

Abstract
Myhre syndrome is a developmental disorder characterized by reduced growth, generalized muscular hypertrophy, facial dysmorphism, deafness, cognitive deficits, joint stiffness, and skeletal anomalies. Here, by performing exome sequencing of a single affected individual and coupling the results to a hypothesis-driven filtering strategy, we establish that heterozygous mutations in SMAD4, which encodes for a transducer mediating transforming growth factor β and bone morphogenetic protein signaling branches, underlie this rare Mendelian trait. Two recurrent de novo SMAD4 mutations were identified in eight unrelated subjects. Both mutations were missense changes altering Ile500 within the evolutionary conserved MAD homology 2 domain, a well known mutational hot spot in malignancies. Structural analyses suggest that the substituted residues are likely to perturb the binding properties of the mutant protein to signaling partners. Although SMAD4 has been established as a tumor suppressor gene somatically mutated in pancreatic, gastrointestinal, and skin cancers, and germline loss-of-function lesions and deletions of this gene have been documented to cause disorders that predispose individuals to gastrointestinal cancer and vascular dysplasias, the present report identifies a previously unrecognized class of mutations in the gene with profound impact on development and growth.
Main Text
Myhre syndrome (MIM 139210) is the eponym for the disorder originally described 30 years ago in two unrelated males exhibiting distinctive dysmorphic features, short stature, generalized muscular hypertrophy, cryptorchidism, limitation of joints, deafness, and intellectual disability.1 Myhre syndrome is a rare condition with less than 30 affected individuals, mostly males, reported to date.2, 3, 4, 5, 6, 7, 8 Based on the available records, cardinal features of Myhre syndrome include reduced growth, generalized muscular hypertrophy, variable cognitive deficits, a recognizable pattern of facial features (i.e., narrow palpebral fissures, midface hypoplasia, a small mouth with a thin upper lip, and prognathism), and skeletal anomalies (i.e., thick skull bones, platispondyly, large vertebral pedicles, broad ribs, hypoplastic iliac wings, and brachydactyly), joint stiffness, and deafness. Additional recurrent features include a cleft lip and/or palate, congenital heart defects, stiff skin, cryptorchidism, hypertension, and ocular defects. Myhre syndrome has clinical overlap with growth retardation, ocular abnormalities, microcephaly, brachydactyly, and oligophrenia (GOMBO) syndrome (MIM 233270); laryngotracheal stenosis, progressive, with short stature and arthropathy syndrome (LAPS) syndrome (MIM 603391); geleophysic dysplasia (GPHYSD1 [MIM 231050] and GPHYSD2 [MIM 614185]); acromicric dysplasia (ACMICD [MIM 102370]); Weill-Marchesani syndrome (WMS1 [MIM 277600] and WMS2 [MIM 608328]); and Moore-Federman syndrome (MIM 127200); genetic relatedness among these disorders has been suggested.9, 10, 11 Although the molecular basis for some of these clinically related conditions is unknown, increasing evidence supports the relevant role of transforming growth factor β (TGFβ) signaling dysregulation in the pathogenesis of GPHYSD, ACMICD, and WMS.12, 13 Here, an exome sequencing-based approach was used to identify similar to mothers against decapentaplegic (SMAD) family member 4 (SMAD4 [MIM 600993]), previously known as mothers against decapentaplegic homolog 4 (Drosophila) (MADH4), which encodes a protein with a crucial role in TGFβ/bone morphogenetic protein (BMP) signal transduction, as the gene mutated in Myhre syndrome.
Eight subjects with clinical features fitting Myhre syndrome were included in the study (Table 1 and Figure 1A). Clinical data and biological material collection and storage were attained from the participating families in accordance with the ethical standards of the institutional review boards (Ospedale “Bambino Gesù”, Rome; Università di Torino, Turin; Arcispedale Santa Maria Nuova, Reggio Emilia; Ospedale Microcitemico, Cagliari; Università “Federico II”, Naples) and after written informed consent was secured. Genomic DNA was isolated from skin fibroblasts, hair bulb cells, peripheral blood leukocytes, saliva, and/or buccal mucosal epithelial cells with standard protocols. In all subjects, karyotype and subtelomeric fluorescence in situ hybridization (FISH) analyses provided normal results. High-resolution array- comparative genomic hybridization (CGH) or SNP array analyses were performed on three out of the eight subjects and did not disclose any disease-causing genomic rearrangement.
Table 1.
