235.1 | 37.9 | ns |
| 90 d | 18 | 236.3 | 32.1 | ns | 16 | 232.5 | 23.6 | ns | |
| Vigabatrin | 45 d | 8 | 93.9 | 33.1 | ** | 6 | 225.2 | 44.2 | ns |
| 90 d | 20 | 110.1 | 32.4 | *** | 20 | 193.5 | 34.8 | * | |
| Standard Combined ERG | |||||||||
| Females | Males | ||||||||
| n | Mean | SEM | p-value | n | Mean | SEM | p-value | ||
| Control | 45 d | 10 | 393.1 | 56.4 | 10 | 397.1 | 58.9 | ||
| 90 d | 20 | 379.2 | 32.6 | 20 | 362.4 | 62.2 | |||
| CPP-115 | 45 d | 8 | 337.8 | 67.8 | ns | 8 | 311.6 | 49.3 | ns |
| 90 d | 18 | 280.6 | 44.3 | ns | 16 | 309.1 | 30.5 | ns | |
| Vigabatrin | 45 d | 8 | 169.8 | 36.2 | ** | 6 | 280.9 | 69.4 | ns |
| 90 d | 20 | 129.6 | 39.8 | *** | 20 | 235.7 | 44.3 | ns | |
| Photopic (Cone) ERG | |||||||||
| Females | Males | ||||||||
| n | Mean | SEM | p-value | n | Mean | SEM | p-value | ||
| Control | 45 d | 10 | 62.6 | 8.2 | 10 | 73.7 | 14.3 | ||
| 90 d | 20 | 62.2 | 5.1 | 20 | 66.0 | 5.6 | |||
| CPP-115 | 45 d | 8 | 62.8 | 13.9 | ns | 8 | 52.8 | 7.2 | ns |
| 90 d | 18 | 51.7 | 7.9 | ns | 16 | 52.9 | 4.6 | ns | |
| Vigabatrin | 45 d | 8 | 37.8 | 3.5 | * | 6 | 57.6 | 9.5 | ns |
| 90 d | 20 | 26.4 | 5.4 | *** | 20 | 47.6 | 6.9 | * | |
| 15 Hz Flicker ERG | |||||||||
| Females | Males | ||||||||
| n | Mean | SEM | p-value | n | Mean | SEM | p-value | ||
| Control | 45 d | 10 | 33.7 | 3.5 | 10 | 44.4 | 6.3 | ||
| 90 d | 20 | 33.4 | 3.2 | 20 | 36.2 | 3.6 | |||
| CPP-115 | 45 d | 8 | 34.5 | 10.8 | ns | 8 | 27.8 | 4.2 | ns |
| 90 d | 18 | 24.1 | 4.7 | ns | 16 | 27.8 | 3.9 | ns | |
| Vigabatrin | 45 d | 8 | 8.6 | 1.4 | ** | 6 | 26.4 | 7.3 | ns |
| 90 d | 20 | 12.1 | 3.5 | *** | 20 | 21.8 | 4.8 | ** | |
Significant p-values are indicated with
for extremely significant <0.0001,
for very significant 0.0001 to 0.01,
0.01 to 0.05 for significant, and ns >0.05 as not significant.
Figure 3.

The effect of CPP-115 and vigabatrin (GVG in the figure) on the ERG responses. The b-wave amplitudes for the control, CPP-115, and vigabatrin groups, for both males (M) and females (F), for the scotopic (rod) (A), photopic (cone) (B), standard combined (C), and the 15 Hz flicker (D) ERG average response at 45 and 90 days are presented, with the standard error of the mean of each measurement. Statistically significant differences, where observed, are also indicated. Significant p-values are indicated with *** for extremely significant <0.0001, ** for very significant 0.0001 to 0.01, * 0.01 to 0.05 for significant, and ns >0.05 as not significant.
Discussion
Because of the importance of GABAergic effects on a variety of neurological disorders and the inherent complexity of this system resulting from multiple subtypes of receptors and transporters, it is crucial that potential therapeutic compounds are selective for specific components of the GABAergic system. In this study, we evaluated the selectivity profile of a recently described GABA-AT inhibitor, CPP-115.18 We observed that CPP-115 does not affect GABA uptake in recombinantly expressed human and mouse GABA transporters or in mouse cortical astrocytes or neurons (Supporting Information Table 1). Therefore, unlike vigabatrin, which is a GABA reuptake inhibitor,30 CPP-115 does not interfere with GABA transport. Furthermore, CPP-115 displays no affinity for GABAA or GABAB receptors (Supporting Information Table 2) and is neither an agonist nor an antagonist for GABAC receptors (Supporting Information Figure 1). The principal GABAergic site of action of CPP-115 appears to be GABA-AT, the enzyme that catabolizes GABA.31 Because of the effectiveness of vigabatrin, an irreversible inactivator of GABA-AT, on the reversal of specific addictionassociated biochemical and behavioral measures to a variety of drugs of abuse, CPP-115 was investigated for its ability to block cocaine-induced increases in NAc dopamine concentrations by microPET in sedated animals, an indicator of addictive behavior. Further, we extended these biochemical findings to a behavioral measure, the expression of a cocaine-induced conditioned place preference.
