

Abstract
The ε4 allele of the apolipoprotein E (APOE) gene is the strongest genetic risk factor for Alzheimer's disease (AD). Evidence suggests that the effect of apoE isoforms on amyloid-β (Aβ) accumulation in the brain plays a critical role in AD pathogenesis. Like in humans, apoE4 expression in animal models that develop Aβ amyloidosis results in greater Aβ and amyloid deposition than with apoE3 expression. However, whether decreasing levels of apoE3 or apoE4 would promote or attenuate Aβ-related pathology has not been directly addressed. To determine the effect of decreasing human apoE levels on Aβ accumulation in vivo, we generated human APOE isoform haploinsufficient mouse models by crossing APPPS1-21 mice with APOE isoform knock-in mice. By genetically manipulating APOE gene dosage, we demonstrate that decreasing human apoE levels, regardless of isoform status, results in significantly decreased amyloid plaque deposition and microglial activation. These differences in amyloid load between apoE3- and apoE4-expressing mice were not due to apoE4 protein being present at lower levels than apoE3. These data suggest that current therapeutic strategies to increase apoE levels without altering its lipidation state may actually worsen Aβ amyloidosis, while increasing apoE degradation or inhibiting its synthesis may be a more effective treatment approach.
Introduction
Aggregation and accumulation of amyloid β (Aβ) in the brain is a key initiating factor in the pathogenesis of Alzheimer's disease (AD) (Holtzman et al., 2011). Familial AD-causing mutations in amyloid β precursor protein (APP), presenilin 1 (PS1), and PS2 genes have provided strong support for the crucial role of Aβ aggregation in AD pathogenesis (Holtzman et al., 2011). The strongest genetic risk factor for the most common form of late-onset AD is the ε4 allele of the apolipoprotein E (APOE) gene (Strittmatter et al., 1993). Although several mechanisms for the effect of apoE4 on AD pathogenesis have been proposed, the primary pathway appears to be the differential effects of apoE3 and apoE4 isoforms on Aβ aggregation and clearance (Kim et al., 2009a; Castellano et al., 2011).
In addition to isoform status, it appears that the amount of apoE also plays a role in determining the extent of Aβ accumulation in the brain. There has been intense debate as to whether potential AD therapeutics should increase or decrease apoE levels (Ashford, 2002; Rebeck et al., 2002; Teter et al., 2002; Osherovich, 2009). Initial studies clearly indicated that the deletion of the endogenous murine APOE gene causes a dramatic decrease in fibrillar and total Aβ deposition in APP transgenic mouse models (Bales et al., 1997). However, whether decreasing human apoE levels will have the same beneficial effect or not has not yet been answered. Some recent studies indirectly suggest that increasing, rather than decreasing, human apoE levels may be a good therapeutic approach. Therefore, understanding how modulation of human apoE levels will alter AD pathology must be addressed before the development of apoE-targeting drugs.
To directly test the effect of human APOE gene dosage on amyloid pathology, we generated Aβ-depositing mouse models homozygous or hemizygous for the APOE3 or APOE4 gene. APPPS1–21 mice with two copies of APOE (APOE3/3 or APOE4/4) or one copy of APOE (APOE3/− or APOE4/−) were generated by breeding APPPS1-21 mice with human APOE knock-in mice. Compared with the respective homozygous mice, APOE3/− and APOE4/− hemizygous mice had significantly decreased Aβ accumulation, amyloid deposition, and microgliosis. Our results strongly suggest that the reduction of human apoE levels, regardless of the isoform present, will decrease Aβ-related AD pathology.
Materials and Methods
Generation of human APOE isoform mice with APPswe/PS1(L166P) mutant transgenes.
