

Abstract
Angiotensin-converting enzyme inhibitors (ACEis) are known to have antifibrotic effects on the heart and kidney in both animal models and humans. N-acetyl-seryl-aspartyl-lysyl-proline is a natural inhibitor of proliferation of hematopoietic stem cells and a natural substrate of ACEi that was reported to prevent cardiac and renal fibrosis in vivo. However, it is not clear whether N-acetyl-seryl-aspartyl-lysyl-proline participates in the antifibrotic effects of ACEi. To clarify this issue, we used a model of aldosterone-salt–induced hypertension in rats treated with the ACEi captopril either alone or combined with an anti-N-acetyl-seryl-aspartyl-lysyl-proline monoclonal antibody. These hypertensive rats had the following: (1) left ventricular and renal hypertrophy, as well as increased collagen deposition in the left ventricular and the kidney; (2) glomerular matrix expansion; and (3) increased ED1-positive cells and enhanced phosphorylated-p42/44 mitogen-activated protein kinase in the left ventricle and kidney. The ACEi alone significantly lowered systolic blood pressure (P=0.008) with no effect on organ hypertrophy; it significantly lowered left ventricular collagen content, and this effect was blocked by the monoclonal antibody as confirmed by the histological data. As expected, the ACEi significantly decreased renal collagen deposition and glomerular matrix expansion, and these effects were attenuated by the monoclonal antibody. Likewise, the ACEi significantly decreased ED1-positive cells and inhibited p42/44 mitogen-activated protein kinase phosphorylation in the left ventricle and kidney, and these effects were blocked by the monoclonal antibody. We concluded that in aldosterone-salt–induced hypertension, the antifibrotic effect of ACEi on the heart and kidney, is partially mediated by N-acetyl-seryl-aspartyl-lysyl-proline, resulting in decreased inflammatory cell infiltration and p42/44 mitogen-activated protein kinase activation.
Keywords: aldosterone-salt, hypertension, angiotensin-converting enzyme inhibitor, Ac-SDKP, collagen, mitogen-activated protein kinase, macrophage/monocyte infiltration
Angiotensin (Ang)-converting enzyme (ACE) inhibitors (ACEis) are important agents for treatment of hypertension, heart failure, and other cardiovascular and renal diseases. In vivo studies showed that ACEi significantly attenuated cardiac fibrosis in rats with heart failure induced by myocardial infarction,1 spontaneously hypertensive rats,2 and rats with aldosterone (ALDO)-salt hypertension3 and also prevented both cardiac and renal fibrosis in mice given deoxycorticosterone acetate-salt.4 N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP), a natural inhibitor of hematopoietic stem cell proliferation and a natural substrate of ACEi,5,6 was reported to inhibit rat cardiac fibroblast proliferation and collagen synthesis7,8 and mesangial cell proliferation.9 It also prevented left ventricular (LV) and renal fibrosis in hypertensive rats,10,11 as well as renal insufficiency and fibrosis in diabetic db/db mice or rats with established antiglomerular basement membrane nephritis.12,13 The antifibrotic effects of ACEi might be mediated by preventing degradation of endogenous Ac-SDKP and thereby increasing Ac-SDKP in plasma and tissue.6,14,15
ALDO-salt–induced hypertension is characterized by severe fibrosis in the heart and kidney, as well as extensive inflammatory reactions that are central to stimulation of collagen synthesis.11,16,17 It is believed that the mitogen-activated protein kinases (MAPKs), including the p42/44, p38, and JNK signaling pathways, may be involved in cardiac and renal fibrosis.18-21 Using an anti-Ac-SDKP antibody, we tested the hypothesis that the antifibrotic effects of ACEi in ALDO-salt–induced hypertension are at least partially mediated by Ac-SDKP. We examined whether an ACEi (captopril) or exogenous Ac-SDKP would blunt LV monocyte/macrophage infiltration, phosphorylation of MAPK, and collagen deposition in the heart and kidney and whether a blocking monoclonal antibody (mAb) to Ac-SDKP would antagonize the effects of ACEi and Ac-SDKP.
Methods
This study was approved by the Henry Ford Hospital Institutional Animal Care and Use Committee.
Animals and Experimental Design
Ten-week–old male Sprague–Dawley rats weighing 325 to 350 g (Charles River, Wilmington, DE) were anesthetized with sodium pentobarbital (50 mg/kg, IP), and the left kidney was removed. ALDO (Fisher; 0.75 μg/h) or Ac-SDKP (synthesized by Drs Domenico Regoli and Witold Neugebauer, University of Sherbrooke, Sherbrooke, Canada; 800 μg/kg/d) was infused subcutaneously for 6 weeks by osmotic minipump (Alzet 2 ML4), and rats receiving ALDO were given 1% NaCl/0.2% KCl to drink. Captopril (100 mg/kg/d) was given in drinking water, and IgG (Rockland, 400 μg/kg) or anti-Ac-SDKP mAb (Rockland, 400 μg/kg) was given intraperitoneally every other day. Ac-SDKP, captopril, IgG, and mAb were begun simultaneously with ALDO-salt and continued for 6 weeks. Rats were divided into 6 groups: (1) sham+IgG, (2) ALDO-salt +IgG, (3) ALDO-salt+captopril+IgG, (4) ALDO-salt+captopril+mAb, (5) ALDO-salt+Ac-SDKP+IgG, and (6) ALDO-salt+Ac-SDKP+mAb.
Before the study, we tested whether the mAb (1:1000) blocked the effect of Ac-SDKP on collagen synthesis when adult rat cardiac fibroblasts in culture were stimulated with endothelin-1.7 Collagen in control cells was 5.7±0.8 μg/mg of protein; it was 14.3±0.5 with endothelin-1 (10−8 mol/L), 5.3±0.7 with endothelin-1+Ac-SDKP (10−9 mol/L), and 14.2±1.5 with endothelin-1+Ac-SDKP+mAb. We also tested whether the mAb had any effect on collagen content in the heart and kidney of rats with ALDO-salt–induced hypertension and found that there were no significant differences between ALDO-salt alone and ALDO-salt combined with the mAb.
Systolic blood pressure (SBP) was measured by tail cuff every other week for 6 weeks. At the end of the experiment, animals were anesthetized with 50 mg/kg of pentobarbital sodium, the heart was stopped at diastole with an intraventricular injection of 15% KCl and rapidly excised, and the LV (including the septum) and right kidney were weighed and harvested. The samples were kept at −80°C until the assay.