The microdialysis studies demonstrated that pretreatment with vigabatrin or CPP-115 reduced the magnitude of the cocaine challenge-induced increase, or “surge”, in NAcc dopamine levels (Figure 2). This reduced NAcc surge in dopamine levels correlated with elimination of cocaine induced conditioned place preference (Table 1) and an increase in raclopride binding to dopamine receptors (Figure 1), which is known to correlate with reduced dopamine receptor occupancy by dopamine. In fact, we observed that a dose of CPP-115 that is 1/300 (1.0 mg/kg) to 1/600 (0.5 mg/kg) that of vigabatrin (300 mg/kg) reversed cocaine-induced increases in synaptic dopamine as well as in the expression of a cocaine-induced conditioned place preference. The effects of vigabatrin, an irreversible GABA-AT inhibitor, are thought to be activity dependent. That is, while vigabatrin increases presynaptic GABA stores, these stores are not released into the synaptic space (and thus the extracellular environment) unless stimulated by an increase in neuronal cell firing rates. The fact that we do not see changes in either extracellular (microdialysis) or synaptic (microPET) dopamine following vigabatrin treatment is consistent with this reported mechanism of action and suggests that CPP-115 might also be activity dependent. Given the effectiveness of vigabatrin in clinical trials for the treatment of cocaine and/or methamphetamine addiction,10,11,12 in combination with its preclinical efficacy for the treatment of nicotine,7 methamphetamine, heroin, and ethanol8 abuse, it is reasonable that CPP-115 also will be effective in treating these addictive behaviors in humans.
The effects of a 20 mg/kg dose, compared to the 0.5 mg/kg to 1 mg/kg addiction treatment dose, of CPP-115 on retinal function were evaluated by electroretinography. This dose is effective at reducing or eliminating cocaine induced CNS changes associated with addiction and represents a safety factor of 20 to 40 fold over the treatment dose. In contrast, the retinal function effects of vigabatrin, the positive control, evaluated at a 200 mg/kg dose, compared to its effective dose for cocaine addiction of 100 mg/kg to 300 mg/kg, represents a safety factor of only about 1, which could account for its serious visual field defects in humans. Despite the high dose of CPP-115 used in the retinal toxicity studies relative to its therapeutic dose, the effects on ERG parameters were less than those found for vigabatrin. It is also interesting that the vigabatrin induced reductions in ERG response plateau at 45 days (see Figure 3) in the female animals. There is insufficient data from this study to determine the rate profile at which vigabatrin approaches this plateau of reduced retinal function. However, based on the magnitude of the reduced ERG response values for both drugs at both 45 and 90 days, we conclude the rate at which vigabatrin affects retinal function is substantially faster than that of CPP-115.
A substantial sex-based bias in the effect of vigabatrin on retinal function was also noted that has not been previously reported for this model. However, the statistical analysis of the ERG results for CPP-115 and the control showed no statistically significant sex-based differences.
CPP-115 also exhibits remarkable pharmacokinetic characteristics: it is not metabolized, exhibits rapid and complete oral absorption, and is rapidly eliminated, which are all highly favorable for orally delivered drugs.
A remarkable advantage of CPP-115 is its much greater potency relative to vigabatrin, which should markedly reduce its daily dose relative to that of vigabatrin (1–3 g/day). Furthermore, given the greatly reduced retinotoxicity of CPP-115 and the greater safety factor at which this retinotoxicity was evaluated, it is reasonable to conclude that CPP-115 will exhibit greater visual safety in clinical use for both addiction and epilepsy. Since the predominant cause for visual field defects in vigabatrin therapy is believed not to be the result of elevated GABA,15 the mechanistic differences between vigabatrin and CPP-115 with regard to their potential inactivation of GABA-AT would contribute to less visual field defects in the case of CPP-115.
Many clinical studies have examined visual deficits associated with vigabatrin treatment for epilepsy. Because of these potential side effects, an official registry has been initiated to closely monitor and follow-up with patients being treated with vigabatrin.32 Clinical ophthalmic visual testing, including ERGs to examine cone function with flicker analysis, are essential components for defining visual toxicity in vigabatrin treatment.33 In a newly released pediatric study, a subgroup of patients had vigabatrin-induced visual field loss and progressive cone ERG deficits.34 Our animal ERG studies demonstrate that CPP-115 is an alternative, and potentially safer pharmacological agent, to vigabatrin for targeting the deficits in the GABA pathway and in other clinical studies that require visual testing.
Experimental
Materials
Vigabatrin (Sabril®), (R)-baclofen, GABA, isoguvacine, sodium pyruvate, theophylline, gentamycin, and all buffer reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Vigabatrin for the electroretinography study was supplied by Divi’s Laboratories, Andhra Pradesh, India. (1S,3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) was synthesized as reported previously; calculated for C7H9NO2F2: C, 47.46; H, 5.12; N, 7.91. Found: C, 47.16; H, 4.79; N, 7.68.18 CPP-115 was ≥95% pure. [3H]GABA (35 or 40.0 Ci/mmol) and [3H]muscimol (36.6 Ci/mmol) were purchased from PerkinElmer (Boston, MA, USA). All reagents for cell culture techniques were purchased from Invitrogen (Paisley, UK). Cocaine USP was provided by the National Institute on Drug Abuse (NIDA). All animals were adult male Sprague-Dawley rats (200–225 g, supplied by Taconic Farms, Germantown, NY), with the exception of those animals used for the electroretinography study, as described below. The general care and housing of the animals in all experiments met or exceeded the AAALAC standards. The animals were euthanized in accordance with the AVAMA Guidance on Euthanasia.