Clinical Features of the Affected Subjects Included in the Study
| Features |
Subjects |
|||||||
|---|---|---|---|---|---|---|---|---|
| OBG_01 | TO_01 | TO_02 | TO_03 | RE_01 | NA_01 | NA_02 | CA_01 | |
| Nucleotide substitution | 1498A>G | 1499T>C | 1498A>G | 1498A>G | 1499T>C | 1498A>G | 1498A>G | 1499T>C |
| Predicted amino acid change | Ile500Val | Ile500Thr | Ile500Val | Ile500Val | Ile500Thr | Ile500Val | Ile500Val | Ile500Thr |
| Paternal age at birth (years) | 35 | 34 | 38 | 36 | 31 | 33 | 28 | 36 |
| Length at birth (centile) | n.a. | 3rd | <3rd | 3rd | <3rd | n.a. | n.a. | <3rd |
| Weight at birth (centile) | <3rd | 3rd | <3rd | 3rd | <3rd | <3rd | n.a. | <3rd |
| Head circumference at birth (centile) | n.a. | 3rd | <3rd | n.a. | <3rd | n.a. | n.a. | n.a. |
| Age at examination (years) | 42 | 13 | 4.5 | 26 | 13 | 14 | 34 | 13 |
| Gender | male | male | female | female | male | male | female | male |
| Height (centile) | <3rd | <3rd | <3rd | <3rd | <3rd | <3rd | <3rd | <3rd |
| Obesity | + | − | − | + | + | + | + | − |
| Macrocephaly | − | + | − | − | + | − | − | − |
| Facial Features | ||||||||
| Narrow palpebral fissures | + | + | + | + | + | + | + | + |
| Midfacial hypoplasia | + | + | + | + | + | + | + | + |
| Short philtrum | + | + | + | + | + | + | + | + |
| Narrow mouth and/or thin upper lip | + | + | + | + | + | − | − | + |
| Cleft lip and/or palate | − | +a | +b | + | − | − | − | − |
| Prognathism | + | + | + | + | + | + | + | + |
| Ocular Anomalies | ||||||||
| Refractive errors | +c | + | − | + | − | + | − | − |
| Cataract | − | − | − | − | − | n.a. | − | + |
| Hearing loss | +d | +e | +f | +e | +f | +e | +e | +d |
| Cognitive impairment | mild | moderate | +g | −h | +g | + | + | + |
| Autistic behavior | − | − | − | − | − | + | − | + |
| Muscular hypertrophy | + | + | + | + | + | + | + | + |
| Joint stiffness | + | + | + | + | + | + | + | + |
| Skeletal Anomalies | ||||||||
| Thick calvarium | + | + | + | + | + | + | + | + |
| Hypoplastic iliac wings | + | + | + | + | + | + | + | + |
| Platispondyly | + | + | + | + | − | − | + | + |
| Large vertebral pedicles | + | − | n.a. | + | + | + | + | + |
| Fused vertebrae | − | − | n.a. | − | − | − | − | − |
| Broad Ribs | − | + | − | + | + | − | − | + |
| Brachycamptodactyly | + | + | + | + | + | + | + | + |
| Congenital heart defects | +i | − | +j | +k | +l | − | +k | +m |
| Cryptorchidism | − | + | − | + | − | |||
| Hypertension | + | − | − | − | − | − | − | + |
| Skin Anomalies | ||||||||
| Stiff skin | + | + | − | + | + | + | + | − |
| Thick skin | − | + | − | + | + | + | + | + |
| Other features | − | +n | +o | − | +p | − | +q | − |
n.a. is used as an abbreviation for data not available. Subjects TO_3 and NA_01 were previously reported by Rulli et al.5 and Titomanlio et al.45
Unilateral cleft lip.
Submucous cleft palate.
Astigmatism.
Sensorineural deafness.
Mixed hearing loss.
Conductive.
Delayed psychomotor and language development.
Psychological and behavioral problems.
Right ventricular hypertrophy.
Ventricular and atrial septal defects, patent ductus arteriosus.
Aortic valve stenosis.
Aortic coarctation.
Patent ductus arteriosus.
Benign mesencephalic glioma, fibrous dysplasia of the maxilla.
Hirschprung disease diagnosed at birth.
Pericarditis with cardiac tamponade.
Secondary amennorrea.
Figure 1.
