

Abstract
Recent studies suggest that cancer stem cells (CSCs) are responsible for cancer resistance to therapies. We therefore investigated how glioblastoma-derived CSCs respond to the treatment of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Neurospheres were generated from glioblastomas, characterized for CSC properties including self-renewal, cell differentiation and xenograft formation capacity, and analyzed for TRAIL-induced apoptosis, CASP8 genomic status, and caspase-8 protein expression. The neurosphere NSC326 was sensitive to TRAIL-induced apoptosis as evidenced by cell death and caspase-8, -3, and -7 enzymatic activities. In contrast, however, the neurosphere NSC189 was TRAIL-resistant. G-banding analysis identified five chromosomally distinguishable cell populations in the neurospheres. Fluorescence in situ hybridization revealed the variation of chromosome 2 copy number in these populations and the loss of CASP8 locus in 2q33-34 region in a small set of cell populations in the neurosphere. Immunohistochemistry of NSC189 cell blocks revealed the lack of caspase-8 protein in a subset of neurosphere cells. Western blotting and immunohistochemistry of human glioblastoma tumors demonstrated the expression of caspase-8 protein in the vast majority of the tumors as compared to normal human brain tissues that lack the caspase-8 expression. This study shows heterogeneity of glioblastomas and derived CSCs in the genomic status of CASP8, expression of caspase-8, and thus responsiveness to TRAIL-induced apoptosis. Clinic trials may consider genomic analysis of the cancer tissue to identify the genomic loss of CASP8 and use it as a genomic marker to predict the resistance of glioblastomas to TRAIL apoptosis pathway-targeted therapies.
Keywords: Apoptosis, cancer stem cells, caspase-8, glioblastoma, TRAIL
Introduction
Glioblastoma is the most common and malignant brain tumor and current standard therapies provide no curative treatments. Recent studies suggest that targeting of the apoptosis pathway of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) can drive glioblastoma cells into self-destruction, thus providing a novel therapy to this devastating disease (1). TRAIL can trigger apoptosis in cancer cells through binding of its death receptors, DR4 and DR5 on the cell surface that in turn recruit intracellular Fas-associated death domain (FADD) and caspase-8 for the formation of the death-inducing signaling complex (DISC). In the DISC, caspase-8 becomes activated and initiates the apoptotic cascade through cleavage of downstream effector caspases and activation of the mitochondrial apoptosis pathway (2). The TRAIL apoptotic pathway can be targeted by recombinant human TRAIL (rhTRAIL) and its agonistic DR4 and DR5 antibodies. Preclinical studies have demonstrated the presence of the TRAIL apoptotic pathway in cancer cell lines and the therapeutic effects of rhTRAIL and DR4 and DR5 antibodies in treating cell lines-derived xenografts, leading to the clinical trials of TRAIL apoptotic pathway-targeted cancer therapies (2). However, clinical trials of rhTRAIL and DR4/DR5 antibodies have shown that the vast majority of human cancers are resistant to the treatments (3).
Cancer stem cells (CSCs) in glioblastomas have been shown to be responsible for the cancer resistance to treatment (4, 5), raising the possibility that TRAIL apoptosis pathway may be defective in CSCs. The first evidence of the existence of CSCs was reported by Steindler and his colleagues in 2002 who generated neurospheres from glioblastoma tissues (6), following the approach established for normal neural stem cells by Reynolds and Weiss in 1992 (7). Dirks' laboratory reported the presence of CSCs in glioblastomas and isolated them from the tumor tissues through sorting with the neural stem cell surface marker CD133 (8, 9). Vescovi's (10) and Yu's laboratories (11) established the CSC properties of neurospheres while Fine's laboratory showed that these neurospheres retain the genomic properties of the original glioblastomas (12). Rich's laboratory further showed the role of CD133+ cells in tumor radioresistance (4), angiogenesis (13), and development of a hypoxic environment (14), while Yu's laboratory reported the chemoresistance of glioblastoma-derived CD133+ cells (5). However, not every glioblastoma contains CD133+ cells and CD133− cells in the tumor have been shown to have the CSC prosperities (15), raising the controversial issue regarding CD133 as the glioblastoma stem cell marker. Clinical studies have correlated neurosphere formation (16) with chemoresistance and studies of neurospheres have revealed signaling pathways activated in the CSC-enriched cultures such as Sonic Hedgehog and c-Myc and shown that targeting of these pathways may provide novel therapies to glioblastomas (17–19). These studies have established neurospheres as the CSC models of glioblastomas.
In examining TRAIL apoptosis pathways in glioblastoma-derived CSCs, we have generated neurospheres from early passage primary cultures of glioblastomas surgically removed from patients. The results presented here demonstrate the genomic and biologic heterogeneity of the TRAIL apoptosis pathway in CSC-enriched neurospheres. The TRAIL apoptosis pathway is intact in some neurospheres, and targeting this pathway can drive them into apoptosis through caspase-8-initiated apoptosis cascade. However, genomic loss of CASP8 in other neurospheres results in the resistance of the cells to TRAIL-induced apoptosis.
Materials and methods
Glioblastoma Primary Cultures, Cell Clones and Cell lines
Primary cultures were established from the tissues of glioblastomas (World Health Organization Grade IV) surgically removed from patients as reported previously (20). The cultures were grown in DMEM/F-12 (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen). Once established survived in culture, the cells were grown up in one-two passages, then cryopreserved and recovered either for cell cloning or neurosphere culture, based on the earlier report (11). For cell cloning, the cells were plated in 96-well plates at the density of one cell per well in DMEM/F-12 containing 10% FBS. The wells that contained single cells were identified and grown into monolayers in serum-containing culture conditions. These clones were expended from 96-well plates into larger culture dishes. Glioblastoma cell lines LN18 and LN443 were reported previously and grown in DMEM containing 10% FBS (21).