To determine the gene dosage effect of human APOE3 and APOE4 on amyloid deposition, we used knock-in mouse models in which the endogenous murine Apoe gene is replaced with the APOE3 or APOE4 gene. Breeding pairs were gifts from Dr. Patrick Sullivan, Duke University, Durham, NC (Sullivan et al., 1997). APPPS1-21 mice overexpress a human APP with a Swedish mutation (KM670/671NL) and mutant PS1 with the L166P mutation. Breeding pairs were obtained from Dr. Mathias Jucker, University of Tübingen, Tübingen, Germany (Radde et al., 2006). To replace the murine Apoe gene with human APOE isoforms, APPPS1-21 mice were bred with either APOE3/3 or APOE4/4 knock-in mice. APPPS1-21/APOE3/Apoe mice and APOE3/Apoe mice from the first generation were bred with each other to generate APPPS1-21/APOE3/3 and APOE3/3 mice. APPPS1-21/APOE3/3 and APOE3/3 mice were then bred to generate more APPPS1-21/APOE3/3 mice. After successful generation of APPPS1-21/APOE3/3 mice at the third generation, APPPS1-21/APOE3/3 mice were bred with Apoe knock-out mice (Jackson Laboratory). APPPS1-21/APOE3/− mice from the fourth generation were bred with APOE3/3 mice to generate APPPS1-21/APOE3/3 and APPPS1-21/APOE3/− mice. Littermates generated at the fifth generation were used in our analyses. Similarly, APPPS1-21/APOE4/4 and APPPS1-21/APOE4/− mice were generated by using the same mating strategy. All mice used in this study were maintained on a C57BL/6J background.
Quantitative real-time PCR.
mRNAs were extracted from frozen cortical tissue using Trizol and reverse transcribed with High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative real-time PCR (qPCR) was performed in ABI 7500 using the default thermal cycling condition with Power SYBR (Applied Biosystems) with the following primers: forward primer, TTGCTGGTCACATTCCTGG; and reverse primer, AGGTAATCCCAAAAGCGACC. Mouse GAPDH endogenous control was used as a normalization reference. Relative mRNA levels were calculated by comparative Ct method. To confirm the specificity of qPCR reactions, dissociation curves were analyzed at the end of the qPCR assay.
Western blots.
Cortical tissues were gently lysed in PBS and modified RIPA (1% NP-40, 1% sodium deoxycholate, 25 mm Tris-HCl, 150 mm NaCl) in the presence of 1× protease inhibitor mixture (Roche). Tissue homogenates were centrifuged at 18,000 relative centrifugal force (rcf) for 30 min. Equal amounts of protein for each sample were run on 4–12% Bis-Tris XT gels (Bio-Rad) and transferred to PVDF membranes. Blots were probed with the following antibodies: apoE (Academy Biomedical); APP (ZYMED); PS1-NTF (EMD Chemicals); β-secretase 1 (BACE1) (Cell Signaling Technology); synaptophysin (or SYP) (Sigma); glutamate receptor (GluR) 2/3/4 (Cell Signaling Technology); NMDAR2b (Cell Signaling Technology); postsynaptic density protein 95 (PSD-95) (Millipore); and tubulin (Sigma). Tubulin-normalized band intensity was quantified using NIH ImageJ software.
Sandwich ELISA for Aβ and apoE.
Cortical tissues were sequentially homogenized with PBS, modified RIPA, and 5 m guanidine HCl buffer. Tissue homogenates were centrifuged at 18,000 rcf for 30 min after each extraction. The levels of Aβ and apoE were measured by enzyme-linked immunosorbent assay (ELISA). For Aβ ELISA, HJ2 (anti-Aβ35–40) and HJ7.4 (anti-Aβ37–42) were used as capture antibodies, and HJ5.1-biotin (anti-Aβ13–28) as the detection antibody. The anti-apoE monoclonal antibody WUE4 and another anti-apoE antibody (Calbiochem) were used for apoE ELISA.
Histology.
Histology, staining, immunohistochemistry, and quantitative analysis were performed as described except that we used biotinylated mouse monoclonal antibody HJ3.4 (1:1000, targeted against amino acids 1–13 of the human Aβ sequence) to detect Aβ in tissue sections (Kim et al., 2009b).
Statistical analysis.
To determine the statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001), we first tested whether our datasets passed the equal variance test (Levene Median test) and normality test (Kolmogorov–Smirnov test) (SigmaStat 3.5.). After confirmation that the data did not violate the assumptions of parametric testing, a two-tailed Student's t test was used (GraphPad Prism 5). Variance in all graphs represents SEM.
Results
Generation of human APOE haploinsufficient mouse models
To directly examine the effect of decreasing human apoE levels on Aβ and amyloid deposition, we generated mouse models that have either one or two copies of the human APOE3 or APOE4 gene along with the APPswe/PS1(L166P) transgene (APPPS1-21 mice). APOE knock-in mouse models that express human apoE isoforms under the control of endogenous regulatory elements were used, eliminating any potentially confounding effects due to the presence of the endogenous murine Apoe gene.