Ac-SDKP Urine Levels
Urine was collected for 24 hours in vials containing lisinopril (final concentration, 10 μmol/L) and centrifuged at 2000g for 15 minutes. Urine Ac-SDKP was quantified using a competitive enzyme immunoassay kit (SPI-BIO) and expressed as micrograms per 24 hours.22
Hydroxyproline Assay
Collagen content of the LV and kidney was determined by hydroxyproline assay as described previously.11,23 Briefly, tissue was dried, homogenized, and hydrolyzed with 6 n HCl for 16 hour at 110°C. A standard curve of 0 to 5 μg of hydroxyproline was used. Data were expressed as micrograms of collagen per milligram of dry weight, assuming that collagen contains an average of 13.5% hydroxyproline.24
Picrosirius Red Staining for Detection of Collagen in the Heart
Sections measuring 6 μm were deparaffinized, rehydrated, and stained with picrosirius red using a modification of the method of Sweat et al.25 Briefly, sections were postfixed in Bouin’s fluid, followed by iron hematoxylin stain to show the nuclei. They were then stained with 0.1% picrosirius red for 1 hour and washed twice with 0.5% acetic acid. Images were obtained with a computerized digital camera (SPOT, Diagnostic Instruments) and collagen examined under a microscope (Nikon E600) using normal light and analyzed with SigmaScan Pro (Jandel Scientific).
Periodic Acid-Schiff Staining for Determination of Glomerular Matrix
The glomerular matrix was evaluated by periodic acid-Schiff staining (Sigma) following the standard method in the manual.26 The dark pink color in the glomerulus was considered to represent the extracellular matrix. Twenty five to 30 glomeruli on each slide were imaged at ×400 magnification. Images were analyzed by computerized imaging software (Microsuite Biological imaging software). The glomerular matrix was expressed as a percentage of the glomerular area.
Immunohistochemical Staining for Determination of Monocyte/Macrophage Count (ED1)
Sections measuring 6 μm were deparaffinized and rehydrated. To reveal antigen, they were boiled in 10 mmol/L of sodium citrate buffer (pH 6.0) for 10 minutes in a microwave oven. They were washed in distilled H2O and incubated in 3% H2O2 for 10 minutes at room temperature, then preincubated in blocking solution for 30 minutes at room temperature, and finally incubated with an mAb against rat macrophages/monocytes (ED1, 1:1000 dilution; Chemicon) overnight at 4°C. ED1 antigen was assayed with a Vectastain ABC kit (Vector Laboratories). Sections were developed with diaminobenzidine substrate (Vector) and counterstained with hematoxylin. ED1-positive cells in half of the LV were examined. Cells with dark brown staining were counted and expressed as cells per micrometer squared.
Western Blot for Detection of Total and Phosphorylated MAPK in the LV and Kidney
Protein (60 μg) from the LV or kidney extracts was subjected to 12% SDS-PAGE under reducing conditions. Proteins were transferred to a nitrocellulose membrane. The primary antibodies were a rabbit polyclonal antibody against p42/44, p38, or JNK and a rabbit polyclonal antibody against phosphorylated p42/44, p38, or JNK MAPK (1:1000, Cell Signaling Technology). Bands were quantified with a bioscanner; results were normalized for actin and expressed as fold increase compared with sham. The positive bands were both total and phosphorylated p42/44, molecular weight 42/44 kDa; both total and phosphorylated p38, molecular weight 43 kDa; both total and phosphorylated JNK1, molecular weight 46 kDa; and both total and phosphorylated JNK2/3, molecular weight 54 kDa.
Statistical Analysis
SBP was examined using ANOVA with repeated measures and a covariate factor (baseline SBP). All of the other variables were analyzed by 1-way ANOVA. Log transforms of variables were used to stabilize the variances. There were 6 groups: (1) sham+IgG, (2) ALDO-salt+IgG, (3) ALDO-salt+captopril+IgG, (4) ALDO-salt +captopril+mAb, (5) ALDO-salt+Ac-SDKP+IgG, and (6) ALDO-salt+Ac-SDKP+mAb. Seven comparisons were prespecified (1 versus 2, 2 versus 3, 2 versus 4, 2 versus 5, 2 versus 6, 5 versus 6, and 3 versus 4). Significance was judged using Hochberg’s method for posthoc comparisons. Values are expressed as mean±SEM. All Ps<0.05 are reported.
Results
SBP, LV, and Renal Hypertrophy
SBP in the ALDO-salt vehicle group increased significantly (P=0.001) compared with sham. Ac-SDKP alone or combined with the mAb for 4 weeks had no significant effect on hypertension. However, SBP was significantly lowered in the captopril group compared with vehicle (P=0.008); the mAb did not significantly block the lowering effect of ACEi on SBP (Table). The ratios LV weight/body weight and kidney weight/body weight were significantly increased in the ALDO-salt vehicle group (P=0.001), and neither captopril nor Ac-SDKP (with or without the mAb) significantly decreased LVH or renal hypertrophy (Table).
Table.
SBP, LV Weight/Body Weight Ratio, Right Kidney Weight/Body Weight Ratio, Right Ventricular Collagen Content, and Urinary Ac-SDKP Content in ALDO-Salt Hypertensive Rats Treated With ACEi Either Alone or Combined With an Anti-Ac-SDKP mAb
| Parameters | ALDO-Salt
|
|||||
|---|---|---|---|---|---|---|
| Sham (n=8) | Vehicle (n=10) | ACEi (n=7) | ACEi+mAb (n=9) | Ac-SDKP (n=10) | Ac-SDKP+mAb (n=8) | |
| SBP, mm Hg | 107±3 | 197±7* | 176±7*† | 184±4* | 193±8* | 184±7* |
| LVW/BW, mg/100 g | 178.8±4.1 | 281.8±10.8* | 271.5±11.1* | 250.6±10.1* | 273.1±27.6* | 254.2±8.2* |
| RKW/BW, mg/100 g | 426.2±17.4 | 809.7±39.8* | 738.8±68.9* | 786.0±39.3* | 825.5±51.5* | 759.0±65.6* |
| RV collagen, μg/mg of dry RV | 22.6±3.0 | 43.7±2.0* | 22.4±5.1† | 35.5±2.4‡ | 26.7±2.5† | 36.5±3.2§ |
| Urine Ac-SDKP, μg/24 hours | 1.2±0.2 | 3.1±0.7 | 11.4±1.0† | 11.5±1.7† | 6.8±1.5† | 4.8±0.9 |
LVW indicates LV weight; BW, body weight, RKW, right kidney weight; RV, right ventricle. Data are mean±SE.
P<0.005 vs sham;
P<0.05 vs ALDO-salt+vehicle;
P<0.05 vs ALDO-salt+ACEi;
P<0.05 vs ALDO-salt+Ac-SDKP.
Ac-SDKP Urine Levels
Urine Ac-SDKP excretion did not increase significantly after chronic ALDO-salt treatment (Table). ACEi increased Ac-SDKP 4-fold (P=0.0001), and infusion of Ac-SDKP elevated urine Ac-SDKP (P=0.018) compared with ALDO-salt vehicle. Chronic mAb treatment tended to decrease urine Ac-SDKP in the Ac-SDKP–treated rats but did not reach significance. Chronic mAb had no effect on urine Ac-SDKP in the ACEi-treated rats (Table).