Neurosphere Cultures
Neurospheres were generated from the first one-two passages of glioblastoma primary cultures according to the protocol as reported by Yu's group (11). In brief, cells were plated in uncoated plastic dishes at a clonal density of 3,500 cells/cm2 in neurobasal medium (Invitrogen) supplemented with N2 (0.5×; Invitrogen), B27 (0.5×; Invitrogen), 1 mM L-Glutamine, epidermal growth factor (EGF; 50 ng/ml; Peprotech Inc, Rocky Hill, NJ) and fibroblast growth factor 2 (FGF2; 50 ng/ml; Peprotech Inc). The cultures were fed every 7 days by changing half of the medium. Once the neurospheres reached approximately 200–300 cells in size, they were dissociated by repeatedly triturating and then grown at the clonal density of 3,500 cells/cm2 in the same medium again for passage.
Neurosphere formation assay
Neurospheres were examined for the CSC properties in the following three assays. First, the self-renewing capacity was tested in a neurosphere formation assay. Dissociated cells from neurospheres were plated at 200 cells per well in 24-well plates and grown in the neurosphere culture conditions as described above for 14 days. The neurospheres formed were counted and presented as the percentage of the neurosphere forming cells over the total 200 cells plated.
Immunofluorescent staining of neurospheres
Neurospheres were freely floated in 96-well plates and fixed with freshly prepared fixative containing 4% paraformaldehyde and 0.4% Triton X-100 in PHEMO buffer [PIPES (0.068 mol/L), HEPES (0.025 mol/L), EGTANa2 (0.015 mol/L), MgC12_6H20 (0.003 mol/L), and DMSO (10% v/v), pH adjusted to 6.8] for 10 min at room temperature. The neurospheres were then incubated with primary mouse monoclonal antibody to nestin (Chemicon), CD133 (Milteny Biotech) and rabbit antibody to glial fibrilary acid protein (GFAP; Chemicon), then stained with Tex-Red or FITC-conjugated secondary anti-mouse and rabbit IgG (Jackson ImmunoResearch) and counterstained with 4',6-diamidino-2-phenylindole (DAPI; Vector Laboratories).
Differentiation assay
The neurospheres were dissociated by repeating triturating and plated on laminin-coated glass coverslips at 3 × 104 cells/cm2. The cells were grown in DMEM/F-12 medium supplemented with 5% fetal bovine serum (FBS) for seven days. The cells were fixed in a buffer containing 4% paraformaldehyde and 0.4% Triton X-100 and labeled with rabbit antibody to astrocytic marker GFAP and mouse monoclonal antibody to neuronal marker Tuj1. The cells were then stained with Tex-Red or FITC-conjugated anti-mouse and rabbit IgG (Jackson ImmunoREsearch) and finally counterstained with DAPI.
Mouse intracranial xenograft
The neurospheres were dissociated and stereotactically injected into the striatum of the right side of brains of five to six weeks old female NOD-SCID mice (National Cancer Institute). The mice were anesthetized with an intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) and immobilized on a stereotactic frame. A small incision was made in the scalp sagittal middle and a 2 mm burr hole was created. Two μl of serum-free culture medium containing dissociated cells were introduced through a 10 μl Hamilton syringe 1 mm anterior and 3 mm lateral to the bregma at 3 mm depth; and then the needles were removed and the scalp was sutured closed. The mice were then maintained to allow for the development of the orthotopic xenografts. The mice were sacrificed and brains were removed and submerged in 10% neutrally buffered formalin for histology and immunohistochemistry as described in the Materials and Methods. The animal studies were approved by Emory University Institutional Animal Care and Use Committee (IACUC ID#: 226-2008).
Cell Death Assay
Cell clones in serum culture and neurospheres in neurobasal medium were dissociated, respectively by trypsin treatment and repeating triturating, and plated in 96-well plates at 5 × 103 cells per well. Cell clones were grown in serum culture condition whereas neurospheres were grown in neurosphere culture conditions overnight. For cell death, cells were untreated or treated with 300 ng/ml of rhTRAIL (PeproTech, Inc, Rocky Hill, NJ) for 24 hours and then assessed by quantitation of the ATP present using Cell Titer-glo Luminescent Cell Viability Assay kit according to the manufacture's protocol (Promega). Cell death was calculated based on the formula: 1 − (luminescent density of cells treated / luminescent density of cells untreated) × 100.
Caspase Activity Assay
Caspase-8, -3 and -7 activities were examined using the Caspase-Glo®8 and Caspase-Glo®3/7 kits according to the manufacturer's protocol (Promega, Madison, WI). In brief, dissociated neurospheres were plated and grown overnight in 96-well plates (5 × 103 cells/well) and untreated or treated with rhTRAIL for 24 hours in the presence or absence of caspase-8 inhibitor, carbobenzyloxy-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl ketone (z-IETD-fmk; R & D Systems, Minneapolis, MN). The cells were then incubated with 100μl of Caspase-Glo®8 or Caspase-Glo®3/7 reagent per well at room temperature for one hour. The caspase activity was determined by caspase substrate luminescence as recorded by a Dynex MLX® luminometer.
Western Blotting for Protein Expression and Caspase Cleavage
For protein expression, neurospheres were lysed in a cell lysis buffer (50 mM Tris, 150 mM NaCl, 2mM EDTA, 10% glycerol, 1% Triton X-100, 1% protease inhibitor mixture and 1mM PMSF). Fifty micrograms of total protein from each lysate were separated through SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with the primary antibodies against DR4 (Diaclone, Besancon, France), DR5 (pROsCI. Inc., Poway, CA), FADD, caspase-8 (Medical & Biological Laboratories, Nagoya), Bcl-2 inhibitory BH3-domain-containing protein (Bid; Biosource International, Inc., Camarillo, CA), Bak (Upstate Biotechnology, Lake Placid, NY), Bax, β-actin (Santa Crux Biotechnology, Santa Cruz, CA), Smac (Biomol, Plymouth Meeting, PA), and caspase-9 (Cell Signaling).