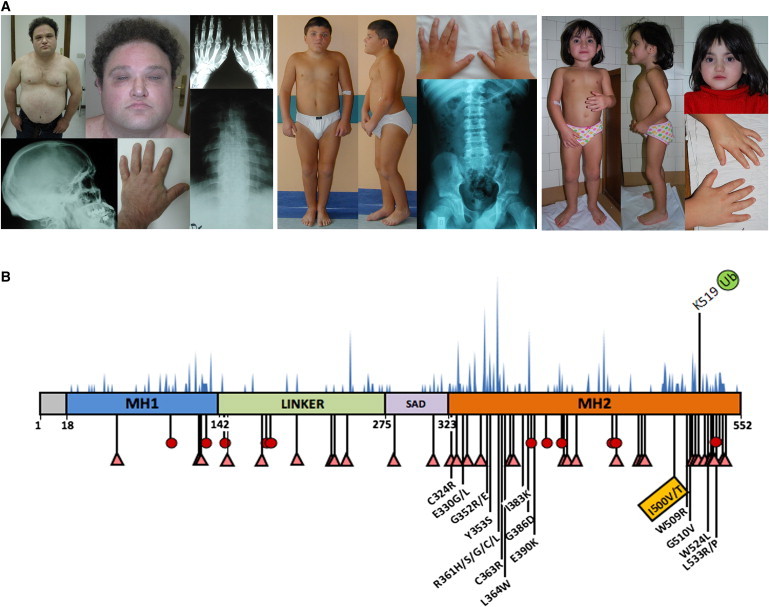
A Restricted Spectrum of Germline Missense Mutations in SMAD4 Underlies Myhre Syndrome
(A) Clinical features of representative subjects (from left, OBG_01, TO_01, and TO_02) carrying the heterozygous SMAD4 mutations. Pictures illustrate the distinctive facial features, muscular build, joint stiffness, camptodactyly, and brachydactyly. Radiographs show the thickening of the skull bones, vertebrae with large pedicles and irregular endplates, wide ribs, and hypoplastic iliac wings.
(B) The location of Ile500 is shown below the SMAD4 domain structure scheme. SMAD4 comprises an N-terminal MAD homology 1 domain (MH1) connected by a linker to the SMAD4 activation domain (SAD) and the C-terminal MAD homology 2 domain (MH2). Numbers below the domain structure indicate the amino acid boundaries of those domains. Nonsense (red circle) and truncating (pink triangle) mutations reported in juvenile polyposis syndrome (JPS) and JPS-hereditary hemorrhagic telangiectasia are shown below the illustration. The distribution of somatic SMAD4 mutations occurring in cancer (blue-filled peaks) is shown above the illustration. The location of Lys519, which is the residue subject to ubiquination, is also shown.
Targeted enrichment and parallel sequencing experiments were performed on genomic DNA extracted from circulating leukocytes of a single affected subject (OBG_01). After fragment size selection, GS FLX Titanium adaptors (Roche) were ligated to the sheared genomic fragments to construct a shotgun sequencing library that was hybridized with a Sequence Capture Human Exome 2.1M Array v.2 (NimbleGen). Captured fragments were then amplified through emPCR and sequenced with Titanium reagents in a two-lane gasket PicoTiterPlate device (70X75) with a GS FLX instrument (Roche 454). Image analysis and base calling were performed with the GS FLX pipeline software (version 2.5.3). Approximately 1.2 Gb of genomic sequence from two GS FLX runs was obtained. The average read length was 380 bp, and fragment sizes ranged from 40 to 1,100 bp. Sequencing data were initially processed with the GSMapper software package v.2.5.3 (Roche), and high-quality sequencing reads were aligned to the human genome reference sequence (USCS hg19, GRCh37). Ninety-four percent of sequence reads mapped to the human reference genome. Targeted sequence coverage was 98%, and there was an average sequence depth of 10×. Sequence alignment and functional annotation of the identified variants data were obtained with BEDTools v.2.1214 or extracted from reference databases with batch queries. Variant detection was performed with a combination of stringency parameters with the GSMapper software, resulting in the generation of a high-confidence variants detection gene list of 72,426 variants. Among the 2,946 novel variants (filtered against the dbSNP build 131 database), 665 changes mapped within coding regions. Nonsynonymous and splice-site variants were further filtered (UniProtKB/Swiss-Prot, HapMap and in-house exome variants databases) and stringently aligned to the human genome (BLAST NCBI) allowing the selection of 185 nonsynonymous and 18 splice-site valid variants. PolyPhen-215 and SIFT16 analyses predicted a potential deleterious effect on protein structure and/or function in 40% of those variants.