LV, Right Ventricle, and Kidney Collagen Content
LV collagen was significantly increased in the ALDO-salt vehicle group (20.90±1.03 μg/mg dry LV weight) compared with sham (12.05±0.65; P=0.001), and this increase was significantly blunted by either captopril (14.88±0.7; P=0.002) or Ac-SDKP (13.97±1.1; P=0.001). The mAb significantly blunted the effect of captopril (P=0.029) and Ac-SDKP (P=0.039; Figure 1). Right ventricular collagen and kidney collagen content showed a tendency similar to the LV (Table and Figure 1).
Figure 1.
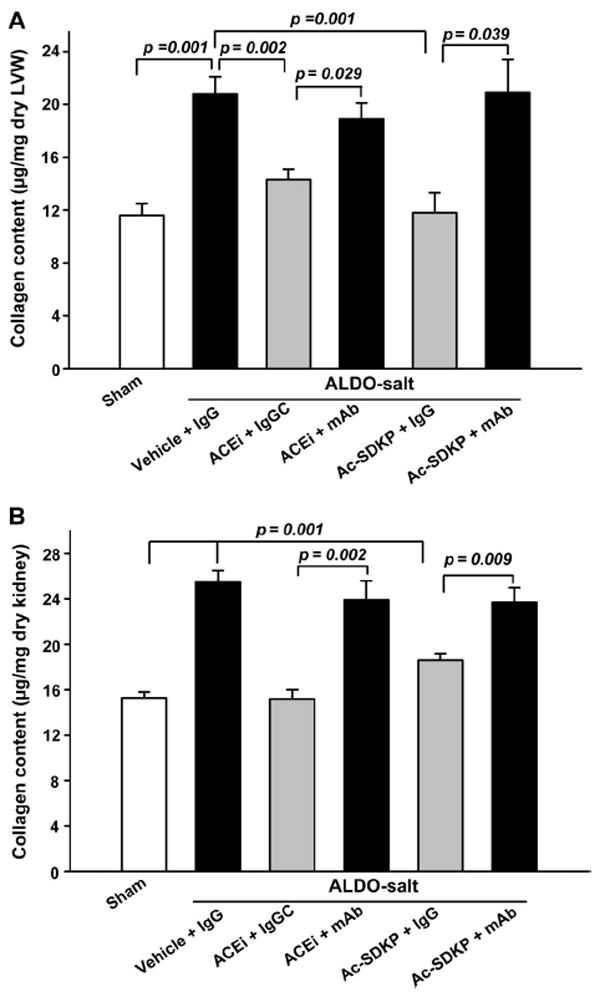
Collagen deposition in the LV (A) and kidney (B) in rats with ALDO-salt–induced hypertension after 6 weeks of ACEi or Ac-SDKP. Both prevented cardiac and renal fibrosis, and this effect was blunted by an anti-Ac-SDKP mAb (n=7 to 10).
LV Interstitial and Perivascular Collagen
LV interstitial collagen fraction was significantly increased in the ALDO vehicle group (9.86±0.99%) compared with control (4.73±0.14; P=0.0001), and this increase was significantly inhibited by either captopril (6.01±0.46; P=0.001) or Ac-SDKP (5.36±0.23; P=0.0001; Figure 2). The mAb significantly blocked the effect of captopril (P=0.023) and Ac-SDKP (P=0.0001; Figure 2). The ratio of perivascular collagen area to luminal area of the coronary arteries (PVCA/LA) was 4.94±0.56 in the sham group and increased significantly to 18.22±2.69 in the ALDO-salt hypertensive rats (P=0.0001; Figure 3). ACEi and Ac-SDKP significantly attenuated the increase in perivascular collagen, with PVCA/LA falling to 6.01±0.63 (P=0.0001) and 5.10±0.76 (P=0.0001), respectively. The mAb significantly blocked the effect of captopril (P=0.017) and Ac-SDKP (P=0.0001).
Figure 2.
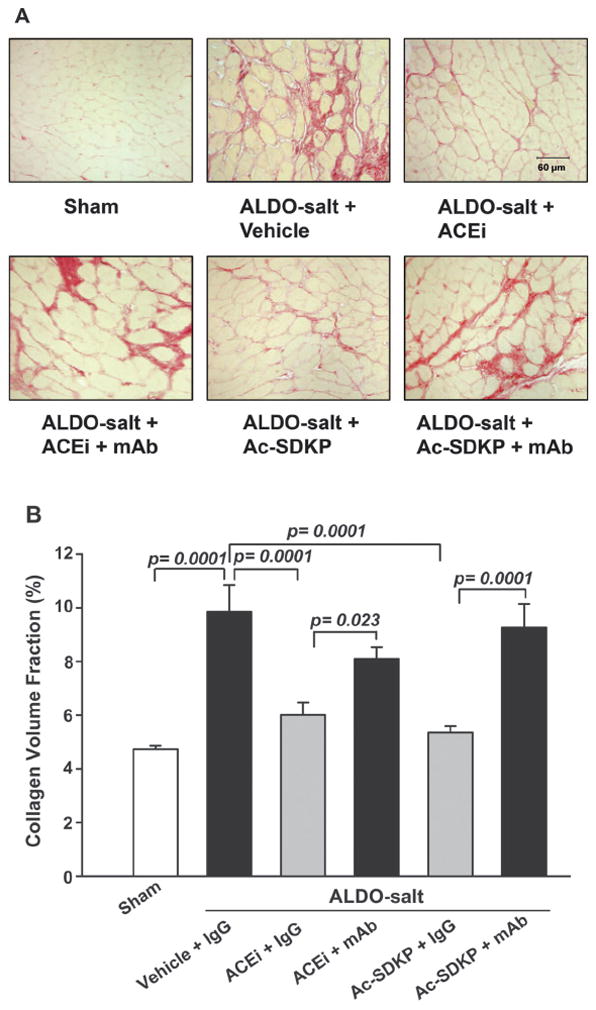
LV interstitial collagen deposition (red staining; A) in rats with ALDO-salt–induced hypertension after 6 weeks of ACEi or Ac-SDKP either alone or combined with an anti-Ac-SDKP mAb. B, Quantitative analysis of LV interstitial collagen. Both the ACEi and Ac-SDKP blunted cardiac fibrosis, and this effect was diminished by an anti-Ac-SDKP mAb (n=7).
Figure 3.