First, we assessed the effect of APOE gene haploinsufficiency on apoE mRNA and protein levels in the cortex. As expected, apoE mRNA levels decreased by ∼50% in the hemizygous APOE3/− and APOE4/− mice (Fig. 1A). Western blot analysis indicated that apoE protein levels were also significantly decreased in the APOE hemizygous compared with APOE homozygous mice (Fig. 1B,C). Consistent with the Western blot data, a similar decrease in apoE levels was observed by apoE ELISA for both APOE3 and APOE4 isoform mice (Fig. 1D,E). There were no significant alterations in the levels of proteins involved in Aβ production (Fig. 1F).
Figure 1.

Reduction of apoE levels in human APOE haploinsufficient mice. Cortex from APOE homozygous (APOE3/3 and APOE4/4) and hemizygous (APOE3/− and APOE4/−) mice were used to measure apoE mRNA and protein levels. A, ApoE mRNA levels were measured by quantitative real-time PCR. B, Levels of PBS-soluble apoE were assessed by probing a membrane with anti-apoE antibody. C, Quantitative analyses of Western blots were performed with tubulin normalization (n = 4 per genotype). D, E, To validate the Western blot data, apoE levels were also measured by using an apoE-specific ELISA from APOE3/3 and APOE3/− mice (D) and APOE4/4 and APOE4/− mice (E) (n = 9–18 per genotype). F, Levels of APP, APP CTFs, PS1 NTF, and BACE1 proteins were measured by Western blotting (n = 4 per genotype). Levels of these proteins after normalizing with tubulin signals were not significantly altered by APOE haploinsufficiency. All graphs represent values in mean ± SEM.
Haploinsufficiency of human APOE dramatically inhibits Aβ accumulation
To determine the effect of APOE genotype and dosage on Aβ, we analyzed Aβ accumulation in APOE homozygous and hemizygous mice. Significant reductions in insoluble Aβ40 and Aβ42 levels were observed in APOE3/− mice, compared with APOE3/3 mice (Fig. 2A,B). APOE4/− mice also had a dramatic decrease in insoluble Aβ40 and Aβ42 levels compared with APOE4/E4 mice (Fig. 2C,D). We also analyzed PBS-soluble Aβ levels in mice of all genotypes. Soluble Aβ levels were ∼100-fold less than PBS-insoluble Aβ levels. PBS soluble Aβ levels, except Aβ42 levels between APOE4/4 and APOE4/− mice, were not altered by APOE haploinsufficiency (APOE3/3: Aβ40, 5.82 ± 0.31 pg/mg; Aβ42, 0.77 ± 0.068 pg/mg; APOE3/−: Aβ40, 5.27 ± 0.32 pg/mg; Aβ42, 0.77 ± 0.099 pg/mg; APOE4/4: Aβ40, 7.49 ± 0.88 pg/mg; Aβ42, 1.39 ± 0.13 pg/mg; APOE4/−: Aβ40, 7.90 ± 0.51 pg/mg; Aβ42, 1.12 ± 0.058 pg/mg).
Figure 2.

Decreased Aβ accumulation in human APOE haploinsufficient mice. Cortical tissues from 3-month-old APOE homozygous and hemizygous mice were sequentially homogenized with PBS and RIPA. Aggregated forms of Aβ in the RIPA-insoluble pellet were solubilized with 5 m guanidine HCl buffer. A, B, RIPA-insoluble Aβ40 (A) and Aβ42 levels (B) were measured from APOE3/3 and APOE3/− mice. C, D, Similarly, RIPA-insoluble Aβ40 (C) and Aβ42 levels (D) were measured from APOE4/4 and APOE4/− mice. (n = 12–20 per genotype).