For cleavage of caspase-8, -3 and DNA fragmentation factor 45 (DFF45), cell clones in the serum culture were grown untreated or treated with TRAIL for 6 hours and then lysed in the same lysis buffer. Fifty micrograms of total protein from each lysate were separated through SDS–PAGE and transferred to nitrocellulose membranes and the membranes were incubated overnight with mouse monoclonal antibody to caspase-8 (Medical & Biological Laboratories, Nagoya, Japan) and rabbit antibody to caspase-3 and DFF45 (StressGen, Victoria, British Columbia, Canada). The membranes were incubated for one hour with HRP-conjugated goat anti-mouse and anti-rabbit antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) and developed by chemiluminescence (Pierce).
Chromosome and Fluorescence in Situ Hybridization (FISH) Analysis
The cell clones in the monolayer culture and neurospheres were dissociated by trypsin treatment and repeating triturating, respectively and then subjected to chromosome analysis by trypsin-Giemsa banding (GTG banding) according to the standard procedure at the University of Oklahoma University Genetic Lab. For FISH analysis, the chromosome region-specific Bac clone for caspase-8 (RPCI11-575C6) and centromere DNA-specific probe CEP 2 for chromosome 2 (Vysis, Inc., Des Plaines, IL) were labeled respectively with SpectrumGreen-dUTP and SpectrumOrange-dUTP (Vysis) by nick translation. The procedures of denaturation, hybridization, and signal detection were carried out based on the standard procedure at the University of Oklahoma University Genetic Lab. Hybridization signals were captured and analyzed using a FISH workstation (Perceptive Scientific Instrument, Inc., League City, Tx).
SNP Array Analysis
One microgram of genomic DNA was analyzed from each clone and neurosphere using the Illumina HumanOmniExpress array which contains 733,202 markers that can be used for both SNP detection and assessment of copy number. Samples were prepared by the Emory's Cancer Genomics Shared Resource according to manufacturers protocol and arrays were scanned using the Illumina HiScan instrument. SNP genotypes and copy number were analyzed using the Illumina GenomeStudio Genotyping model. Additional copy number analysis was performed using Illumina KaryoStudio software.
Cell Block Preparation
Cell blocks were prepared from cell clones and neurospheres according to the standard procedure at the Emory Medical Laboratory. In brief, cell clones and neurospheres were suspended in medium and spun down in centrifuge tubes coated with collodion (Spectrum). The cell clone and neurosphere containing collodion bags were removed from the centrifuge tubes, placed in cassettes, fixed in formalin, dehydrated, and embedded in paraffin. Five-micron think sections were prepared from the paraffin blocks for histology and immunohistochemistry at the Emory Medical Laboratory.
Glioblastoma Tumor Tissues
Frozen tissues and matched paraffin sections of glioblastoma were kindly provided by the London (Ontario) Brain Tumor Tissue Bank (London Health Sciences Center, London, Ontario, Canada) as reported previously (22). Frozen normal brain tissues, surgically removed from epileptic brains, were also obtained from the same Bank. Total proteins were extracted from the tissues by homogenization in 1% Triton X-100 lysis buffer and subjected to Western blotting. Paraffin sections were subjected to histology and immunohistochemistry.
Histology and Immunohistochemistry
For histology, five-micron thick paraffin section were deparaffinized, rehydrated and stained with hematoxylin and eosin (H&E) according to the standard procedure at the Emory Medical Laboratory. For immunohistochemistry, five-micron thick paraffin sections were deparaffinized, rehydrated in deionized water, and then heated in citrate buffer (pH 6.0), using an electric pressure cooker for 3 minutes at 12–15 pounds per square inch (approximately 120°C). All slides were loaded onto an automated system (Dako AutoStainer plus) and exposed to 3% hydrogen peroxide for 5 minutes before incubation with goat-anti-human caspase-8 antibody at 1:80 (Santa Cruz Biotechnolocy) for 30 minutes. The slides were then exposed to labeled polymer (Envision+dual−link) for 30 minutes, 3,3'-diaminobenzidine chromogen for 5 minutes, and hematoxylin counter staining for 5 minutes at the Emory Medical Laboratory.
Results
Functional heterogeneity of glioblastoma-derived cell clones in TRAIL-induced apoptosis
Our earlier studies have shown the heterogeneity of glioblastoma cell lines in the response to TRAIL (23). To investigate how glioblastomas in patients might respond to TRAIL, we established primary cultures from glioblastoma tissues surgically removed from patients (20). Yu's laboratory has shown that primary cultures of early passages maintain CSC population (11) and, therefore, we sought to examine how the CSC and non-stem cell populations in the primary cultures responded to TRAIL treatment. To this end, we first generated non-stem cell clones from TRAIL-sensitive primary culture 326 and TRAIL-resistant primary culture 189 through limiting dilutions under serum culture (SC) conditions. Individual cells were identified per well, grown up, and treated with 300 ng/ml rhTRAIL for 24 hr. Cell death analysis revealed heterogeneous responses of the clones to TRAIL treatment (Fig. 1A). Western blot analysis revealed the presence of the DISC proteins including DR4, DR5, FADD and caspase-8 and mitochondrial apoptotic pathway proteins such as Bid, Bak, Bax, Smac and caspase-9 in all the clones from primary culture 326 (Fig. 1B). These proteins were also expressed in the majority of the clones from the primary culture 189; however, the expression of apoptosis-initiating caspase-8 was absent in the c9, c10, and c13 clones of primary culture 189. The clones of primary culture 189 and 326 were treated with 300 ng/ml rhTRAIL for 6 hr and examined by Western blot. The results showed that TRAIL treatment triggered caspase-8-initiated cleavage of caspase-3, a downstream substrate of caspase-8, and DNA fragmentation factor 45 (DFF45), a caspase-3 substrate, in the following clones: SC326 c4, c14, c16 and SC189 c11, c15 and c17, but not in SC326 c9, c11, c12 and SC189 c8, c9, c10 and c13 (Fig. 1C). This study indicates the heterogeneity of the cell clones derived from the primary culture 326 and 189, and that the levels of caspase-8 expression in part defined the sensitivity of the SC189 clones to TRAIL-induced apoptosis.