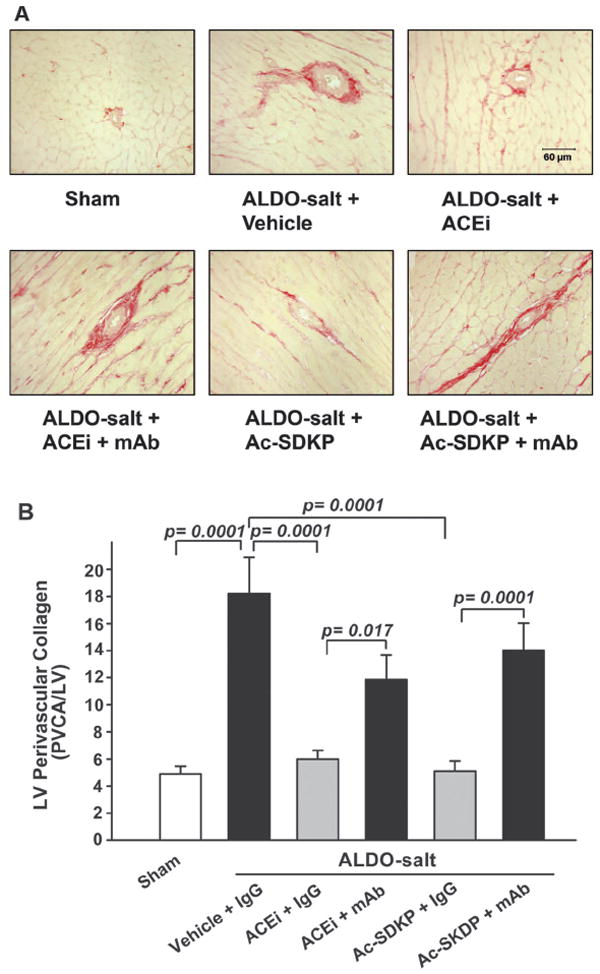
LV perivascular collagen deposition (red staining; A) in rats with ALDO-salt–induced hypertension after 6 weeks of ACEi or Ac-SDKP either alone or combined with an anti-Ac-SDKP mAb. B, Quantitative analysis of LV perivascular collagen, expressed as the ratio of perivascular collagen area to luminal area of the coronary arteries (PVCA/LV). Both blunted perivascular fibrosis, and this effect was diminished by an anti-Ac-SDKP mAb (n=7).
Glomerular Matrix
Glomerular matrix expansion was observed in ALDO-salt hypertensive rats compared with sham (22.2±1.7 versus 11.1±2.0; P<0.005). Both ACEi and Ac-SDKP significantly prevented glomerular matrix expansion in rats with ALDO-salt hypertension (P<0.05), whereas the mAb blocked the effect of the ACEi or Ac-SDKP (P<0.05; Figure 4).
Figure 4.
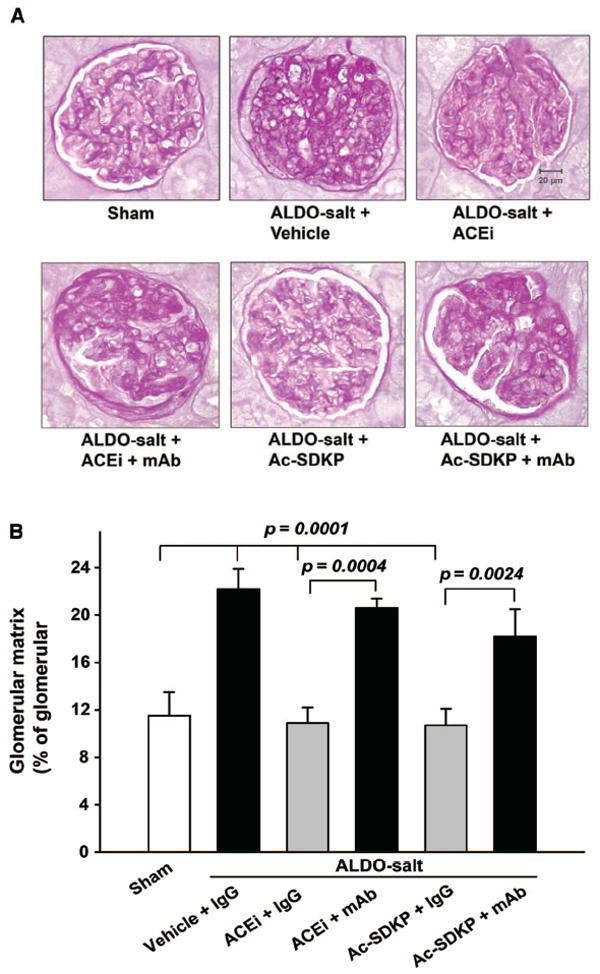
Representative periodic acid-Schiff staining (A) and quantitative data (B) for glomerular matrix in rats with ALDO-salt–induced hypertension after 6 weeks of ACEi or Ac-SDKP either alone or combined with an anti-Ac-SDKP mAb (n=6).
Macrophages/Monocytes in the LV and Kidney
ED1-positive cells (a marker for monocytes/macrophages) were localized to the interstitial space of the LV and different compartments of the kidney. The number of ED1-positive cells was significantly increased in the ALDO-salt vehicle group compared with control (P=0.001). Treatment with captopril and Ac-SDKP significantly reduced the number of ED1-positive cells in the LV and kidney (P<0.005; Figure 5), and the mAb blocked the anti-inflammatory effect of ACEi or Ac-SDKP.
Figure 5.
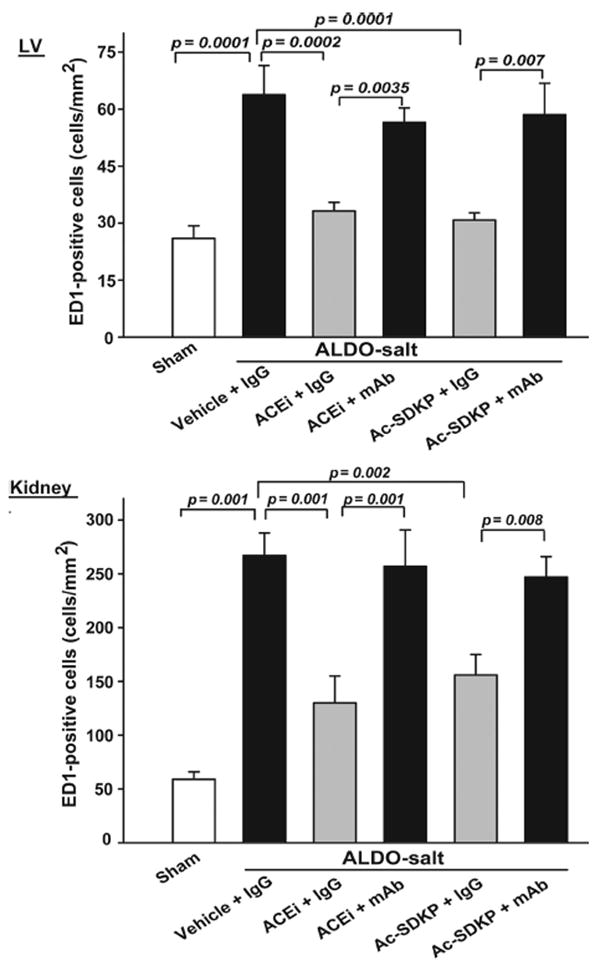
Quantitative analysis of ED1-positive cells (monocytes/macrophages) in rats with ALDO-salt–induced hypertension after 6 weeks of ACEi or Ac-SDKP treatment in the presence or absence of an anti-Ac-SDKP mAb. LV and renal inflammatory cell infiltration were increased in rats given ALDO-salt+vehicle compared with sham. These effects were blunted by the ACEi or Ac-SDKP, whereas the anti-Ac-SDKP mAb blocked both the ACEi and Ac-SDKP (n=6).