Reduction of Aβ and fibrillar Aβ plaque load in human APOE hemizygous mice
To determine the extent of Aβ deposition in APOE homozygous and hemizygous mice, brain sections from 3-month-old mice were immunostained with a biotinylated anti-Aβ antibody, HJ3.4 (Fig. 3A–F). Quantitative analyses of anti-Aβ immunostaining demonstrated that Aβ plaque load was significantly decreased in APOE3/− mice compared with APOE3/3 mice (Fig. 3C). The level of Aβ deposition in both APOE4/4 and APOE4/− mice was also analyzed in the same way (Fig. 3D,E). While APOE4+/+ and APOE4+/− mice had significantly greater Aβ deposition than APOE3+/+ and APOE3+/− mice respectively, APOE4/− mice had a dramatic reduction in Aβ plaque load, compared with APOE4/4 mice (Fig. 3F). To further characterize the nature of the deposited Aβ plaques, brain sections were stained with X-34 dye that selectively detects only fibrillar, but not diffuse, Aβ deposits. Consistent with the anti-Aβ antibody staining, APOE3/− and APOE4/− hemizygous mice had significantly less X-34-positive fibrillar plaque load, compared with APOE3/3 and APOE4/4 homozygous mice (Fig. 3G–L). Together, our results clearly demonstrate that haploinsufficiency of APOE decreases both total Aβ and fibrillar amyloid deposition.
Figure 3.
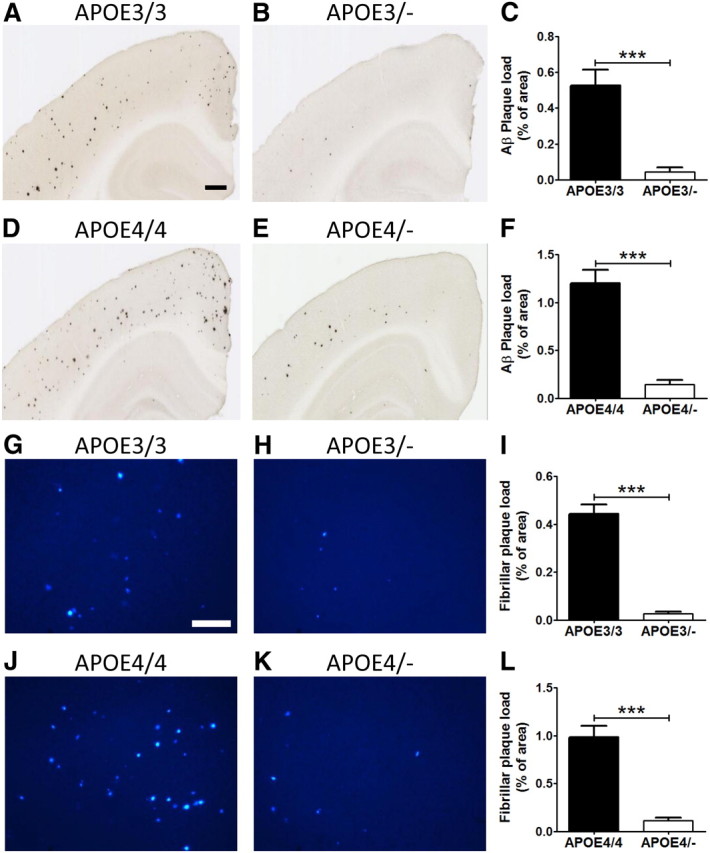
Haploinsufficiency of human APOE leads to reduction of Aβ and fibrillar Aβ plaque deposition. A, B, D, E, Brain sections from 3-month-old APOE homozygous (A, D) and hemizygous (B, E) mice were immunostained for amyloid with anti-Aβ antibody (HJ3.4-biotin). Scale bars, 400 μm. A–C, The extent of Aβ deposition detected by HJ3.4-biotin antibody was quantified (C) from cortex of APOE3/3 (A) and APOE3/− mice (B). D–F, The extent of Aβ deposition detected was quantified (F) from cortex of APOE4/4 (D) and APOE4/− mice (E). G, H, J, K, Brain sections from 3-month-old APOE homozygous (G, J) and hemizygous (H, K) mice were stained with X-34 dye that recognizes only fibrillar plaques. Scale bar, 200 μm. G–I, Fibrillar plaque load detected by X-34 dye was quantified (I) from cortex of APOE3/3 homozygous (G) and APOE3/− mice (H). J–L, The extent of fibrillar plaque load was also analyzed (L) from cortex of APOE4/4 (J) and APOE4/− mice (E). (n = 12–20 per genotype).