Fig. 1.

TRAIL-induced apoptosis in non-stem cell clones derived from SC326 and SC189 primary culture. A Each of the clones (indicated to the right) was treated with 1:2 serial dilutions of rhTRAIL starting at 300 ng/ml for 24 hr and the percentage of cell death was assessed by a luminescent cell viability assay. Data are mean +/− SEM (n = 6). B Western blot analysis of the expression of TRAIL apoptotic proteins (indicated to the left) in SC326 and SC189-derived non-stem cell clones. Actin was used as protein-loading control. C Western blot detection of cleavage of caspase-8 (Casp-8), caspase-3 (Casp3) and DFF45 (indicated to the left) in SC326 and SC189 clones after treatment with 300 ng/ml TRAIL for 6 hr.
Genomic heterogeneity of glioblastoma-derived cell clones
To explore the genomic basis for the functional heterogeneity of the SC189 clones, we first carried out chromosomal analysis of TRAIL-resistant SC189 c13 and TRAIL-sensitive SC189 c17. G-banding analysis revealed chromosomal alterations in both SC189 c13 and c17 with each of the clones having distinguishable chromosomal alterations different from each other. To our surprise, each clone contained at least two cell populations as distinguished by chromosomal abnormalities, suggesting the possibility that the clones might undergo mitotic non-disjunction events resulting in the generation of new clones after the initial plating of the clone during culture. Of the two populations in clone SC189c13, one had two copies and another showed one copy of chromosome 2 (Fig. 2A). Both populations in the SC189c17 clone contained two copies of chromosome 2 (Fig. 2C).
Fig. 2.

Chromosomal alterations in SC189 clones. A G-banding detection of two cell populations with distinguishable chromosomal alterations in clone SC189c13 with one having one copy and another containing two copies of chromosome 2. B FISH detection of one copy of CASP8 (green signal) and one copy of chromosome 2 (red signal) in one cell population and two copes of CASP8 and chromosome 2 in another cell population in SC189c13 at metaphase and interphase (inset). C G-banding findings for two cell populations based on their distinguishing chromosomal alterations including three copies of chromosome 2 in SC189c17. D FISH findings of three and two copes of CASP8 and chromosome 2, respectively, in two cell populations in SC189c17 at metaphase and interphase (inset).
Next, we examined the status of CASP8 by FISH using centromere DNA-specific probe CEP 2 for chromosome 2 and the 2q33-q34 region Bac clone for CASP8 gene which is located in the chromosomal region 2q33-q34 (24). The results showed that 70% of cells had loss of one copy of chromosome 2 and a copy of the CASP8 gene whereas 30% of cells showed two copies of chromosome 2 and the CASP8 gene in the TRAIL-resistant SC189 c13 clone (Fig. 2B). In the TRAIL-sensitive SC189 c17 clone, however, FISH identified 4% of the cells having two copies of chromosome 2 and the CASP8 gene and 96% of the cells with three copies of chromosome 2 and CAPS8 gene (Fig. 2D), indicating CASP8 amplification in the vast majority of the cell populations correlating with the overexpression of caspase-8 protein in this clone. These studies indicate that the genomic status of the CASP-8 gene defines the expression of caspase-8 protein in the clones and thereby the clone sensitivity to TRAIL-induced apoptosis.
Heterogeneity of CSC-enriched neurospheres in response to TRAIL-induced apoptosis
To examine how the CSC populations in primary cultures respond to TRAIL, we generated neurospheres (Fig. 3A, left panel) by placing the earlier passages of primary cultures 326 and 189 in neurosphere culture (NSC) conditions based on Yu's protocol (11). In addition, we attempted to generate single cell clones from the primary cultures under the same NSC culture condition and sought to determine whether single cells had the ability to form neurospheres. In brief, the primary cultures were dissociated and plated in 96-well plates at a density of one cell per well. The wells that contained single cells were identified, the cells were grown under NSC culture conditions for up to a month, but the single cells failed to grow up into neurospheres. Interestingly enough, neurospheres can be formed from a primary glioblastoma culture but not its-derived single cells under the same NSC culture conditions. We therefore used primary culture-derived neurospheres as CSC models in this study.
Fig. 3.

Characterization of NSC for CSC properties. A A neurosphere (left panel) and differentiated cells (right panel) from the 3rd passage of NSC189 (NSC189c3). B Neurosphere formation analysis of two passages (p3, p7) of NSC189 and NSC326 in 1:2 dilutions starting with 200 cells per well in 24-well plates with percentage of neurosphere forming cells presented. Data are mean +/− SEM (n = 6). C Immunofluorescent stainings of NSC189 neurospheres with antibody to stem cell marker nestin, astrocytic marker GFAP, and neuronal marker β-tubulin III counterstained with nuclear dye DAPI. D The differentiated cells, as shown above in A (right panel) on coverslips were examined by immunofluorescent stainings using astrocytic marker GAFP and neuronal marker β-tubulin III.
Once established, neurospheres were characterized for CSC properties in the following four steps. First, the self-renewing capacity of neurospheres was examined by neurosphere formation assay, showing that approximately 5–7% of cells had the capacity of forming neurosphere in NSC189 at passage 3 and 7 (NSC326p3/p7) and 15–20% cells in the same passages of NSC326 (NSC326p3/7) (Fig. 3B). Second, the stemness of the neurospheres was examined by immunofluorescent staining of neurospheres with the stem cell marker nestin. Results showed that the vast majority of the cells were positive for nestin (Fig. 3C). Thirdly, the multipotent property of neurospheres was tested by a differentiation assay. The differentiated neural cells were observed under phase contract microscope (Fig. 3A, right panel) and differentiated astrocytes and neurons were revealed by immunofluorescent staining using GFAP as an astrocyte marker and β-tubulin III as a neuronal marker (Fig. 3D). Finally, the tumorigenic capacity of neurospheres was confirmed by xenograft formation assay, in which dissociated neurosphere cells were injected into NOD-SCID mouse brains (Fig. 4). These series of studies demonstrated that NSC326 and NSC189 retained the CSC properties.