MAPK Expression in the LV and Kidney
Phospho-p42/44 MAPK was highly expressed in the LV of rats with ALDO-salt hypertension (2.7±0.5-fold increase; P=0.001). The ACEi (1.5±0.2; P=0.0013) or Ac-SDKP (1.2±0.2; P=0.031) lowered p42/44 MAPK phosphorylation, whereas the mAb blocked the effect of the ACEi or Ac-SDKP on phospho-p42/44. p42/44 MAPK protein content in the LV did not change in ALDO-salt hypertension and was not affected by either the ACEi or Ac-SDKP (Figure 6A). Phospho-p42/44 MAPK in the kidney showed a tendency similar to the LV (Figure 7). Phosphorylated p38 and C-jun N-terminal protein kinase (JNK) were not detectable with Western blot in either the LV or kidney among the 6 groups.
Figure 6.
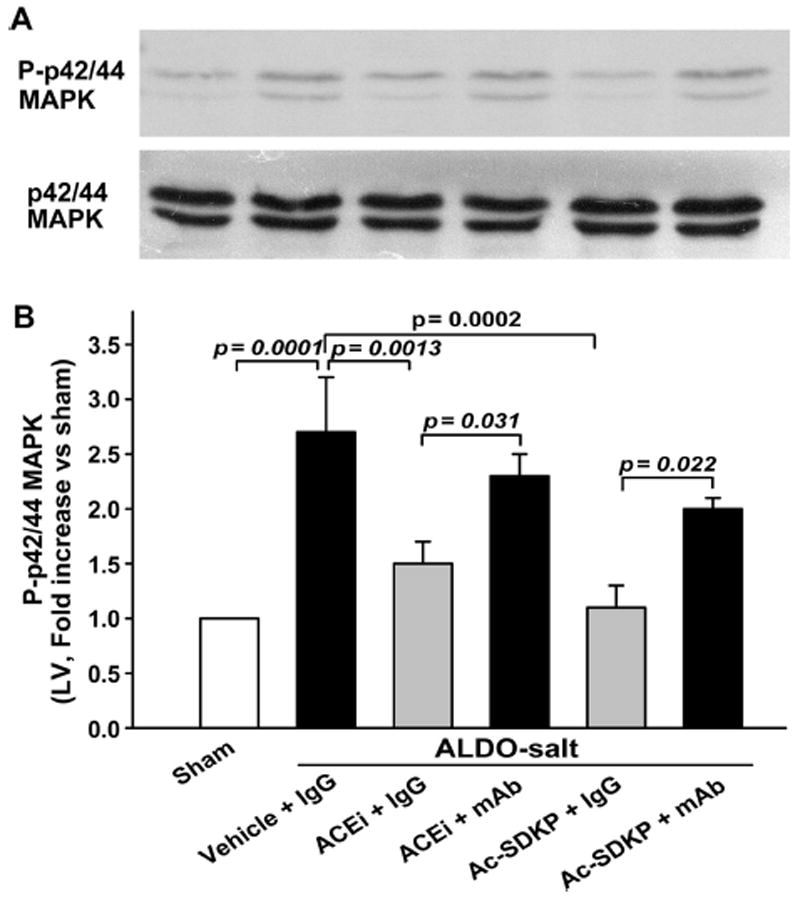
Representative Western blot (A) and corresponding data (B; n=6 to 7) illustrating phospho(P)-p42/44 MAPK and p42/44 MAPK protein expression in the LV as 2 bands of 42 and 44 kDa. Total p42/44 MAPK expression was equally apparent in all of the groups. P-p42/44 MAPK was highly expressed in the LV of rats with ALDO-salt–induced hypertension. Either ACEi or Ac-SDKP lowered P-p42/44 MAPK expression, whereas an anti-Ac-SDKP mAb blocked both the ACEi and Ac-SDKP.
Figure 7.
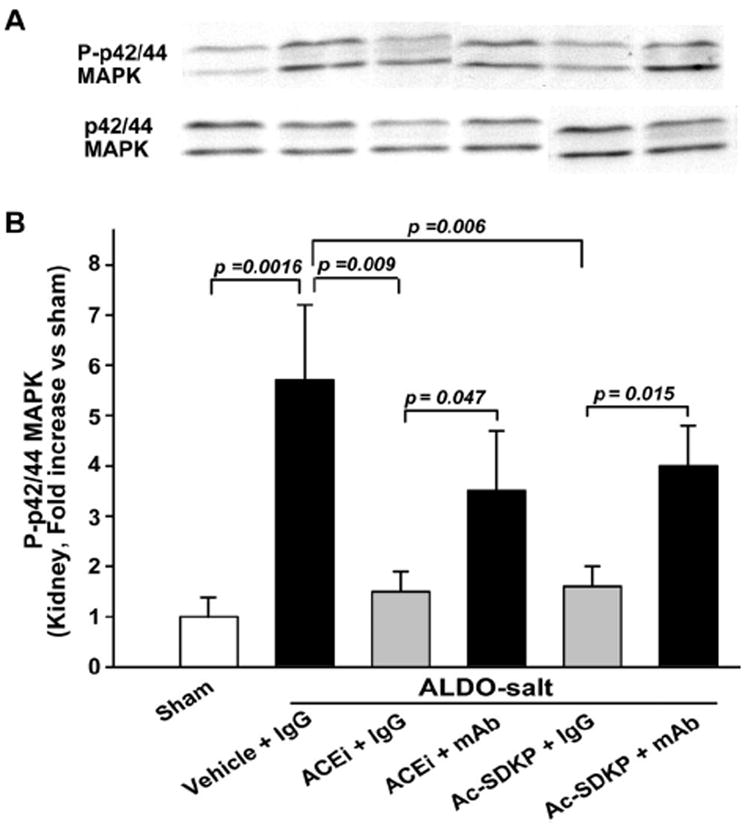
Representative Western blot (A) and corresponding data (B; n=6 to 7) illustrating phospho(P)-p42/44 MAPK and p42/44 MAPK protein expression in the kidney of rats with ALDO-salt–induced hypertension after 6 weeks of ACEi or Ac-SDKP treatment in the presence or absence of an anti-Ac-SDKP mAb.