GABA uptake assay
[3H]GABA uptake assay at human GABA transporters
The tsA201 cells were cultured in GlutaMAX-I DMEM supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. The plasmids encoding hGAT-1, hBGT-1, hGAT-2, and hGAT-3,35 respectively, were transfected into tsA201 cells using PolyFect according to the protocol of the manufacturer (Qiagen, West Sussex, UK). The next day, the tsA201 cells transiently expressing each of the four human GABA transporter subtypes were split into poly-D-lysine-coated white 96-well plates (PerkinElmer). The pharmacological assays were performed 36–48 hours after transfection exactly as described previously.36 In short, assay buffer supplemented with 30 nM [3H]GABA and test compounds was added to the cells, and the uptake of [3H]GABA was determined after incubation at 37 °C for 3 min. Quantification was performed using Microscint™20 scintillation fluid (PerkinElmer) and a Packard TopCount microplate scintillation counter.
[3H]GABA uptake assay at mouse GABA transporters
Cortical astrocytes were cultured essentially as previously described.37 The neopallium was removed from new born NMRI mice (Taconic, Denmark) and passed through an 80 μm nylon sieve and cultured in modified Dulbecco’s modified Eagle’s medium with fetal calf serum. The calf serum was lowered from 20% to 10% over three weeks, and finally the astrocytes were allowed to differentiate using 0.25 mM dibutyryl cyclic AMP during the last week of growth. Cortical neurons were cultured essentially as previously described38 by removing the neopallium of 15-day old NMRI embryos by dissection followed by mild trypsination. The neurons were cultured in 10% fetal calf serum and, after 48 hours, cytosine arabinoside was added to a final concentration of 20 μM to prevent glial proliferation. Four cultures of stably transfected Human Embryonic Kidney (HEK)-293 cells expressing mGAT1-4 were prepared by the method previously reported.39 The stable cell lines are under the selection pressure of blasticidin-S at 5 μg/mL. Determinations of the IC50 values were conducted as described earlier.40 In brief, [3H]GABA uptake was assessed at 37 °C for 3 minutes on desired cells in PBS buffer containing 1 μM GABA, 13 nM [3H]GABA, and test compound. Radioactivity was measured using Microscint™20 scintillation fluid (PerkinElmer) and a Packard TopCount microplate scintillation counter.
GABA receptor binding assays
Receptor preparations
GABAA and GABAB binding assays were performed using rat brain synaptic membranes of cortex and the central hemispheres from adult male Sprague-Dawley rats with tissue preparation as earlier described.41 On the day of the assay, the membrane preparation was quickly thawed, suspended in 40 volumes of ice cold 50 mM Tris-HCl buffer (pH 7.4) using an UltraTurrax homogenizer and centrifuged at 48,000 g for 10 minutes at 4°C. This washing step was repeated four times. The final pellet was resuspended in incubation buffer and the binding assay carried out as detailed below.
GABAA receptor activity assay
Rat brain synaptic membranes (100 μg protein/aliquot) prepared above in Tris-HCl buffer (50 mM, pH 7.4) were incubated with [3H]muscimol (5 nM) and 100 μM of compound CPP-115 at 0°C for 60 minutes in a total volume of 250 μl. GABA (1 mM) was used to define non-specific binding. The binding reaction was terminated by rapid filtration through GF/B unifilters (PerkinElmer) using a 96 well Packard FilterMate cell-harvester, followed by washing with 3 × 250 μl of ice cold binding buffer, drying, and adding scintillation fluid, as described for the [3H]GABA uptake assay.
GABAB receptor binding assay
For [3H]GABA binding to the GABAB receptors, rat brain synaptic membranes (200 μg protein/aliquot) were suspended in Tris-HCl buffer (50 mM + 2.5 mM CaCl2, pH 7.4) and incubated with [3H]GABA (5 nM), isoguvacine (40 μM), and 100 μM of compound CPP-115 at 25°C for 45 minutes in 1 ml total volume. Isoguvacine serves to saturate GABAA receptors.42 Non-specific binding was determined using 100 μM (R)-baclofen. Binding was terminated by filtration through Whatman GF/C filters, using a Brandell M-48R Cell Harvester; filters were washed with 3 × 3 ml of ice cold buffer, and filter-bound radioactivity was counted in a Packard Tricarb 2100 liquid scintillation analyzer using 3 ml of Opti-fluor scintillation fluid (PerkinElmer).
Electrophysiology
Expression of ρ1 in Xenopus leavis oocytes
Human ρ1 cDNA encapsulated in pcDNA1.1 was linearized with Not1 restriction endonuclease. Linearized cDNA was transcribed to mRNA using the T7 “mMESSAGE mMACHINE” kit (Ambion Inc. Austin, Texas, USA) as previously described.43 GABAC receptor activity assays were performed in oocytes harvested from Xenopus laevis (housed in the Department of Veterinary Science at the University of Sydney) and defolliculated. The oocytes were stored in ND96 solution (in mM) NaCl (96), KCl (2), MgCl2 (1), CaCl2 (1.8), HEPES (hemi-Na salt; 5) supplemented with sodium pyruvate (2.5), theophylline (0.5), and 50 μg/ml−1 gentamycin for 2–5 days post-injection.