APOE dosage-dependent modulation of neuroinflammatory response
Aberrant inflammatory responses, such as activation of microglia, are common pathological features in the brains of AD patients. To identify activated microglia in the brain, mouse brain sections were stained with anti-CD45 antibody, a marker of microgliosis. Plaque-associated microglial activation was evident around Aβ deposits in APPPS1-21 mice with APOE isoforms (Fig. 4A–D). Quantitative analysis of microgliosis indicated an ∼95% decrease in the CD45-positive activated microglial load in APOE3/− mice, compared with APOE3/3 littermates (Fig. 4E). A similar dramatic phenotype was observed between APOE4/4 and APOE4/− mice (Fig. 4C,D). There was an ∼97% reduction in microgliosis in APOE4/− mice (Fig. 4F). These findings indicate that the reduction of Aβ and amyloid formation by APOE haploinsufficiency is closely associated with an attenuation of the microglial neuroinflammatory response. We also analyzed the levels of presynaptic and postsynaptic proteins. Analysis of synaptophysin, GluR2/3/4, NMDA receptor 2b, and PSD-95 levels indicated there was no significant alteration in these proteins between genotypes or APOE dosage levels (Fig. 4G).
Figure 4.
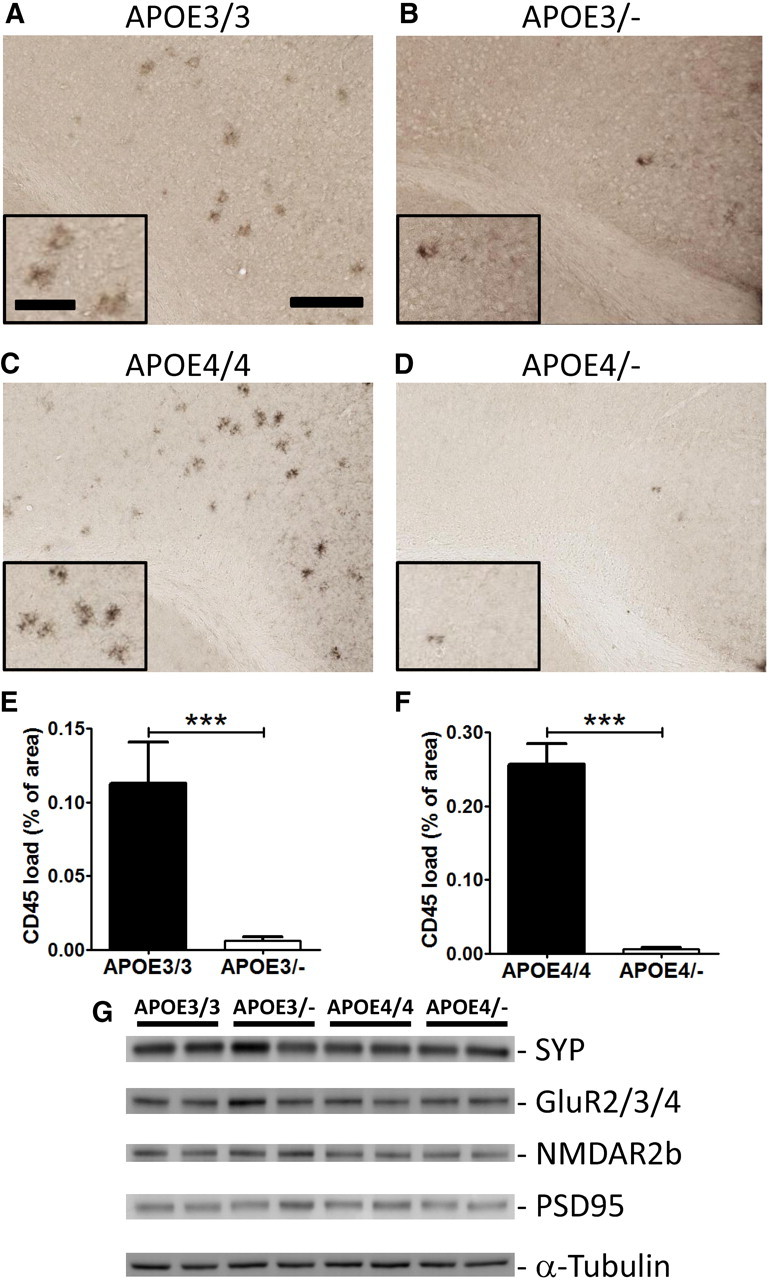
Attenuation of microgliosis in human APOE haploinsufficient mice. A–D, Brain sections from 3-month-old male APOE homozygous (A, C) and male hemizygous (B, D) mice were immunostained with an antibody against activated microglial CD45. E, The percentage area covered by CD45 staining was quantified from the cortex of APOE3/3 and APOE3/− mice. F, The extent of microgliosis was also quantified from the cortex of APOE4/4 and APOE4/− mice. Scale bars: lower magnification, 200 μm; higher magnification, 90 μm. n = 6–12 male mice per genotype. G, Levels of synaptophysin, GluR2/3/4, NMDA receptor 2b, and PSD-95 levels were measured by Western blotting (n = 4 per genotype). Levels of these proteins after normalizing with tubulin signals were not significantly altered by APOE genotype or haploinsufficiency.