Discussion
ACEis are commonly used to treat hypertension and congestive heart failure, to avert remodeling after myocardial infarction, and to ameliorate diabetic nephropathy and other renal diseases related to fibrosis.27-30 ACE is known to hydrolyze a number of peptides, and the therapeutic effects of ACEi have been attributed mainly to inhibition of both conversion of Ang I to II and kinin hydrolysis. Recently we showed that another peptide, Ac-SDKP, has antifibrotic effects on the heart and kidney10,11 and mediates some of the effects of ACEi in hearts of hypertensive rats,31 because ACEi prevented degradation of endogenous Ac-SDKP and significantly raised its circulating levels in Ang II–induced hypertension.15 However, we could not be sure whether Ac-SDKP also mediates the antifibrotic effects of ACEi in Ang II–independent hypertension. To clarify this issue, we used an ALDO-salt hypertensive rat model and a more direct approach, blocking the effect of endogenous Ac-SDKP with an Ac-SDKP–specific mAb to test our hypothesis that the anti-inflammatory and antifibrotic effects of ACEi in Ang II–independent hypertension are partially mediated by Ac-SDKP.
In ALDO-salt–induced hypertension, both ACEi and Ac-SDKP prevented the following: (1) collagen deposition in the heart and kidney, (2) glomerular matrix expansion, (3) LV and renal monocyte/macrophage infiltration, and (4) increased phosphorylation of p42/44 MAPK in the heart and kidney. Both the ACEi and Ac-SDKP were significantly blocked by an mAb against Ac-SDKP. Thus, we concluded that in ALDO-salt–induced hypertension, the beneficial effects of ACEi are partially mediated by Ac-SDKP. The ACEi alone significantly lowered SBP, confirming our previous observation.32 Because blocking Ac-SDKP with an mAb did not blunt the blood pressure–lowering effect of ACEi, and Ac-SDKP treatment had no effect on SBP, we ruled out the possibility that Ac-SDKP might alter the effect of ACEi. We and others have demonstrated previously that B2 kinin receptors mediate the antihypertensive effect of ACEi in mineralocorticoid-induced hypertension.32,33 ACEi or Ac-SDKP had no effect on organ hypertrophy. Urinary Ac-SDKP increased 4- and 2-fold in rats treated with the ACEi and Ac-SDKP, respectively. Increased Ac-SDKP in the urine tended to be decreased by the mAb only in rats given Ac-SDKP but not the ACEi for reasons that are not clear, and further studies focusing on the role of the kidneys in production and degradation of Ac-SDKP are needed to clarify this matter. It is known that, in humans, Ac-SDKP is found in plasma and circulating mononuclear cells.22 In mice, it is distributed in several tissues, including the lung, kidney, and heart.34 Therefore, one might speculate that the renal tubule is the main source of Ac-SDKP, because an IgG antibody cannot travel through the glomeruli and neutralize Ac-SDKP excreted by the renal tubule. Thymosin-β4 is the most likely precursor of the tetrapeptide, because its N-terminus contains Ac-SDKP. We have reported that the enzyme responsible for release of Ac-SDKP is prolyl oligopeptidase.35 Ac-SDKP is cleaved to an inactive form by the NH2-terminal catalytic domain of ACE.6 It has a 4.5-minute half-life in the circulation and is probably released continuously.14
In vitro Ac-SDKP inhibits cardiac fibroblast proliferation and collagen synthesis, as well as mesangial cell proliferation.7,9 We have shown that this tetrapeptide has an antifibrotic effect on the heart and kidney in rats with ALDO-salt11 or 2-kidney, 1-clip hypertension.10 Ac-SDKP prevented macrophage/monocyte infiltration and decreased transforming growth factor-β and connective tissue growth factor expression.36 These studies clearly indicate that exogenous administration of Ac-SDKP in vitro and in vivo has an antifibrotic effect. In our present work, as well as another recent study,31 we found that the antifibrotic effect of ACE inhibition was partially mediated by endogenous Ac-SDKP. This is further supported by a study37 showing that transgenic rats with cardioselective overexpression of ACE exhibited marked cardiac fibrosis, whereas cardiac Ang II concentrations were normal, suggesting that development of fibrosis in this model involves a decrease in Ac-SDKP.
In the present study we questioned how ACE inhibition via Ac-SDKP may decrease fibrosis, focusing on the effects of ACEi on monocyte/macrophage infiltration and activation of MAPK (p42/44, p38, and JNK) signaling. ALDO-salt hypertension causes an inflammatory reaction leading to extensive cardiac and renal fibrosis.11,16,17 Previously we showed that in vivo Ac-SDKP prevented LV macrophage/monocyte infiltration in Ang II–induced hypertension15 and in rats with myocardial infarction.36 It was reported recently that Ac-SDKP ameliorated the progression of renal dysfunction and fibrosis in rats with established antiglomerular basement membrane nephritis by reducing macrophage accumulation in the glomeruli and tubulointerstitium.13 Here we confirmed these findings and demonstrated that Ac-SDKP participates in the anti-inflammatory effect of ACEi in the heart and kidney, because it was partially blocked by the mAb.
The p42/44, p38, and JNK MAPK pathways are distinct serine-threonine kinase cascades involved in fibrogenic processes.38,39 In vitro studies showed that ALDO stimulated proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling.40 Our previous in vitro study showed that Ac-SDKP blunted p42/44 MAPK activity in serumstimulated cardiac fibroblasts, and a selective inhibitor for p42/44 but not p38 and JNK MAPK significantly prevented endothelin-1–induced collagen synthesis, suggesting that p42/44 MAPK plays a major role in this process.7 The expression of fibronectin in mesangial cells induced by connective tissue growth factor reportedly involves Src, p42/44 MAPK, and protein kinase B pathways21; in type 2 diabetic KKAy/Ta mice characterized by mesangial matrix accumulation and tubulointerstitial fibrosis, monocyte chemoattractant protein-1 expression and extracellular signalregulated kinase 1/2 and p38 MAPK phosphorylation were significantly increased,20 suggesting that phosphorylation of p42/44 MAPK or inflammatory cell infiltration may be involved in the pathophysiological changes of renal fibrosis. The present study clearly showed that phosphorylated p42/44 MAPK was significantly increased in the LV and kidney, whereas both the ACEi and Ac-SDKP decreased phosphorylated p42/44 MAPK but had no effect on its protein; these effects were blocked by the mAb, suggesting that Ac-SDKP can block the activation of p42/44 MAPK in vivo and blocking phosphorylation of p42/44 MAPK may greatly account for the antifibrotic effects of ACEi (via Ac-SDKP). Surprisingly, the phosphorylated p38 and JNK MAPK were not detectable with Western blot, and neither ACEi nor Ac-SDKP had any effect on p38 and JNK MAPK protein, suggesting that p42/44 MAPK phosphorylation mediates ALDO-induced fibrogenic processes not only in vitro but also in vivo.40 LV p38 MAPK and renal p38 MAPK phosphorylation were upregulated in myocardial infarction18 and type 2 diabetic KKAy/Ta mice,20 respectively, but not in ALDO-salt–induced hypertension, suggesting that specific MAPK pathways are involved in different pathophysiological processes. Thus, inhibition of p42/44 MAPK phosphorylation and prevention of macrophage/monocyte infiltration by Ac-SDKP and ACEi (acting via endogenous Ac-SDKP) in ALDO-salt hypertensive rats may be important factors in mediating the antifibrotic effect of ACEi.