Fig. 4.
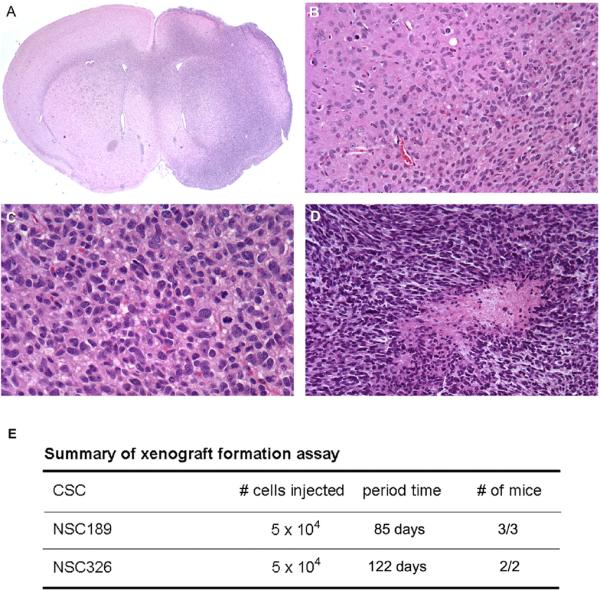
Xenograft formation assay of NSC189p3 showing the xenograft in the right side of the brain (A), infiltrating nature of the xenograft (B), mitotic activities (C), and a necrosis (D) in the xenograft. E Summary of xenograft formation assay of NSC189p3 and NSC326p3 with the numbers of cells injected, the period of time from the injection to xenograft formation and the number of mice with xenografts over total number of mice injected with the cells.
Neurospheres were then examined for TRAIL sensitivity. Neurospheres were dissociated, treated with 300 ng/ml of TRAIL and examined for TRAIL-induced apoptosis. Cell death analysis revealed that TRAIL treatment led to cell death more in NSC326p3/p7 than in NSC189p3/p7 (Fig. 5A), consistent with TRAIL sensitivity of their corresponding primary cultures (Fig. 1A). Caspase activity assay further demonstrated caspase-8, -3 and -7 activity more in NSC326p3/P7 than in NSC189p3/p7. The presence of either the caspase-8 inhibitor, z-IEDT or the pan-caspase inhibitor, z-VAD eliminated TRAIL-induced activation of caspase-8 (Fig. 5B) and caspase-3 and -7 in NSC326p3/p7 (Fig. 5C). Consistent with this cell death analysis was the Western blot finding that caspase-8 expression was lower in NSC189p3/p7 than in NSC326p3/p7 (Fig. 5 E). Taken together, these data indicate the heterogeneity of glioblastoma-derived neurospheres in the expression of caspase-8 protein and the response to TRAIL-induced apoptosis.
Fig. 5.

Heterogeneous response of neurospheres to rhTRAIL treatment. A Cell viability analysis of TRAIL-induced cell death in two passages (p3, p7) of NSC189 and NSC326 after treatment with 300 ng/ml rhTRAIL for 24 hr. Data are mean +/− SEM (n = 6). B–C Caspase-8 and Caspase-3/-7 activity measured in two passages of NSC326 and NSC189 after rhTRAIL treatment for 24 hrs in the presence or absence of caspase-8 inhibitor Z-IEDT and pan-caspase inhibitor z-VAD. Data are mean +/− SEM (n = 3). D Western blot analysis of the expression of DR5, FADD, caspase-8 (Casp-8) in the p3 and p7 passage of NSC326 and NSC189 with primary cultures (p) and clones (c12, c16, c13, c17) included as controls (the molecular weights are indicated to the right). Actin was used as the protein loading control.
SNP arrays establishing the common origin of glioblastoma-derived neurospheres and clones
To compare the genomic alterations of glioblastoma-derived neurosphere and serum-grown clones, we carried out genomic SNP arrays to determine the copy number and heterozygosity of these heterogeneous cultures. The copy number and heterozygosity (B allele frequency) of chromosome 2 obtained from the neurosphere NSC189p3 and the serum-grown clones SC189c13 and SC189c17 were analyzed. No copy number changes of chromosome 2 were detected in NSC-189; in contrast, however, whole chromosome loss was detected in SC189c13 (Fig. 6A). A complex pattern was observed for SC189c17 and composed of the gain of whole chromosome 2 with heterozygous segmental loss of the terminal portion of the short arm of chromosome 2 and an interstitial segment of the long arm which does not contain the CASP8 locus (Fig. 6B). Detection of an approximately 1.3 Megabase (Mb) loss of an interstitial portion of 2q37.3 in NSC189p3 and SC189c17 provided evidence that NSC189p3 and SC189c17 are derived from the same precursors. This copy number variant (CNV) has not been reported previously in the database of genomic variants (http://projects.tcag.ca/variation/). This CNV was not detected in NS189c13 and, instead, SNP arrays of this clone revealed a loss of the chromosome 2 containing the CNV on 2q37.3.
Fig. 6.

SNP array analysis of the neurosphere NSC189 and the clones SC189c13/c17. A Copy number (LogR Ratio) and heterozygosity (B allele frequency) data for chromosome 2 in NSC189 and SC189c13 and SC189c17. B Detection of an approximately 1.3 Megabase (Mb) loss of an interstitial portion of 2q37.3 in NSC189 and SC189c17. C Scatter plots of SNP Theta values comparing NSC-189 with SC189c13 and SC189c17. These data are consistent with all three cell populations being related to a common origin. D Disparate copy number profiles for NSC189, SC189c13, SC189c17 compared to a normal human DNA control. Blue (gain), yellow (heterozygous loss) and red (homozygous loss) are used to indicate copy number change. This figure was generated using the cnvPartition 3.1.6 plug-in in GenomeStudio.