Based on the evidence that all affected subjects were born to apparently healthy nonconsanguineous parents, we considered heterozygosity for a de novo mutation as the most likely event underlying the trait. This hypothesis was also supported by advanced paternal age in the majority of the reported cases as well as in subjects of the present cohort (Table 1). In Myhre syndrome, the differential diagnosis usually considers GPHYSD. Based on the causative link of TGFβ signaling to the latter,12, 13 we performed a filtering strategy directed to select gene candidates functionally linked to this signaling network. For this, data mining analyses were performed with the DAVID17 algorithm, an integrated biological knowledgebase and analytic tool aimed at systematically extracting biological meaning from large gene lists. We used the functional annotation table tool to query corresponding pathway annotation for each gene and selected candidates whose products were functionally related to TGFβ signaling (Kyoto Encyclopedia of Genes and Genomes [KEGG] pathway). This approach allowed the identification of three genes, i.e., PITX2 (MIM 601542), SMAD4 and THBS3 (MIM 18862). Genes harboring novel variants were also ranked via a text mining algorithm, Génie,18 that evaluates gene function based on analysis of the literature. By using combinations of terms from the OMIM clinical synopsis of Myhre syndrome (e.g., short stature, joint stiffness, muscular hypertrophy, brachydactyly, deafness, and mental retardation) as biological topics, and a p value cutoff < 0.005 for the selection specificity of the abstracts and the false discovery rate for the gene selections, the Génie algorithm's evaluation of available MEDLINE records allowed ranking of candidate genes and selection of a panel of ten genes (CRB1 [MIM 604210], EFHC1 [MIM 608815], HSPG2 [MIM 142461], MYH2 [MIM 160740], PKD2 [MIM 173910], PITX2, SETX [MIM 608465], MRE11A [MIM 600814], SMAD4, and USH1C [MIM 605242]) as significantly linked. Given the crucial role of SMAD4 in BMP/TGFβ signaling, its ubiquitous expression, and its relevant function in early and late developmental processes,19 we considered this gene as the most promising candidate. Sanger sequencing of the relevant coding exon confirmed heterozygosity for the c.1498A>G (p.Ile500Val) change in the affected subject, and sequencing of parental DNAs revealed only reference alleles, evidence for its de novo origin. STR genotyping (PowerPlex ESX16 System [Promega]) confirmed paternity. The defect was documented in the proband's skin fibroblasts as well as hair bulb and buccal mucosal epithelial cell specimens, strongly arguing against the possibility of a somatic event (Figure S1, available online). Consistent with the notion that this SMAD4 de novo change was the causative mutation, the other confirmed candidate variants turned out to be inherited from one of the two healthy parents. These findings strongly pointed to SMAD4 as the gene mutated in Myhre syndrome.
To confirm the causal involvement of SMAD4 in Myhre syndrome, the entire coding sequence of the gene (NM_005359) was scanned for mutations in DNA obtained from circulating leukocytes from an additional seven unrelated affected subjects by direct sequencing with an ABI Prism 3500 Genetic Analyzers (Applied Biosystems) and the ABI BigDye Terminator Sequencing Kit v.3.1 (Applied Biosystems). Primer pairs designed to amplify exons and their intron boundaries are listed in Table S1. Mutation analysis identified heterozygous SMAD4 mutations in all individuals. Among the seven subjects, four shared the c.1498A>G nucleotide change (p.Ile500Val), whereas the c.1499T>C change, predicting a different amino acid substitutions at the same codon (p.Ile500Thr), was identified in the three remaining individuals (Figure S1). In all family trios, parental DNA genotyping demonstrated the de novo origin of each mutation, and STR genotyping confirmed paternity. Fibroblasts and/or epithelial cells were available for three of the seven subjects, and sequencing documented the defect, supporting the germline origin of these changes. Consistent with these findings, the c.1498A>G and c.1499T>C substitutions were not observed in more than 400 population-matched unaffected individuals scanned by high-resolution melting analysis and direct sequencing.
The Ile500 residue lies in the MAD homology 2 (MH2) domain, a region at the C terminus of the protein that is conserved among SMAD4 orthologs and paralogs (Figures S2 and S3) and known to represent the mutational hot spot for somatic and germline lesions occurring in malignancies and cancer-prone disorders (Figure 1B).20 The MH2 domain mediates interaction with a wide variety of proteins, provides specificity and selectivity to SMAD function, and is indispensable for SMAD oligomerization,21 which is required for TGFβ/BMP signal transduction. Flow through this signaling network occurs through ligand-induced heteromeric receptor complexes consisting of type I and type II transmembrane serine/threonine receptors that recruit and phosphorylate receptor-regulated SMADs (R-SMADs), which in turn heterotrimerize with the common mediator, SMAD4. SMAD complexes then accumulate into the nucleus to regulate expression of target genes.22 In the structure of the SMAD4 MH2 domain, Ile500 is located close to the protein surface, but it is solvent accessible only for 6% of the residue surface, because it is surrounded by and interacts with the hydrocarbon part of the side chains of residues Arg496, Arg497, Arg502, Glu526, and His528 (Figure 2). With the exception of Arg496, these residues are highly conserved in SMAD4 orthologs and paralogs and constitute a binding site for anions.23 The solvent-exposed location of this region and its highly charged character make it a likely candidate for protein-protein interactions and point to disturbed SMAD4 binding to signaling partners at this site as the possible effect promoted by the identified mutations.