GABAC receptor electrophysiological assay
Electrophysiological methods were performed as previously described.36 Stage V-VI oocytes were injected with 10 ng 50 nl−1 of ρ1 mRNA and then stored at 16 °C. Recordings of receptor activity were obtained for 2–5 days by a two-electrode voltage clamp by means of a Geneclamp 500 amplifier (Axon Instruments Inc., Foster City, CA), a MacLab 2e recorder (AD Instruments, Sydney, NSW), and Chart version 3.6.3 program. Oocytes were voltage clamped at −60 mV, and the preparation was continually perfused with ND96 solution at room temperature. Compound CPP-115 (100 μM) dissolved in ND96 was applied in the absence and presence of GABA, respectively, until maximum current was reached, at which time the oocytes were washed for 5 to 10 minutes to allow complete recovery of response to GABA (1 μM). CPP-115 was tested on three oocytes from at least two harvests.
Off-target assays
Tests of inhibition of human liver cytochrome P450s by CPP-115 were carried out at XenoTech (Lenexa, Kansas). The hERG channel test was done at ChanTest (Cleveland, Ohio). Carcinogenicity tests (Ames and chromosomal aberration tests) were done at BioReliance (Rockville, MD). The blood sample collection for the PK analyses was done by Sinclair Research (Columbia, MO) and the sample analysis was done by Cyanta Analytical Services (Maryland Heights, MO).
Aspartate aminotransferase or alanine aminotransferase (0.2 units) was incubated in a total volume of 2000 μL with 6.6 mM α-ketoglutarate, malic dehydrogenase (for Asp-AT) or lactate dehydrogenase (for Ala-AT) (5 units), 3.5 μM NADH and various concentrations of CPP-115 (0 – 4 mM) in 100 mM potassium phosphate, pH 7.4, for 1 h at room temperature. L-Aspartate or L-alanine (125 mM) was added to the incubation mixture and changes in absorbance at 340 nm were determined for 30 min, monitoring NADH degradation.
Succinic semialdehyde dehydrogenase (SSADH) (5 nM) was incubated in a total volume of 500 μL with an excess amount of CPP-115 (5 mM) in 100 mM potassium phosphate, pH 7.4, at room temperature. At several time points, (1, 2, 4, 8, and 20 h) a 100 μL aliquot was removed and added to 500 μL of potassium phosphate buffer (100 mM) containing 2 mM succinic semialdehyde and 3 mM NADP+. Changes in absorbance were measured at 340 nm for 10 min.
Cocaine-induced conditioned place preference
A non-biased approach was used for all conditioned place preference studies. Specifically, animals were pretested in the conditioned place preference chambers for a pre-existing chamber bias. Any animals that spent more than 70% of their time in any chamber were eliminated from the study. Thus, only animals that demonstrated no pre-existing chamber bias were used in the study.
In all rodent studies (n = 8/group) animals were allowed to acclimate to the animal housing facility for at least 5 days prior to beginning the experiments. Conditioned place preference chambers were used as previously described44 except instead of one chamber being entirely white and the other black, one chamber was entirely light blue with a stainless steel floor, and the second chamber was light blue with horizontal black stripes (2.5 cm wide) spaced 3.8 cm apart with a smooth Plexiglass floor. In all conditioned place preference studies with CPP-115, the saline volume was (1 ml/kg), the cocaine doses were 20 mg/kg, and the dose of CPP-115 was 1.0 mg/kg. The saline, cocaine, and compound CPP-115 were all injected intraperitoneally (i.p.). The conditioning procedure for the acquisition phase consisted of 12 sessions carried out consecutively over 12 days. The conditioned place preference pairings were: 1) saline/saline; 2) saline/cocaine; 3) CPP-115/saline, and 4) saline/cocaine + CPP-115.
Animals in each group were randomly assigned to a 2 × 2 factorial design with one factor being the pairing chamber and the other factor being the order of conditioning. Animals that received either saline or cocaine were injected and confined to the appropriate compartment for 30 min. CPP-115 injections were given 2.5 hours prior to saline or cocaine injections. This was done as it has been shown that GABA levels reach maximal values 3 to 4 hours following the administration of CPP-115. On the test day (day 12) neither saline nor drugs was administered, and the animals were allowed to move freely between both chambers for 15 minutes. The amount of time spent in each chamber was recorded using an automated infrared beam electronically coupled to a timer. For the expression phase of CPP to cocaine, the animals were habituated and conditioned to cocaine as described in the acquisition studies, but no animals in the expression studies were given CPP-115 on conditioning days. On the test day, the animals being tested received either saline or CPP-115 2.5 hours prior to their being placed in the apparatus and allowed free access to both chambers for 15 minutes. A time period of 2.5 hours was selected because previous studies1 demonstrated that this was the optimal pretreatment interval allowing for a maximal increase in GABA concentrations.
MicroPET imaging studies
Using separate adult animals (male Sprague-Dawley rats, n = 2) microPET studies were performed using a Concorde Microsystems R4. Baseline [11C]-raclopride binding was examined in anesthesized (ketamine/xylazine) animals. [11C]-Raclopride (20.4 min half-life) is selective for the dopamine family of receptors and competes directly with dopamine for receptor binding. Thus, drug-induced increases in brain dopamine produce a decrease in [11C]-raclopride binding while dopamine depletion produces an increase in binding. Approximately 2 hours following these baseline scans, animals received an intravenous injection of cocaine (5 mg/kg) followed 5 minutes later by a second injection of [11C]-raclopride. Approximately 2 hours following this scanning session, animals received CPP-115 (0.5 mg/kg). Approximately 2.5 hours following the administration of CPP-115, animals received a second intravenous dose of cocaine (5.0 mg/kg) followed 5 minutes later with a third injection of [11C]-raclopride (this was the procedure used for vigabatrin so they could be compared). Imaging studies show that in an acute, drug naïve setting, where animals (adult female baboons and/or rats) are tested up to 4 times with cocaine, methylphenidate, or methamphetamine, the reproducibility of the response (<8%) is consistent with the test/retest variability in [11C]-raclopride binding.