Discussion
We tested whether decreasing human apoE levels will increase or decrease Aβ accumulation in vivo. Given the importance of this question, there have been numerous attempts to investigate the effects of modulating apoE levels on AD pathogenesis (Ashford, 2002; Rebeck et al., 2002; Teter et al., 2002). However, studies ranging from in vitro Aβ aggregation, ex vivo Aβ assays, and pharmacological approaches have provided conflicting results (Kim et al., 2009a). Most in vivo studies also provided limited information regarding the effect of human apoE modulation, since they indirectly inferred the effect of human apoE based on the manipulation of mouse apoE. To overcome some of the limitations of previous studies, we took a genetic approach by generating human APOE homozygous or hemizygous mice without the presence of the confounding endogenous mouse Apoe gene. Aβ-depositing mouse models with two copies (APOE3/3 or APOE4/4) or one copy (APOE3/− or APOE4/−) of each APOE isoform were successfully generated. APOE3/− and APOE4/− mice had significantly less Aβ accumulation, amyloid deposition, and microgliosis compared with the respective homozygous littermates. Thus, decreasing human apoE levels may be an attractive therapeutic strategy for attenuating Aβ deposition and its downstream effects in AD.
Given the critical role of Aβ in AD pathogenesis, the effect of apoE isoforms on Aβ deposition has been extensively investigated. APOE isoform-dependent amyloid deposition has been consistently observed in humans and Aβ-depositing mouse models (Holtzman et al., 2000; Bales et al., 2009; Reiman et al., 2009; Morris et al., 2010). Consistent with previous studies with other APP mouse models, we also observed APOE isoform-dependent effects on Aβ deposition in the APPPS1-21 mouse model (E4 > E3). Since apoE levels differ between APOE4/4 and APOE3/3, with levels of apoE4 being lower than apoE3, it is tempting to speculate that the difference in Aβ accumulation between the apoE isoforms is due to differences in apoE3 versus apoE4 levels. However, the structures of apoE3 and apoE4 differ significantly from each other (Mahley et al., 2006), and the observed difference in Aβ deposition cannot be simply attributed to apoE4 levels being lower than apoE3. For example, APOE4/E4 mice have greater amyloid deposition than APOE3/− mice despite the fact that apoE4 levels are higher in the APOE4/E4 than in the APOE3/− mice. APOE gene dosage-dependent amyloid deposition could be due to an effect of an alteration in Aβ clearance and/or aggregation. Several studies demonstrated that apoE isoforms can differentially affect Aβ clearance (Deane et al., 2008; Jiang et al., 2008; Castellano et al., 2011). ApoE isoforms may be slowing Aβ clearance in an apoE-isoform-dependent manner (E4 > E3 > E2). This idea is supported by the fact that mice lacking apoE have accelerated Aβ clearance from the brain relative to apoE-expressing mice (DeMattos et al., 2004). Another potentially important mechanism is direct facilitation of Aβ fibrillogenesis by an ApoE and Aβ interaction. While there are a few conflicting reports (Kim et al., 2009a), several groups have reported that all three apoE isoforms increase Aβ fibrillogenesis, with the effect being most exacerbated with ApoE4 and the least with ApoE2 (Ma et al., 1994; Wisniewski et al., 1994; Castano et al., 1995).