To determine whether these heterogeneous neurospheres and clones are genomically related, a scatter plot analysis of SNP array data was carried out and demonstrated a statistically significant correlation between SNP Theta values comparing NSCp189 with SC189c13 and SC189c17 (Fig. 6C). These data are consistent with all three cell populations being related to a common origin. Disparate copy number profiles for each of the three cultures with an unrelated normal human DNA sample as a control illustrated the gain, heterozygous and homozygous loss in all the chromosomes (Fig. 6D).
Genomic heterogeneity of the cell populations in neurospheres
To our surprise, SNP arrays detected no genomic abnormality of chromosome 2 and CAPS8 loci in TRAIL-resistant NSC189p3. Since SNP arrays examine a pooled DNA from all the cell populations in the culture with the limit of detection for CNV of 20–30% of the cell population, we sought to further examine the individual cell populations in NSC189p3 by G-banding and FISH. Interestingly enough, G-banding identified five genomically distinguishable cell populations in this neurosphere: one population showed gains of chromosome 1, 3, and 9 but losses of chromosome 4, 11, 18 and Y; the second population had gains of chromosome 1, 3, 5, 21 and losses of chromosomal 2 and Y; the third had gains of chromosome 3 and 5 and losses of chromosome 15, 17, X and Y; the fourth had gains of chromosome 1, 3, 6, 9, and 21 but losses of chromosome 4, 11, 16, 18, and Y (Fig. 7A), and the fifth showed the gains of two copies of chromosome 1, one copy of chromosome 3, 9, 12, 21 and loss of one copy of chromosome 5, 10, 11, and two copies of chromosome Y (data not shown).
Fig. 7.

Genomic heterogeneity of NSC189. A G-banding detection of multiple genomically distinguished cell populations in NSC189p3. B FISH analysis of NSC189p3 at metaphase and interphase (inset) and detection of two copies of chromosome 2 (identified by CEP2 in red) and CASP8 (in green) in one cell population but one copy of chromosome 2 and CASP8 in another cell population. C Summary of the percentage of cells in NSC189p3 with copy numbers of chromosome 2 and CASP8 gene. D–E H&E sections (left) and immunohistochemistry (right) of NSC189p3 (D) and SC189c17 (E) showing the heterogeneous cell populations of NSC189p3 as determined by the cytoplasmic expression of caspase-8.
Consistent with the G-banding finding, FISH identified five cell populations based on the status of chromosome 2 and the CAPS8 gene (Fig. 7B): 51% of cells showed two copies of chromosome 2 and the CASP8 gene; 24% cells had one copy of chromosome 2 and the CASP8 gene; 13% cells revealed two copies of chromosome 2 but three copies of the CASP8 gene; 4% cells had two copies of chromosome 2 but four copies of the CASP8 gene; and 3% cells showed three copies of both chromosome 2 and the CASP8 gene (Fig. 7C).
In examining the histological features and caspase-8 expression in the heterogeneous cell populations in neurospheres, we prepared paraffin cell blocks from NSC189p3 neurospheres. H&E sections confirmed the heterogeneous populations of cells within the neurosphere as evident by distinguishable cellular features (Fig. 7D). Immunohistochemistry further revealed the heterogeneity of caspase-8 protein expression in the neurospheres in that the majority of the cells were negative for caspase-8 whereas a few cells were positive for caspase-8. In contrast, TRAIL-sensitive SC189c17 cell blocks, which were included as positive controls, showed cytoplasmic expression of caspase-8 protein in the vast majority of the cells (Fig. 7E). These results indicate the heterogeneity of the cell populations within a neurosphere. They further demonstrate that the genomic loss of CASP8 in CSC-enriched neurospheres results in the lack of the caspase-8 protein expression and the neurosphere resistance to TRAIL-induced apoptosis.
CASP8 genomic status and protein expression in glioblastomas
In validating these results in patients' glioblastomas, we first checked the genomic status of CASP8 in glioblastomas as reported in the National Cancer Institute's Repository for Molecular Brain Neoplasia Data (REMBRANDT) (https://caintegrator.nci.nih.gov/rembrandt). Based on the database, CASP8 was amplified in 11–31 of 201 cases (5–16%), and deleted in 3–32 cases (1–16%) depending on the stringency of the cut-off criteria used; however, there was no correlation between CASP8 genomic status and patients' survival (Fig. 8A). Analysis of caspase-8 mRNA using Affymetrix gene expression data from the REMBRANDT revealed 2-fold upregulation of caspase-8 mRNA in 26 of 101 (26%) glioblastomas (Fig. 8B). To examine the expression of caspase-8 protein, we analyzed glioblastoma tumor tissues as compared to normal human brain tissue by western blot. The results showed the caspase-8 protein expression in eight of nine glioblastoma tumors examined. However, no caspase-8 protein expression was detected in all five samples of normal human brain tissue from five different individuals (Fig. 8C). The caspase-8 expression status in each glioblastoma was confirmed by immunohistochemistry, which revealed the cytoplasmic expression of caspase-8 protein in all glioblastomas except 1125 (Fig.8D). These results indicate the heterogeneity of glioblastoma genomic status of CASP8 and the expression of caspase-8 mRNA and proteins. The data identify the deletion of CASP8 and the lack of caspase-8 mRNA and protein expression in a small subset of glioblastomas.
Fig. 8.

CASP8 genomic status and protein expression in glioblastomas. A–B REMBRANDT data showing CASP8 gene amplification and deletion (A) and caspase-8 mRNA expression (B) in glioblastoma tissues with no correlation to patients' survival. C Demonstration by western blot of the expression of caspase-8 (Casp-8) in glioblastoma tissues but not normal human brain tissues. Glioblastoma cell line LN443 and LN18 were included as positive controls and actin was used as protein loading control. D H&E sections of glioblastoma 1109 and 1125 (left) and immunohistochemistry (IHC, right) demonstrating caspase-8 expression in 1109 but not in 1125 consisting with the findings in western blots as presented above in C.