In vivo microdialysis studies of extracellular dopamine levels in freely moving rats
In vivo microdialysis studies were performed as previously described.8 All drugs (saline, vigabatrin, and CPP-115) were administered by intraperitoneal injection 60 minutes prior to cocaine (20 mg/kg; n = 8/group).
Evaluation of retinotoxicity by electroretinography (ERG)
Materials and Methods for ERG
These studies were carried out at the Sinclair Research Center, LLC (Columbia, MO). Forty-five male and female Wistar albino rats (Charles River Laboratories), 9 weeks of age at the start of dosing, were acclimated, and placed into one of three treatment groups (vehicle, CPP-115, or vigabatrin). Animals received a single intraperitoneal injection of vehicle or test formulations once daily for either 45 or 90 consecutive days at 0, 20, or 200 mg/kg for the vehicle, CPP-115, and vigabatrin treatment groups, respectively. The formulations for each treatment group were prepared fresh weekly in 0.9% normal saline.
All animals were acclimated for 7 days to the test facility prior to the start of dosing and were housed in individual polycarbonate cages. The cage environment uses a standard 12 hour/12 hour light/dark cycle, with light intensity of 7.7 lum/ft2 or 82.9 lux during the light cycle. During the course of the study, the animals were monitored for mortality, moribundity, clinical signs of illness, feed intake, and body weight change. Any animals found in distress for more than 24 hours were humanely euthanized. Due to the sedative effects of both CPP-115 and vigabatrin, some animals received special food supplements and/or intraperitoneal fluids in the first 3 weeks of dosing. By the end of the third week of the dosing, the sedative effects of both drugs decreased, and no special food supplements or i.p. fluids were required. At the conclusion of the dosing phase (5 of each sex for 45 days and 10 of each sex for 90 days), the animals entered a 5 to 7 day “washout” period, after which electroretinograms (ERGs) were measured for both eyes. The animals were then humanely euthanized by CO2 asphyxiation, in accordance with AVMA guidelines on euthanasia, for post-mortem pathology examinations.
Electroretinogram Recordings
Following a dark adaptation period of at least 12 hours, each eye was dilated with tropicamide (1 drop of 1% solution) and phenylepherine (1 drop of 10% solution), an anesthetic dose of ketamine HCl (up to 55 mg/kg) and xylazine HCl (up to 12 mg/kg) was administered i.m. or i.p. and, just prior to the ERG, a topical anesthesia (0.5% proparacaine HCl) was applied to the eye. The types of ERG measurements made and the ERG testing parameters are found in Table 3. The ERGs were measured with an Espion2 ERG system with ColorDome Ganzfeld type illuminator by Diagnosys, Inc. All light stimuli were white light. The ground electrode was a Grass needle electrode inserted subcutaneously at the base of the tail. The reference electrode was a Grass gold disc electrode placed on the tongue. The eye electrode was a gold wire. Data were collected on a dual channel system (1 channel per eye) at 1000 samples per second with a 2nd order 0.3 Hz to 300 Hz band pass Bessel filter and also a 60 Hz notch filter to remove interference from the line power.
Table 3.
Electroretinography (ERG) experimental parameters
| ERG type | Flashes/interval | Flash Mode | Pulse Period | Intensity |
|---|---|---|---|---|
| Rod | 5 @ 500ms | Pulse | 4ms | 0.02cd•s/m2 |
| Standard Combined | 5 @ 40000ms | Pulse | 4ms | 7cd•s/m2 |
| Light Adaptation | 0 @ 900sec | N/A | N/A | 25cd/m2 |
| Single Flash – Cone | 5 @ 15000ms | Pulse | 4ms | 6cd•s/m2 |
| 10Hz Flicker | 20 | Continuous | 4ms | 3cd•s/m2 |
| 15Hz Flicker | 20 | Continuous | 4ms | 3cd•s/m2 |
The ERG data were analyzed with Prism Sigmaplot, version 5.2. It is generally well known that ERG data are not normally distributed. Therefore, Kruskal-Wallis one-way ANOVA by ranks and Dunn’s post test were employed. Significant P values are indicated with *** for extremely significant (p<0. 001), ** for very significant (0. 001 < p < 0.01, * for significant (0.01 < P < 0.05), and ns (p > 0.05) as not significant in Table 2.
Supplementary Material
Acknowledgments
The authors are grateful to the National Institutes of Health (grants GM066132 and DA030604 to R.B.S., grant DA022346 to S.L.D., grant EY015851 to C.M.C., and grant EY030140 to DEI Core), the Lundbeck Foundation (grant 113/06 to H.B.-O.), the Danish Medical Research Council (grant 271-06-0094 to P.W.), and the Drug Research Academy (to T.K.) for financial support of this research. Professor Povl Krogsgaard-Larsen and Dr. Bolette Kragholm are gratefully acknowledged for fruitful discussions and Katherine Fehlhaber Jones for ERG statistical analysis.