Given the isoform-dependent effect on AD pathology, one therapeutic approach is to convert apoE4 to an apoE3-like protein by using a small molecule (Mahley et al., 2006). Disrupting the apoE–Aβ interaction could be another attractive therapeutic approach to prevent apoE-mediated Aβ aggregation (Sadowski et al., 2004). Previous studies suggested that modulation of apoE levels and lipidation state, independent of APOE genotype, may be another potential strategy to decrease Aβ accumulation (Cao et al., 2006; Wahrle et al., 2008; Kim et al., 2009b). Interestingly, deletion of ATP-binding cassette, subfamily A1 (Abca1) and liver x receptor (Lxr) genes decreased apoE levels but increased or had no effect on Aβ deposition (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005; Zelcer et al., 2007). In addition, several studies have shown that activation of Lxr increases mouse apoE and decreases Aβ levels (Jiang et al., 2008). However, it is difficult to interpret the effect of altering apoE level in these studies, since modulation of Abca1 and Lxr also influences the lipidation state of apoE. Whether reducing human apoE levels without affecting its lipidation state will promote or attenuate Aβ accumulation was not clear from previous studies. Our study demonstrates the beneficial effect of decreasing apoE levels on Aβ accumulation. Consistent with our finding, low levels of apoE were strongly associated with low cerebral Aβ load in humans (Darreh-Shori et al., 2011). APOE gene promoter studies also suggest that higher levels of apoE, regardless of isoform status, increase the risk of developing AD (Laws et al., 2003). APOE may also influence risk for AD by mechanisms that are not related to Aβ. We found no clear differences in presynaptic and postsynaptic proteins in APPPS1-21 mice of different APOE genotypes and doses. This, however, does not rule out the possibility the structural synaptic changes might be present that have been observed by others (Dumanis et al., 2009). Our results in which apoE levels are altered by changing gene dosage suggest that strategies to decrease apoE levels in the brain should be further explored for the prevention/treatment of AD. Changing apoE levels in an adult in the context of two copies of APOE is different than having a lifelong genetic alteration of APOE dosage as in our experiments. It will be important to see in the former context whether altering apoE levels in an adult with two endogenous copies of APOE has similar or different effects as we report herein.
Footnotes
This work was supported by NIH Grants AG13956 (D.M.H.) and P30NS069329 (J.K.), American Health Assistance Foundation grants (J.K., D.M.H.), NIH Neuroscience Blueprint Center Core Grant P30NS057105, and the Alafi Neuroimaging Laboratory Human. APOE knock-in and APPPS1-23 mice breeding pairs were gifts from Dr. Patrick Sullivan (Duke University, Durham, NC) and Dr. Mathias Jucker (University of Tübingen, Tübingen, Germany), respectively.
References
- Ashford JW. ApoE4: is it the absence of good or the presence of bad? J Alzheimers Dis. 2002;4:141–143. doi: 10.3233/jad-2002-4303. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D, Fukuchi K, Wan H, Kim H, Li L. Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol Aging. 2006;27:1632–1643. doi: 10.1016/j.neurobiolaging.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, Frangione B. Fibrillogenesis in Alzheimer's disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306:599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darreh-Shori T, Forsberg A, Modiri N, Andreasen N, Blennow K, Kamil C, Ahmed H, Almkvist O, Langstrom B, Nordberg A. Differential levels of apolipoprotein E and butyrylcholinesterase show strong association with pathological signs of Alzheimer's disease in the brain in vivo. Neurobiol Aging. 2011;32:2320, e15–32. doi: 10.1016/j.neurobiolaging.2010.04.028. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Maia LF, Burgess BL, Blain JF, Naus KE, McIsaac SA, Parkinson PF, Chan JY, Tansley GH, Hayden MR, Poirier J, Van Nostrand W, Wellington CL. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280:43243–43256. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009a;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A[beta] clearance. Neuron. 2009b;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Laws SM, Hone E, Gandy S, Martins RN. Expanding the association between the APOE gene and the risk of Alzheimer's disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem. 2003;84:1215–1236. doi: 10.1046/j.1471-4159.2003.01615.x. [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherovich L. The APOE4 conundrum. SciBX. 2009;2:1–3. [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW, Kindy M, LaDu MJ. Apolipoprotein E and Alzheimer's disease: the protective effects of ApoE2 and E3. J Alzheimers Dis. 2002;4:145–154. doi: 10.3233/jad-2002-4304. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, DeKosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, Mathews PM, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J Pathol. 2004;165:937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- Teter B, Raber J, Nathan B, Crutcher KA. The presence of apoE4, not the absence of apoE3, contributes to AD pathology. J Alzheimers Dis. 2002;4:155–163. doi: 10.3233/jad-2002-4305. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Castaño EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc Natl Acad Sci U S A. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]