Figure 2.
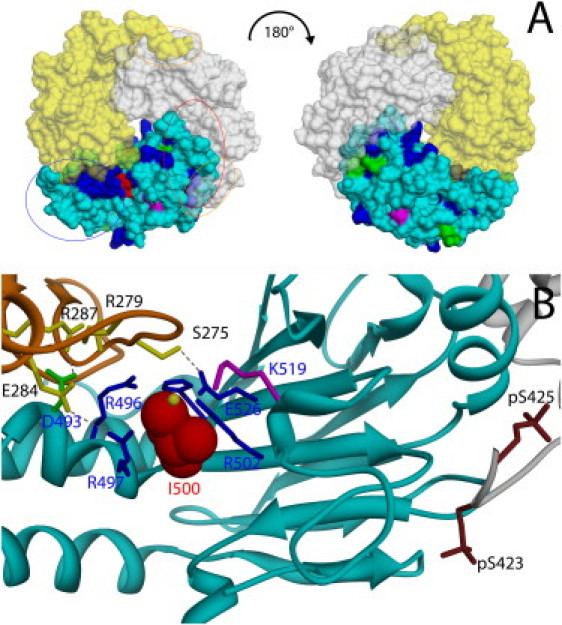
Location of Ile500 in the Spatial Structure of the SMAD3/SMAD4 MH2 Domain Complex
(A) Solvent accessible surface representation of the SMAD4/SMAD3 MH2 domain complex (Protein Data Bank [PDB] ID codes 1U7F). The SMAD4 monomer is shown in cyan, whereas the two SMAD3 subunits are colored in yellow and white. The solvent accessible surface of residues affected by somatic (blue) and germline (magenta) mutations and lesions occurring as both germline and somatic events (green) are shown. The residue mutated in Myhre syndrome, Ile500, is shown in red. The red and blue ovals indicate the location of the loop/helix and helix bundle SMAD4 subdomains, respectively. Orange ovals indicate the position of the serine residues that are phosphorylated in activated SMAD3.
(B) Cα ribbon trace (PDB ID codes 1U7F) of SMAD4 MH2 domain (cyan) and those of the two interacting SMAD3 subunits (orange and gray). Ile500 is represented in space-filling mode (red). Residues interacting with Ile500 (Arg496, Arg497, Arg502, Glu526, and His528) are shown as blue sticks. Asp493 (green), which is important for interdomain interactions, and Lys519 (violet), which is subject to ubiquination, are also shown. SMAD3 residues directly contributing to interactions with the evidenced SMAD4 residues (Ser275, Arg279, Glu284, and Arg287) are shown as yellow sticks, together with the phosphorylated SMAD3 serine residues (brown). The anion binding site located in proximity of Ile500, as indicated by the structure of the SMAD4 homotrimer (pdb code 1DD1), is shown by a yellow sphere. Visualization and analysis of the molecular structure was performed with the program UCSF Chimera.46
The best characterized complexes formed by SMAD4 are those resulting from its association with SMAD2 (MIM 601366) or SMAD3 (MIM 603109). Biochemical and crystallographic evidence indicates that these complexes are trimeric units comprising one SMAD4 and two phosphorylated R-SMAD subunits. The complex is characterized by three nonidentical interfaces, two of which involve separate solvent-exposed surfaces of the SMAD4 MH2 domain.24 According to the crystal structure of the SMAD2/SMAD4 and SMAD3/SMAD4 heterotrimeric complexes,24 Ile500 is located within one of the two MH2 surfaces involved in R-SMAD binding, and the residues surrounding it participate directly in the intermolecular interaction. Particularly, Arg497 and Asp493 form ion pairs with acidic residues of the R-SMAD subunit (Figure 2B). Although a number of missense mutations affecting conserved residues of this region (e.g., Asp493), which were documented or predicted to disrupt SMAD4 homo- and heteromerization, has been identified as somatic and/or germline lesions in malignancies and cancer-prone diseases (Catalogue of Somatic Mutations in Cancer, [COSMIC] and Human Gene Mutation Database [HGMD]) (Figure 1B and Figure S2), no missense mutation affecting Ile500 has previously been reported in human disease.