Abbreviations
- ERG
electroretinograhic
- GABA
γ-aminobutyric acid
- GABA-AT
γ-aminobutyric acid aminotransferase
- hERG
human ether-à-go-go-related gene
- HPBL
human peripheral blood lymphocytes
- NAc
nucleus accumbens
- PET
positron emission tomography
- SSADH
succinic semialdehyde dehydrogenase
- VFD
visual field defect
Footnotes
Catalyst Pharmaceutical Partners compound number assignment
Supporting Information Available: Table of effects of GABA and CPP-115 on GABA uptake in neurons, astrocytes, and human and mouse GABA transporter-expressing cells, table of the ability of CPP-115 to displace [3H]GABA binding to ionotropic GABAA receptors or metabotropic GABAB receptors in rat brain cortical homogenate, table of in vitro off-target pharmacology summary results using binding assays, table of in vitro off-target pharmacology summary results using enzyme assays, effects of GABA and CPP-115 on GABAC. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dewey SL, Morgan AE, Ashby CR, Jr, Horan B, Kushner SA, Logan J, Volkow ND, Fowler JS, Gardner EL, Brodie JD. A novel strategy for the treatment of cocaine addiction. Synapse. 1998;30:119–129. doi: 10.1002/(SICI)1098-2396(199810)30:2<119::AID-SYN1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Silverman RB. Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology. I and II CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 3.Silverman RB. Mechanism-based enzyme inactivators. Methods Enzymol. 1995;249:40–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 4.Lippert B, Metcalf BW, Jung MJ, Casara P. 4-Aminohex-5-enoic acid a selective catalytic inhibitor of 4-aminobutyric aminotransferase in mammalian brain. Eur J Biochem. 1977;74:441–445. doi: 10.1111/j.1432-1033.1977.tb11410.x. [DOI] [PubMed] [Google Scholar]
- 5.Karila L, Gorelick D, Weinstein A, Noble F, Benyamina A, Coscas S, Blecha L, Lowenstein W, Martinot JL, Reynaud M, Lepine JP. New treatments for cocaine dependence: a focused review. Int J Neuropsychopharmacol. 2008;11:425–438. doi: 10.1017/S1461145707008097. [DOI] [PubMed] [Google Scholar]
- 6.Peng X-Q, Li X, Gilbert JG, Pak AC, Ashby CR, Jr, Brodie JD, Dewey SL, Gardner EL, Xi Z-X. Gamma-vinyl GABA inhibits cocaine-triggered reinstatement of drug-seeking behavior in rats by a non-dopaminergic mechanism. Drug Alcohol Depend. 2008;97:216–225. doi: 10.1016/j.drugalcdep.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dewey SL, Brodie JD, Gerasimov J, Horan B, Gardner EL, Ashby CR., Jr A pharmaceutical strategy for the treatment of nicotine addiction. Synapse. 1999;31:76–86. doi: 10.1002/(SICI)1098-2396(199901)31:1<76::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 8.Gerasimov MR, Madina R, Ashby CR, Jr, Gardner EL, Mills MJ, Brodie JD, Dewey SL. γ-vinyl GABA inhibits methamphetamine heroin or ethanol-induced increases in nucleus accumbens dopamine. Synapse. 1999;34:11–19. doi: 10.1002/(SICI)1098-2396(199910)34:1<11::AID-SYN2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Stromberg MF, Mackler SA, Volpicelli JR, O’Brien CP, Dewey SL. The effect of γ-vinyl-GABA on the consumption of concurrently available oral cocaine and ethanol in the rat. Pharmacol Biochem Behav. 2001;68:291–299. doi: 10.1016/s0091-3057(00)00456-1. [DOI] [PubMed] [Google Scholar]
- 10.Brodie JD, Figueroa E, Dewey SL. Treating cocaine addiction: from preclinical to clinical trial experience with γ-vinyl GABA. Synapse. 2003;50:261–265. doi: 10.1002/syn.10278. [DOI] [PubMed] [Google Scholar]
- 11.Brodie JD, Figueroa E, Laska EM, Dewey SL. Safety and efficacy of γ-vinyl GABA (GVG) for the treatment of methamphetamine and/or cocaine addiction. Synapse. 2005;55:122–125. doi: 10.1002/syn.20097. [DOI] [PubMed] [Google Scholar]
- 12.Brodie JD, Case BG, Figueroa E, Dewey SL, Robinson JA, Wanderling JA, Laska EM. Randomized, double-blind, placebo-controlled trial of vigabatrin for the treatment of cocaine dependence in Mexican parolees. Am J Psychiatry. 2009;166:1269–1277. doi: 10.1176/appi.ajp.2009.08121811. [DOI] [PubMed] [Google Scholar]
- 13.Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields D. Vigabatrin: 2008 update. Epilepsia. 2009;50:163–173. doi: 10.1111/j.1528-1167.2008.01988.x. [DOI] [PubMed] [Google Scholar]
- 14.Wild JM, Chiron C, Ahn H, Baulac M, Bursztyn J, Gandolfo E, Goldberg I, Goni FJ, Mercier F, Nordmann J-P, Safran AB, Schiefer U, Perucca E. Visual field loss in patients with refractory partial epilepsy treated with vigabatrin. CNS Drugs. 2009;23:965–982. doi: 10.2165/11317650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Izumi Y, Ishikawa M, Benz AM, Izumi M, Zorumski CF, Thio LL. Acute vigabatrin retinotoxicity in albino rats depends on light but not GABA. Epilepsia. 2004;45(9):1043–1048. doi: 10.1111/j.0013-9580.2004.01004.x. [DOI] [PubMed] [Google Scholar]
- 16.Roberts JE. Ocular phototoxicity. J Photochem Photobiol B. 2001;64(2–3):136–143. doi: 10.1016/s1011-1344(01)00196-8. [DOI] [PubMed] [Google Scholar]
- 17.Jammoul F, Wang Q, Nabbour R, Coriat C, Duboc A, Simonutti M, Dubus E, Craft CM, Ye W, Collins SD, Dulac O, Chiron C, Sahel JA, Picaud S. Taurine deficiency is a cause of vigabatrin-induced retinal phototoxicity. Ann Neurol. 2009;65:98–107. doi: 10.1002/ana.21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y, Qiu J, Silverman RB. Design, synthesis and biological activity for a difluoro-substituted, conformationally-rigid vigabatrin analogue as a potent γ-aminobutyric acid aminotransferase inhibitor. J Med Chem. 2003;46:5292–5293. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]
- 19.US Labeling for Sabril®. http://www.lundbeckinc.com/USA/products/CNS/Sabril/sabril_PI_CPS.pdf.