Discussion
Extensive studies of glioblastoma cell lines and primary cultures in serum-supplemented culture conditions have shown the heterogeneity of these lines and cultures in response to TRAIL-induced apoptosis. In this study, we further identify the heterogeneity of cell clones generated from individual glioblastoma primary cultures under serum-supplemented culture conditions. While studies of cell lines and primary cultures have established signaling pathways in TRAIL-induced apoptosis, recent studies have suggested that traditionally grown cultures using serum-supplementation do not adequately recapitulate the genetic properties of glioblastoma tumors in patients and may therefore not be optimal for glioblastoma research (12). Instead, CSC-enriched neurospheres derived from glioblastomas share the genomic properties of the original cancers and should therefore be the research models for studying cancer signaling pathways and screening pathway-targeted drugs. By generating CSC-enriched neurospheres, we show here the genomic and biologic heterogeneity of these CSC-enriched cultures and suggest that genomic alterations define the responsiveness of glioblastoma-derived CSCs to TRAIL-induced apoptosis.
The cytotoxicity of rhTRAIL in glioblastoma cell lines and its therapeutic effect in treating cell lines-derived xenografts were first reported by Ashkenazi and Weller's laboratories (25, 26). TRAIL-induced cytotoxicity was further shown to occur through induction of the apoptotic cascade in glioblastoma cell lines by Ashkenazi's (27), Hawkins' (28) and our laboratory (23). In the meantime, Zhou's laboratory reported for the first time that TRAIL apoptotic pathway can be targeted in glioblastoma cell lines by an agonistic DR5 antibody (29). In delineating the TRAIL apoptotic pathway, we have shown that TRAIL-induced apoptosis occurs through DR4/DR5-mediated recruitment of FADD and caspase-8 for the subsequent assembly of the DISC and the cleavage and activation of caspase-8 in the DISC (30). In this study, we have further shown that the TRAIL apoptotic pathway is intact in glioblastoma-derived CSC-enriched neurospheres and therefore provided evidence in support of the development of TRAIL-based therapies targeting CSC populations.
TRAIL-induced apoptosis, however, occurs only in a small set of glioblastoma-derived CSC-enriched cultures. In examining eight glioblastoma-derived neurospheres, we have identified only one neurosphere (NSC326) that undergoes apoptosis after treatment with TRAIL (data not shown). In examining the remaining TRAIL-resistant neurospheres, we have demonstrated the lack of caspase-8 expression in one neurosphere (NSC189). Caspase-8 is an apoptosis-initiating caspase (31) that consists of two death effector domains and a protease domain made of two subunits, p18 and p12 (32). In the DISC, caspase-8 zymogens become prototypically cleaved to form enzymatically active p18 and p10 tetramers (33–35) that cleave downstream effector caspases (30). The CASP8 gene is located in chromosomal region 2q33-q34 (24) and our earlier study has revealed chromosomal loss of the region 2q33-q34 in the TRAIL resistant LN215 and U373MG cell lines (36). In this study, we have identified the loss of one copy of chromosome 2 and the CASP8 gene-containing chromosomal loci in neurosphere NSC189 and shown that genomic loss of CASP8 results in the failure of TRAIL-induced caspase-8-initiated caspase cascade in the CSC-enriched cultures. Taken together, these results suggest that these genomic alterations define the functional integrity of the TRAIL apoptotic pathway in both CSC and non-stem cells.
Genomic alterations of CASP8 have been identified in human tumors. CASP8 is silenced by DNA methylation in neuroblastomas, resulting in tumor resistance to TRAIL treatment.(37, 38) Somatic inactivating mutations of CASP8 have been reported in a small fraction of colorectal and gastric carcinoma tumor tissues (39, 40); however, the functional status of these CASP8 mutants remain unknown. The data from the REMBRANDT database indicates that the deletion of CASP8 is present in a small subset of glioblastomas, consistent with our findings by western blotting that one of nine glioblastomas lacks of the expression of caspase-8 proteins. While the data from the REMBRANDT database indicates no correlation between the genomic status of CASP8 and the expression of caspase-8 mRNA, the data presented here indicates that the genomic loss of CASP8 and the lack of caspase-8 protein expression may result in the tumor resistance to TRAIL apoptotic pathway-targeted therapies.
Our study has also shown for the first time the heterogeneity of cell populations within each of CSC-enriched neurospheres. In addition, we have also generated multiple chromosomally distinguished non-stem cell clones from each of glioblastoma-derived primary cultures. This study has further enhanced our understanding that glioblastomas are genomically heterogeneous not only among different tumors but also among CSC and non-stem cell populations within each tumor. The genomic heterogeneity suggests the presence of multiple mechanisms in TRAIL resistance in both CSC and non-stem cells. Studies of glioblastoma cell lines have indeed revealed multiple molecular models in TRAIL resistance, in which the caspase-8 inhibition in the DISC appears to be the most upstream event in TRAIL resistance. Caspase-8 cleavage, for instance, can be inhibited by adaptor proteins such as phosphorprotein enriched in astrocytes-15 kDa/diabetes (PEA-15/PED) (30), cellular FADD-like interleukin-1β-converting enzyme-inhibitory protein (c-FLIP) (30, 41, 42), and receptor-interacting protein (1) that are recruited by FADD to the DISC where they interact with caspase-8, resulting in the inhibition of caspase-8 in the DISC and the cell resistance to TRAIL treatment. Targeting of these caspase-8 inhibitory proteins can overcome TRAIL-resistance in the cell lines (1), which supports the current clinical strategies for development of TRAIL-based combination therapies (3). This study has clearly demonstrated that the genomic and biologic integrity of the TRAIL apoptotic pathway is necessary for glioblastoma-derived CSC and non-stem cells to respond to the treatment of TRAIL apoptotic pathway-targeted therapies. Future clinical trials of TRAIL apoptotic pathway targeted therapies may consider genomic analysis of tumor tissue to identify the genomic status of TRAIL apoptotic genes such as CASP8 and use it as a genomic marker to predict tumor responsiveness to TRAIL apoptotic pathway-targeted therapies.