In summary, ACE inhibition increased circulating Ac-SDKP as reflected by urine Ac-SDKP levels, which, in turn, blocked LV and renal inflammatory cell infiltration, p42/44 MAPK activation, and collagen deposition. This was confirmed by the fact that neutralizing Ac-SDKP function in vivo with an anti-Ac-SDKP mAb significantly blunted these effects. These findings suggest that Ac-SDKP prevents cardiac and renal fibrosis by blocking collagen production in ALDO-salt hypertensive rats, most likely by suppressing inflammation and decreasing activation of the p42/44 MAPK signaling pathway.
Perspectives
ACEi increase plasma14 and tissue Ac-SDKP41 and decrease cardiac and renal fibrosis.1-3,42,43 We conclude that Ac-SDKP may be an important mediator of the anti-inflammatory and antifibrotic effects of ACEi in hypertension. In the future, Ac-SDKP agonists that are resistant to ACE might be used to prevent inflammation and fibrosis in the heart and kidney.
Acknowledgments
Sources of Funding This study was supported by National Institutes of Health grants HL 71806-01 (N-E.R.) and HL 28982 (O.A.C.).
Footnotes
Disclosures None.
References
- 1.Liu Y-H, Yang X-P, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks WW, Bing OH, Robinson KG, Slawsky MT, Chaletsky DM, Conrad CH. Effect of angiotensin-converting enzyme inhibition on myocardial fibrosis and function in hypertrophied and failing myocardium from the spontaneously hypertensive rat. Circulation. 1997;96:4002–4010. doi: 10.1161/01.cir.96.11.4002. [DOI] [PubMed] [Google Scholar]
- 3.Sun Y, Ratajska A, Zhou G, Weber KT. Angiotensin-converting enzyme and myocardial fibrosis in the rat receiving angiotensin II or aldosterone. J Lab Clin Med. 1993;122:395–403. [PubMed] [Google Scholar]
- 4.Peng H, Carretero OA, Alfie ME, Masura JA, Rhaleb N-E. Effects of angiotensin-converting enzyme inhibitor and angiotensin type 1 receptor antagonist in deoxycorticosterone acetate-salt hypertensive mice lacking Ren-2 gene. Hypertension. 2001;37:974–980. doi: 10.1161/01.hyp.37.3.974. [DOI] [PubMed] [Google Scholar]
- 5.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A. 1989;86:779–782. doi: 10.1073/pnas.86.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azizi M, Rousseau A, Ezan E, Guyene T-T, Michelet S, Grognet J-M, Lenfant M, Corvol P, Meénard J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. doi: 10.1172/JCI118484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhaleb N-E, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. doi: 10.1161/01.hyp.37.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pokharel S, Rasoul S, Roks AJM, van Leeuwen REW, van Luyn MJA, Deelman LE, Smits JF, Carretero O, van Gilst WH, Pinto YM. N-acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of Smad2 in cardiac fibroblasts. Hypertension. 2002;40:155–161. doi: 10.1161/01.hyp.0000025880.56816.fa. [DOI] [PubMed] [Google Scholar]
- 9.Kanasaki K, Haneda M, Sugimoto T, Shibuya K, Isono M, Isshiki K, Araki S, Uzu T, Kashiwagi A, Koya D. N-acetyl-seryl-aspartyl-lysyl-proline inhibits DNA synthesis in human mesangial cells via up-regulation of cell cycle modulators. Biochem Biophys Res Commun. 2006;342:758–765. doi: 10.1016/j.bbrc.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 10.Rhaleb N-E, Peng H, Yang X-P, Liu Y-H, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation. 2001;103:3136–3141. doi: 10.1161/01.cir.103.25.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb N-E. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. doi: 10.1161/01.hyp.37.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibuya K, Kanasaki K, Isono M, Sato H, Omata M, Sugimoto T, Araki S, Isshiki K, Kashiwagi A, Haneda M, Koya D. N-acetyl-seryl-aspartyl-lysyl-proline prevents renal insufficiency and mesangial matrix expansion in diabetic db/db mice. Diabetes. 2005;54:838–845. doi: 10.2337/diabetes.54.3.838. [DOI] [PubMed] [Google Scholar]
- 13.Omata M, Taniguchi H, Koya D, Kanasaki K, Sho R, Kato Y, Kojima R, Haneda M, Inomata N. N-acetyl-seryl-aspartyl-lysyl-proline ameliorates the progression of renal dysfunction and fibrosis in WKY rats with established anti-glomerular basement membrane nephritis. J Am Soc Nephrol. 2006;17:674–685. doi: 10.1681/ASN.2005040385. [DOI] [PubMed] [Google Scholar]
- 14.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. doi: 10.1161/01.hyp.30.5.1015. [DOI] [PubMed] [Google Scholar]
- 15.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhuo J, Sanchez-Mendoza A, Brigstock DR, Rhaleb N-E. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J Hypertens. 2004;22:593–603. doi: 10.1097/00004872-200403000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blasi ER, Rocha R, Rudolph AE, Blomme EAG, Polly ML, McMahon EG. Aldosterone/salt reduces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003;63:1791–1800. doi: 10.1046/j.1523-1755.2003.00929.x. [DOI] [PubMed] [Google Scholar]
- 17.Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart. Role of oxidative stress. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y-H, Wang D, Rhaleb N-E, Yang X-P, Xu J, Sankey SS, Rudolph AE, Carretero OA. Inhibition of p38 mitogen-activated protein kinase protects the heart against cardiac remodeling in mice with heart failure resulting from myocardial infarction. J Card Fail. 2005;11:74–81. doi: 10.1016/j.cardfail.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Xie Z, Singh M, Singh K. ERK1/2 and JNKs, but not p38 kinase, are involved in reactive oxygen species-mediated induction of osteopontin gene expression by angiotensin II and interleukin-1beta in adult rat cardiac fibroblasts. J Cell Physiol. 2004;198:399–407. doi: 10.1002/jcp.10419. [DOI] [PubMed] [Google Scholar]
- 20.Hagiwara S, Makita Y, Gu L, Tanimoto M, Zhang M, Nakamura S, Kaneko S, Itoh T, Gohda T, Horikoshi S, Tomino Y. Eicosapentaenoic acid ameliorates diabetic nephropathy of type 2 diabetic KKAy/Ta mice: involvement of MCP-1 suppression and decreased ERK1/2 and p38 phosphorylation. Nephrol Dial Transplant. 2006;21:605–615. doi: 10.1093/ndt/gfi208. [DOI] [PubMed] [Google Scholar]