- 20.Duboc A. Vigabatrin, the GABA-Transaminase Inhibitor, Damages Cone Photoreceptors in Rats. Ann Neurol. 2004;55:695–705. doi: 10.1002/ana.20081. [DOI] [PubMed] [Google Scholar]
- 21.(a) Høg S, Greenwood JR, Madsen KB, Larsson OM, Frolund B, Schousboe A, Krogsgaard-Larsen P, Clausen RP. Structure-activity relationships of selective GABA uptake inhibitors. Curr Top Med Chem. 2006;6:1861–1882. doi: 10.2174/156802606778249801. [DOI] [PubMed] [Google Scholar]; (b) Liu Q-R, Lopez-Corcuera B, Mandiyan S, Nelson H, Nelson N. Molecular characterization of four pharmacologically distinct α-aminobutyric acid transporters in mouse brain. J Biol Chem. 1993;268:2106–2112. [PubMed] [Google Scholar]; (c) Madsen KK, Clausen RP, Larsson OM, Krogsgaard-Larsen P, Schousboe A, White HS. Synaptic and extrasynaptic GABA transporters as targets for anti-epileptic drugs. J Neurochem. 2009;109(Suppl 1):139–144. doi: 10.1111/j.1471-4159.2009.05982.x. [DOI] [PubMed] [Google Scholar]
- 22.(a) Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johnston GAR, Chebib M, Hanrahan JR, Mewett KN. Neurochemicals for the Investigation of GABAC Receptors. Neurochem Res. 2010;35:1970–1977. doi: 10.1007/s11064-010-0271-7. [DOI] [PubMed] [Google Scholar]
- 23.Molina PE, Ahmed N, Ajmal M, Dewey S, Wolkow N, Fowler J, Abumrad N. Co-administration of γ-vinyl GABA and cocaine: preclinical assessment of safety. Life Sci. 1999;65:1175–1182. doi: 10.1016/s0024-3205(99)00351-3. [DOI] [PubMed] [Google Scholar]
- 24.Foletti GB, Delisle MC, Bachmann C. Reduction of plasma alanine aminotransferase during vigabatrin treatment. Epilepsia. 1995;36:804–809. doi: 10.1111/j.1528-1157.1995.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 25.Hammerstad JP, Gronke L, Nutt J, Casey D. The effect of γ-vinyl GABA on amphetamine stereotypy in rats. Brain Res Bull. 1980;5(Suppl 2):609–612. [Google Scholar]
- 26.Gibson JP, Yarrington JT, Loudy DE, Gerrig CG, Hurst GH, Newberne JW. Chronic Toxicity Studies with Vigabatrin, A GABA-Transaminase Inhibitor. Toxicol Pathol. 1990;18(2):225–238. doi: 10.1177/019262339001800201. [DOI] [PubMed] [Google Scholar]
- 27.Ashby CR, Jr, Rohatgi R, Ngosuwan J, Borda T, Gerasimov MR, Morgan AE, Kushner S, Brodie JD, Dewey SL. Implication of the GABAB receptor in γ-vinyl GABA’s inhibition of cocaine-induced increases in nucleus accumbens dopamine. Synapse. 1999;31:151–153. doi: 10.1002/(SICI)1098-2396(199902)31:2<151::AID-SYN8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 28.Valdizan EM, Armijo JA. Effects of Single and Multiple Increasing Doses of Vigabatrin on Brain GABA Metabolism and Correlation with Vigabatrin Plasma Concentration. Biochem Pharmacol. 1992;43(10):2143–2150. doi: 10.1016/0006-2952(92)90173-g. [DOI] [PubMed] [Google Scholar]
- 29.Morgan AE, Dewey SL. Effects of Pharmacologic Increases in Brain GABA Levels on Cocaine-Induced Changes in Extracellular Dopamine. Synapse. 1998;28(1):60–65. doi: 10.1002/(SICI)1098-2396(199801)28:1<60::AID-SYN7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.Sills GJ, Butler E, Thompson GG, Brodie MJ. Vigabatrin and tiagabine are pharmacologically different drugs. A pre-clinical study. Seizure. 1999;8:404–411. doi: 10.1053/seiz.1999.0326. [DOI] [PubMed] [Google Scholar]
- 31.Sherif FM, Ahmed SS. Basic aspects of GABA transaminase in neuropsychiatric disorders. Clin Biochem. 1995;28:45–54. doi: 10.1016/0009-9120(94)00074-6. [DOI] [PubMed] [Google Scholar]