Acknowledgements
We thank Diane Lawson for her technical support. This work was supported by National Institutes of Health grant CA129687 (C. H.) and Southeastern Brain Tumor Foundation research award (C.H.). C.H. was a Georgia Cancer Coalition Distinguished Scholar.
Footnotes
Ling Qi and Anita C. Bellail contributed equally to this study.
References
- 1.Bellail AC, Tse MC, Song JH, et al. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J Cell Mol Med. 14:1303–1317. doi: 10.1111/j.1582-4934.2009.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 3.Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials. 2009;4:34–41. doi: 10.2174/157488709787047530. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 5.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 8.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 9.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 10.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 11.Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Bao S, Wu Q, Sathornsumetee S, et al. Stem Cell-like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 16.Laks DR, Masterman-Smith M, Visnyei K, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27:980–987. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanamori M, Kawaguchi T, Nigro JM, et al. Contribution of Notch signaling activation to human glioblastoma multiforme. J Neurosurg. 2007;106:417–427. doi: 10.3171/jns.2007.106.3.417. [DOI] [PubMed] [Google Scholar]
- 18.Bar EE, Chaudhry A, Lin A, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H, Gomez-Manzano C, Aoki H, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–1414. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 20.Song JH, Song DK, Pyrzynska B, Petruk KC, Van Meir EG, Hao C. TRAIL triggers apoptosis in malignant glioma cells through extrinsic and intrinsic pathways. Brain Pathol. 2003;13:539–553. doi: 10.1111/j.1750-3639.2003.tb00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol (Berl) 2002;103:171–178. doi: 10.1007/s004010100448. [DOI] [PubMed] [Google Scholar]
- 23.Hao C, Beguinot F, Condorelli G, et al. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001;61:1162–1170. [PubMed] [Google Scholar]
- 24.Kischkel FC, Kioschis P, Weitz S, Poustka A, Lichter P, Krammer PH. Assignment of CASP8 to human chromosome band 2q33-->q34 and Casp8 to the murine syntenic region on chromosome 1B-proximal C by in situ hybridization. Cytogenet Cell Genet. 1998;82:95–96. doi: 10.1159/000015073. [DOI] [PubMed] [Google Scholar]
- 25.Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth W, Isenmann S, Naumann U, et al. Locoregional Apo2L/TRAIL eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem Biophys Res Commun. 1999;265:479–483. doi: 10.1006/bbrc.1999.1693. [DOI] [PubMed] [Google Scholar]
- 27.Pollack IF, Erff M, Ashkenazi A. Direct stimulation of apoptotic signaling by soluble Apo21/tumor necrosis factor-related apoptosis-inducing ligand leads to selective killing of glioma cells. Clin Cancer Res. 2001;7:1362–1369. [PubMed] [Google Scholar]
- 28.Knight MJ, Riffkin CD, Muscat AM, Ashley DM, Hawkins CJ. Analysis of FasL and TRAIL induced apoptosis pathways in glioma cells. Oncogene. 2001;20:5789–5798. doi: 10.1038/sj.onc.1204810. [DOI] [PubMed] [Google Scholar]
- 29.Ichikawa K, Liu W, Zhao L, et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954–960. doi: 10.1038/91000. [DOI] [PubMed] [Google Scholar]
- 30.Xiao C, Yang BF, Asadi N, Beguinot F, Hao C. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J Biol Chem. 2002;277:25020–25025. doi: 10.1074/jbc.M202946200. [DOI] [PubMed] [Google Scholar]
- 31.Bodmer JL, Holler N, Reynard S, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 32.Scaffidi C, Medema JP, Krammer PH, Peter ME. FLICE is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J Biol Chem. 1997;272:26953–26958. doi: 10.1074/jbc.272.43.26953. [DOI] [PubMed] [Google Scholar]
- 33.Boatright KM, Renatus M, Scott FL, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 34.Chang DW, Xing Z, Capacio VL, Peter ME, Yang X. Interdimer processing mechanism of procaspase-8 activation. Embo J. 2003;22:4132–4142. doi: 10.1093/emboj/cdg414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donepudi M, Mac Sweeney A, Briand C, Grutter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell. 2003;11:543–549. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 36.Li YC, Tzeng CC, Song JH, et al. Genomic alterations in human malignant glioma cells associate with the cell resistance to the combination treatment with tumor necrosis factor-related apoptosis-inducing ligand and chemotherapy. Clin Cancer Res. 2006;12:2716–2729. doi: 10.1158/1078-0432.CCR-05-1980. [DOI] [PubMed] [Google Scholar]
- 37.Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315–4319. [PubMed] [Google Scholar]
- 38.Eggert A, Grotzer MA, Zuzak TJ, et al. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–1319. [PubMed] [Google Scholar]
- 39.Kim HS, Lee JW, Soung YH, et al. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology. 2003;125:708–715. doi: 10.1016/s0016-5085(03)01059-x. [DOI] [PubMed] [Google Scholar]
- 40.Soung YH, Lee JW, Kim SY, et al. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 2005;65:815–821. [PubMed] [Google Scholar]
- 41.Panner A, James CD, Berger MS, Pieper RO. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol Cell Biol. 2005;25:8809–8823. doi: 10.1128/MCB.25.20.8809-8823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panner A, Murray JC, Berger MS, Pieper RO. Heat shock protein 90alpha recruits FLIPS to the death-inducing signaling complex and contributes to TRAIL resistance in human glioma. Cancer Res. 2007;67:9482–9489. doi: 10.1158/0008-5472.CAN-07-0569. [DOI] [PubMed] [Google Scholar]