- 21.Crean JK, Finlay D, Murphy M, Moss C, Godson C, Martin F, Brady HR. The role of p42/44 MAPK and protein kinase B in connective tissue growth factor induced extracellular matrix protein production, cell migration, and actin cytoskeletal rearrangement in human mesangial cells. J Biol Chem. 2002;277:44187–44194. doi: 10.1074/jbc.M203715200. [DOI] [PubMed] [Google Scholar]
- 22.Pradelles P, Frobert Y, Creéminon C, Liozon E, Masseé A, Frindel E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun. 1990;170:986–993. doi: 10.1016/0006-291x(90)90489-a. [DOI] [PubMed] [Google Scholar]
- 23.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 24.Chiariello M, Ambrosio G, Cappelli-Bigazzi M, Perrone-Filardi P, Brigante F, Sifola C. A biochemical method for the quantitation of myocardial scarring after experimental coronary artery occlusion. J Mol Cell Cardiol. 1986;18:283–290. doi: 10.1016/s0022-2828(86)80410-2. [DOI] [PubMed] [Google Scholar]
- 25.Sweat F, Puchtler H, Rosenthal SI. Sirius red F3BA as a stain for connective tissue. Arch Pathol. 1964;78:69–72. [PubMed] [Google Scholar]
- 26.Melhem MF, Craven PA, Liachenko J, DeRubertis FR. A-Lipoic acid attenuates hyperglycemia and prevents glomerular mesangial matrix expansion in diabetes. J Am Soc Nephrol. 2002;13:108–116. doi: 10.1681/ASN.V131108. [DOI] [PubMed] [Google Scholar]
- 27.Solomon SD, Skali H, Bourgoun M, Fang J, Ghali JK, Martelet M, Wojciechowski D, Ansmite B, Skards J, Laks T, Henry D, Packer M, Pfeffer MA for the OVERTURE Investigators. Effect of angiotensin-converting enzyme or vasopeptidase inhibition on ventricular size and function in patients with heart failure: the Omapatrilat Versus Enalapril Randomized Study of Utility in Reducing Events (OVERTURE) echo-cardiographic study. Am Heart J. 2005;150:257–262. doi: 10.1016/j.ahj.2004.09.056. [DOI] [PubMed] [Google Scholar]
- 28.McQueen MJ, Lonn E, Gerstein HC, Bosch J, Yusuf S. The HOPE (Heart Outcomes Prevention Evaluation) study and its consequences. Scand J Clin Lab Invest. 2005;240(suppl):143–156. doi: 10.1080/00365510500236366. [DOI] [PubMed] [Google Scholar]
- 29.HOPE/HOPE-TOO Study Investigators. Long-term effects of ramipril on cardiovascular events and on diabetes. Results of the HOPE study extension. Circulation. 2005;112:1339–1346. doi: 10.1161/CIRCULATIONAHA.105.548461. [DOI] [PubMed] [Google Scholar]
- 30.Amann B, Tinzmann R, Angelkort B. ACE inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care. 2003;26:2421–2425. doi: 10.2337/diacare.26.8.2421. [DOI] [PubMed] [Google Scholar]
- 31.Peng H, Carretero OA, Vuljaj N, Liao T-D, Motivala A, Peterson EL, Rhaleb N-E. Angiotensin-converting enzyme inhibitors. A new mechanism of action. Circulation. 2005;112:2436–2445. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhaleb N-E, Yang X-P, Nanba M, Shesely EG, Carretero OA. Effect of chronic blockade of the kallikrein-kinin system on the development of hypertension in rats. Hypertension. 2001;37:121–128. doi: 10.1161/01.hyp.37.1.121. [DOI] [PubMed] [Google Scholar]
- 33.Chen K, Zhang X, Dunham EW, Zimmerman BG. Kinin-mediated anti-hypertensive effect of captopril in deoxycorticosterone acetate-salt hypertension. Hypertension. 1996;27:85– 89. doi: 10.1161/01.hyp.27.1.85. [DOI] [PubMed] [Google Scholar]
- 34.Pradelles P, Frobert Y, Créminon C, Ivonine H, Frindel E. Distribution of a negative regulator of haematopoietic stem cell proliferation (AcSDKP) and thymosin b4 in mouse tissues. FEBS Lett. 1991;289:171–175. doi: 10.1016/0014-5793(91)81062-d. [DOI] [PubMed] [Google Scholar]
- 35.Cavasin MA, Rhaleb N-E, Yang X-P, Carretero OA. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension. 2004;43:1140–1145. doi: 10.1161/01.HYP.0000126172.01673.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang F, Yang X-P, Liu Y-H, Xu J, Cingolani O, Rhaleb N-E, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–236. doi: 10.1161/01.HYP.0000107777.91185.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pokharel S, van Geel PP, Sharma UC, Cleutjens JPM, Bohnemeier H, Tian X-L, Schunkert H, Crijns HJGM, Paul M, Pinto YM. Increased myocardial collagen content in transgenic rats overexpressing cardiac angiotensin-converting enzyme is related to enhanced breakdown of N-acetyl-Ser-Asp-Lys-Pro and increased phosphorylation of Smad2/3. Circulation. 2004;110:3129–3135. doi: 10.1161/01.CIR.0000147180.87553.79. [DOI] [PubMed] [Google Scholar]
- 38.Pagès G, Lenormand P, L’Allemain G, Chambard JC, Meloche S, Pouysségur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci U S A. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schorb W, Conrad KM, Singer HA, Dostal DE, Baker KM. Angiotensin II is a potent stimulator of MAP-kinase activity in neonatal cardiac fibroblasts. J Mol Cell Cardiol. 1995;27:1151–1160. doi: 10.1016/0022-2828(95)90051-9. [DOI] [PubMed] [Google Scholar]
- 40.Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol. 2003;284:H176–H184. doi: 10.1152/ajpheart.00421.2002. [DOI] [PubMed] [Google Scholar]
- 41.Junot C, Nicolet L, Ezan E, Gonzales M-F, Menard J, Azizi M. Effects of angiotensin-converting enzyme inhibition on plasma, urine, and tissue concentrations of hemoregulatory peptide Acetyl-Ser-Asp-Lys-Pro in rats. J Cardiovasc Pharmacol. 1999;291:982–987. [PubMed] [Google Scholar]
- 42.Kim S, Ohta K, Hamaguchi A, Omura T, Yukimura T, Miura K, Inada Y, Wada T, Ishimura Y, Chatani F, Iwao H. Role of angiotensin II in renal injury of deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 1994;24:195–204. doi: 10.1161/01.hyp.24.2.195. [DOI] [PubMed] [Google Scholar]
- 43.Kaneto H, Morrissey J, McCracken R, Reyes A, Klahr S. Enalapril reduces collagen type IV synthesis and expansion of the interstitium in the obstructed kidney of the rat. Kidney Int. 1994;45:1637–1647. doi: 10.1038/ki.1994.215. [DOI] [PubMed] [Google Scholar]