- 32.Pellock JM, Faught E, Sergott RC, Shields WD, Burkhart GA, Krauss GL, Foroozan R, Wesche DL, Weinberg MA. Registry initiated to characterize vision loss associated with vigabatrin therapy. Epilepsy Behav. 2011 doi: 10.1016/j.yebeh.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 33.(a) Harding GF, Wild JM, Robertson KA, Rietbrock S, Martinez C. Separating the retinal electrophysiologic effects of vigabatrin: treatment versus field loss. Neurology. 2000;55:347–352. doi: 10.1212/wnl.55.3.347. [DOI] [PubMed] [Google Scholar]; (b) Ponjavic V, Anreasson S. Multifocal ERG and full field ERG in patients on longterm vigabatrin medication. Doc Ophthalmol. 2001;102:63–72. doi: 10.1023/a:1017589301855. [DOI] [PubMed] [Google Scholar]
- 34.McCoy B, Wright T, Weiss S, Go C, Westall CA. Electroretinogram Changes in a Pediatric Population With Epilepsy: Is Vigabatrin Acting Alone? J Child Neurol. 2011;26:729–733. doi: 10.1177/0883073810390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kvist T, Christiansen B, Jensen AA, Bräuner-Osborne H. The four human gamma aminobutyric acid (GABA) transporters: pharmacological characterization and validation of a highly efficient screening assay. Comb Chem High Throughput Screen. 2009;12:241–249. doi: 10.2174/138620709787581684. [DOI] [PubMed] [Google Scholar]
- 36.Christiansen B, Meinild AK, Jensen AA, Bräuner-Osborne H. Cloning and characterization of a functional human gamma-aminobutyric acid (GABA) transporter, human GAT-2. J Biol Chem. 2007;282:19331–19341. doi: 10.1074/jbc.M702111200. [DOI] [PubMed] [Google Scholar]
- 37.Hertz L, Juurlink BHJ, Hertz E, Fosmark H, Schousboe A. In: Preparation of Primary Cultures of Mouse (Rat) Astrocytes, in a Dissection and Tissue Culture Manual of the Nervous System. Shahar A, de Vellis J, Vernadakis A, Haber H, editors. Alan R. Liss, Inc; New York: 1989. pp. 105–108. [Google Scholar]
- 38.Hertz E, Yu ACH, Hertz L, Juurlink BHJ, Schousboe A. Preparation of Primary Cultures of Mouse Cortical Neurons. In: Shahar A, de Vellis J, Vernadakis A, Haber B, editors. A Dissection and Tissue Culture Manual of the Nervous System. Alan R. Liss, Inc; New York: 1989. pp. 183–186. [Google Scholar]
- 39.White HS, Sarup A, Bolvig T, Kristensen AS, Petersen G, Nelson N, Pickering DS, Larsson OM, Frolund B, Krogsgaard-Larsen P, Schousboe A. Correlation between anticonvulsant activity and inhibitory action on glial gamma-aminobutyric acid uptake of the highly selective mouse gamma-aminobutyric acid transporter 1 inhibitor 3-hydroxy-4-amino-4,5,6,7-tetrahydro-1,2-benzisoxazole and its N-alkylated analogs. J Pharmacol Exp Ther. 2002;302:636–644. doi: 10.1124/jpet.102.034819. [DOI] [PubMed] [Google Scholar]
- 40.Bolvig T, Larsson OM, Pickering DS, Nelson N, Falch E, Krogsgaard-Larsen P, Schousboe A. Action of bicyclic isoxazole GABA analogues on GABA transporters and its relation to anticonvulsant activity. Eur J Pharmacol. 1999;375:367–374. doi: 10.1016/s0014-2999(99)00263-0. [DOI] [PubMed] [Google Scholar]
- 41.Ransom RW, Stec NL. Cooperative modulation of [3H]MK-801 binding to the N-methyl-D-aspartate receptor-ion channel complex by L-glutamate, glycine, and polyamines. J Neurochem. 1988;51:830–836. doi: 10.1111/j.1471-4159.1988.tb01818.x. [DOI] [PubMed] [Google Scholar]
- 42.Hill DR, Bowery NG. 3H-baclofen and 3H-GABA bind to bicuculline-insensitive GABAB sites in rat brain. Nature. 1981;290:149–152. doi: 10.1038/290149a0. [DOI] [PubMed] [Google Scholar]
- 43.Chebib M, Duke RK, Allan RA, Johnston GAR. The effects of cyclopentane and cyclopentene analogs of GABA at recombinant GABAC receptors. Eur J Pharmacol. 2001;430:185–192. doi: 10.1016/s0014-2999(01)01390-5. [DOI] [PubMed] [Google Scholar]
- 44.Ashby CR, Jr, Paul M, Gardner EL, Gerasimov MR, Dewey SL, Lennon IC, Taylor SC. Systemic administration of 1R, 4S-4-amino-cyclopent-2-ene-carboxylic acid a reversible inhibitor of GABA transaminase blocks expression of conditioned place preference to cocaine nicotine in rats. Synapse. 2002;44:61–63. doi: 10.1002/syn.10052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.