To explore the consequences of the identified mutations on SMAD4 function, we performed structural analyses. We observed that residues Asp493 and Arg497, which are spatially close to Ile500 and are directly involved in intersubunit interactions in SMAD complexes, exhibit a significant conformational flexibility in the available SMAD4 crystallographic structures (Figure S4). Interestingly, in the structures of R-SMADs, where a Thr residue is found at the position corresponding to Ile500, an orientation of the residue corresponding to Arg497 pointing away from the intersubunit interface is consistently observed (Figure S4D). Substitution of Ile500 by Thr or Val in SMAD4 is expected to weaken its hydrophobic interaction with apolar atoms of the surrounding charged residues by introducing a more hydrophilic and/or smaller side chain. This is hypothesized to perturb the arrangement of their side chains, which would affect proper function of the SMAD heterotrimer. Of note, several observations indicate that the region surrounding Ile500 is also critical for interactions with other signaling partners. For instance, the Arg496Ser and Arg502Ser substitutions have been shown to impair the transcriptional activity of the complex without affecting trimer formation.25 Therefore, it is possible that the identified mutations might affect other functionally relevant protein-protein interactions of the mutant SMAD4. It is also of interest that, unlike R-SMADs, SMAD4 is not regulated by reversible phosphorylation. Instead, its function is controlled by monoubiquitination,26 which occurs at Lys519 and has been proposed to sterically hamper the ability of the protein to form a stable complex with activated R-SMADs, resulting in pathway inhibition.27 On the basis of these considerations, it can also be speculated that the subtle conformational change promoted by the Myhre syndrome-causing amino acid substitutions might impair proper SMAD4 ubiquitination and perturb signal flow by increasing the intracellular concentration of nonubiquitinylated SMAD4.
Detailed clinical information was obtained for all of the subjects heterozygous for a SMAD4 mutation (Table 1). Overall, these subjects presented with a homogeneous phenotype (Figure 1A). Cardinal features of the syndrome, including short stature (< third centile), a recognizable facial appearance, generalized muscular hypertrophy, hearing loss, short hands, distinctive skeletal anomalies, and joint stiffness were observed in all individuals. All subjects presented with low birth weight but five became obese with age. Delayed psychomotor and/or language development and variable intellectual disability was documented in all but one individual. One subject exhibited noticeable psychological and behavioral issues. A wide spectrum of congenital heart defects and skin anomalies also recurred in the cohort, while macrocephaly, cleft lip and/or palate, refractive errors, and hypertension were less prevalent. Autistic behavior was documented in two subjects. Of note, none of the eight subjects exhibited any gross vascular anomaly or occurrence of skin, pancreatic, or gastrointestinal malignancies. Benign mesencephalic glioma and uncharacterized fibrous dysplasia of the maxilla, however, were reported in one individual (Table 1).
The TGFβ signaling network controls a vast array of biological processes, including cell proliferation, differentiation, apoptosis, adhesion, and lineage commitment, in a cell type-dependent manner.28, 29, 30 The TGFβ superfamily of cytokines includes more than 30 structurally related proteins belonging to the two major TGFβ/activin and BMP subfamilies.31 Among them, TGFβ family members play a diverse role in embryonic development,32, 33 Similarly, the members of the BMP family coordinate early and late developmental processes, including tissue specification, organogenesis, and skeletal pattering.34, 35, 36, 37 Within the TGFβ/BMP signaling network, SMAD4 represents a central node because it is required as a coactivator to have transcriptionally active SMAD complexes.21 Based on this crucial role, dysregulation of SMAD4 function is expected to have pleiotropic effects, which fits well with the cardinal features of Myhre syndrome that result from perturbation of several developmental pathways controlled by TGFβ/BMP signaling, including those controlling axial and appendicular skeletal structures, muscular tissue, and central nervous and cardiovascular systems.
Germline mutations in SMAD4 are known to cause juvenile polyposis syndrome (JPS [MIM 174900]) and JPS-hereditary hemorrhagic telangiectasia (JPS-HHT [MIM 175050]).38, 39 The former is an autosomal-dominant disorder usually diagnosed within the first decade of life and characterized by hamartomatous polyps of the gastrointestinal tract and an increased risk of gastrointestinal cancer.40 In the latter, the cancer-prone disease is associated with vascular dysplasia affecting multiple organs, consisting of telangiectases of the skin, oral and nasal mucosa, and arteriovenous malformations of the lungs, liver, brain, and gastrointestinal tract, which generally manifests by the fourth decade.41 JPS/JP-HHT causative SMAD4 mutations are found throughout the gene and include nonsense, missense, and splice-site changes, as well as partial and complete gene deletions (Figure 1B and Figure S2) (HGMD),42 supporting a loss-of-function model of action. Consistent with this, loss of SMAD4 expression or function due to somatic inactivating intragenic lesions or gene deletions frequently occurs in carcinomas of the pancreas, gastrointestinal tract, and skin (COSMIC). In contrast, the invariant location and restricted spectrum of Myhre syndrome-causative mutations support the idea that these genetic lesions have distinct and specific consequences on SMAD4 function. Although the characterization of these mutants is required to appreciate the molecular mechanism(s) implicated in perturbation of growth and development driven by this previously unrecognized class of SMAD4 mutants, structural analysis suggests that they are likely to selectively interfere with the interaction of the protein to signaling partners, possibly by perturbing SMAD4 binding specificity and/or affinity.
Heterozygous missense SMAD4 mutations were identified in all affected subjects with Myhre syndrome included in the study. This finding supports the idea that Myhre syndrome is a genetically homogeneous disorder, or, at minimum, that SMAD4 accounts for the preponderance of cases. The restricted spectrum of mutations seems to explain the relatively uniform phenotype characterizing this disorder. On the basis of the different clinical outcome of SMAD4 mutations, however, it cannot be excluded that additional, still unrecognized classes of gene variants might underlie clinically related conditions, including GOMBO syndrome, LAPS syndrome, Moore-Federman syndrome, and mutation-negative GPHYSD and ACMICD.
Recent advances in targeted sequence capture and massively parallel sequencing technologies offer new opportunities for our understanding of the molecular bases of Mendelian disorders,43, 44 particularly those dramatically influencing the fitness of affected individuals. Candidate gene filtering strategies based on the use of ontologies of phenotypic features and cellular networks are predicted to enhance the power of such unbiased approaches even when coupled to the sequencing of single individuals with rare diseases.
Acknowledgments
We are indebted to the families who participated in the study, the physicians who referred the subjects, and Valentina Fodale, Francesca Pantaleoni, Viviana Cordeddu, and Serenella Venanzi (Istituto Superiore di Sanità, Rome, Italy) for experimental support. We thank the colleagues at the SIDBAE (Istituto Superiore di Sanità) for information technology support. This research was funded by grants from Telethon-Italy (GGP10020), Associazione Italiana per la Ricerca sul Cancro Investigator Grant 2009 (8803), and “Collaborazione Italia-USA” to M.T, and Italian Ministry of Health (Ricerca Corrente 2011) to B.D and M.T.
Published online: January 12, 2012
Footnotes
Supplemental Data include four figures and one table and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Catalogue of Somatic Mutations in Cancer (COSMIC), http://www.sanger.ac.uk/genetics/CGP/cosmic/
DAVID Bioinformatics Resources 6.7, http://david.abcc.ncifcrf.gov/
Entrez Gene, http://www.ncbi.nlm.nih.gov/gene/
Human Gene Mutation Database (HGMD), http://www.hgmd.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Protein Data Bank (PDB), http://www.rcsb.org/pdb/home/home.do
UCSF Chimera, http://www.cgl.ucsf.edu/chimera/
Supplemental Data
References
- 1.Myhre S.A., Ruvalcaba R.H.A., Graham C.B. A new growth deficiency syndrome. Clin. Genet. 1981;20:1–5. doi: 10.1111/j.1399-0004.1981.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 2.Burglen L., Héron D., Moerman A., Dieux-Coeslier A., Bourguignon J.P., Bachy A., Carel J.C., Cormier-Daire V., Manouvrier S., Verloes A. Myhre syndrome: New reports, review, and differential diagnosis. J. Med. Genet. 2003;40:546–551. doi: 10.1136/jmg.40.7.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez-Cardona M.G., Garcia-Cruz D., Garcia-Ortiz J.E., Davalos N.O., Feria-Velasco A., Rodriguez-Rojas L.X., Garcia-Cruz M.O., Figuera-Villanueva L.E., Stephens A., Larios-Arceo F., Sanchez-Corona J. Second female case of Myhre syndrome. Clin. Dysmorphol. 2004;13:91–94. [PubMed] [Google Scholar]
- 4.van Steensel M.A.M., Vreeburg M., Steijlen P.M., de Die-Smulders C. Myhre syndrome in a female with previously undescribed symptoms: Further delineation of the phenotype. Am. J. Med. Genet. A. 2005;139A:127–130. doi: 10.1002/ajmg.a.30988. [DOI] [PubMed] [Google Scholar]
- 5.Rulli I., Ferrero G.B., Belligni E., Delmonaco A.G., Defilippi C., Silengo M. Myhre's syndrome in a girl with normal intelligence. Am. J. Med. Genet. A. 2005;134A:100–102. doi: 10.1002/ajmg.a.30444. [DOI] [PubMed] [Google Scholar]
- 6.Becerra-Solano L.E., Díaz-Rodriguez M., Nastasi-Catanese J.A., Toscano-Flores J.J., Bañuelos-Robles O., Figuera L.E., Matute E., de Lourdes Ramírez-Dueñas M. The fifth female patient with Myhre syndrome: Further delineation. Clin. Dysmorphol. 2008;17:113–117. doi: 10.1097/MCD.0b013e3282f52828. [DOI] [PubMed] [Google Scholar]
- 7.Bachmann-Gagescu R., Hisama F.M., Yuen A.L. Myhre syndrome with ataxia and cerebellar Atrophy. Clin. Dysmorphol. 2011;20:156–159. doi: 10.1097/MCD.0b013e3283468043. [DOI] [PubMed] [Google Scholar]
- 8.McGowan R., Gulati R., McHenry P., Cooke A., Butler S., Keng W.T., Murday V., Whiteford M., Dikkers F.G., Sikkema-Raddatz B., et al. Clinical features and respiratory complications in Myhre syndrome. Eur. J. Med. Genet. 2011;54:e553–e559. doi: 10.1016/j.ejmg.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Bottani A., Verloes A. Myhre-GOMBO syndrome: Possible lumping of two “old” new syndromes. Am. J. Med. Genet. 1995;59:523–524. doi: 10.1002/ajmg.1320590423. [DOI] [PubMed] [Google Scholar]
- 10.Figuera L.E. Geleophysic dysplasia vs. Myhre syndrome. Am. J. Med. Genet. 1996;65:361–362. doi: 10.1002/(SICI)1096-8628(19961111)65:4<361::AID-AJMG25>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 11.Lindor N.M. LAPS syndrome and Myhre syndrome: Two disorders or one? Am. J. Med. Genet. A. 2009;149A:798–799. doi: 10.1002/ajmg.a.32719. [DOI] [PubMed] [Google Scholar]
- 12.Le Goff C., Morice-Picard F., Dagoneau N., Wang L.W., Perrot C., Crow Y.J., Bauer F., Flori E., Prost-Squarcioni C., Krakow D., et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 2008;40:1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Goff C., Mahaut C., Wang L.W., Allali S., Abhyankar A., Jensen S., Zylberberg L., Collod-Beroud G., Bonnet D., Alanay Y., et al. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am. J. Hum. Genet. 2011;89:7–14. doi: 10.1016/j.ajhg.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 17.Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Fontaine J.F., Priller F., Barbosa-Silva A., Andrade-Navarro M.A. Génie: Literature-based gene prioritization at multi genomic scale. Nucleic Acids Res. 2011;39(Web Server issue):W455–W461. doi: 10.1093/nar/gkr246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Attisano L., Lee-Hoeflick S.T. The Smads. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-8-reviews3010. S3010.1–3010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waite K.A., Eng C. From developmental disorder to heritable cancer: It's all in the BMP/TGF-β family. Nat. Rev. Genet. 2003;4:763–773. doi: 10.1038/nrg1178. [DOI] [PubMed] [Google Scholar]
- 21.Massagué J., Seoane J., Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 22.Moustakas A., Souchelnytskyi S., Heldin C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 23.Qin B., Lam S.S., Lin K. Crystal structure of a transcriptionally active Smad4 fragment. Structure. 1999;7:1493–1503. doi: 10.1016/s0969-2126(00)88340-9. [DOI] [PubMed] [Google Scholar]
- 24.Chacko B.M., Qin B.Y., Tiwari A., Shi G., Lam S., Hayward L.J., De Caestecker M., Lin K. Structural basis of heteromeric smad protein assembly in TGF-beta signaling. Mol. Cell. 2004;15:813–823. doi: 10.1016/j.molcel.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 25.Chacko B.M., Qin B., Correia J.J., Lam S.S., de Caestecker M.P., Lin K. The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat. Struct. Biol. 2001;8:248–253. doi: 10.1038/84995. [DOI] [PubMed] [Google Scholar]
- 26.Izzi L., Attisano L. Ubiquitin-dependent regulation of TGFbeta signaling in cancer. Neoplasia. 2006;8:677–688. doi: 10.1593/neo.06472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M.J., Soligo S., Morsut L., et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 28.Massagué J., Blain S.W., Lo R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 29.Descotte V., O'Connor M.B., Jadrich J. In: Inborn Errors of Development. Epstein C.J., Erikson R.P., Wynshaw-Boris A., editors. Oxford University Press; Oxford: 2008. The Transforming Growth Factor-β (TGF-β) Signaling Pathway; pp. 358–368. [Google Scholar]
- 30.Massagué J. The transforming growth factor-beta family. Annu. Rev. Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 31.Newfeld S.J., Wisotzkey R.G., Kumar S. Molecular evolution of a developmental pathway: Phylogenetic analyses of transforming growth factor-beta family ligands, receptors and Smad signal transducers. Genetics. 1999;152:783–795. doi: 10.1093/genetics/152.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Proetzel G., Pawlowski S.A., Wiles M.V., Yin M., Boivin G.P., Howles P.N., Ding J., Ferguson M.W., Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanford L.P., Ormsby I., Gittenberger-de Groot A.C., Sariola H., Friedman R., Boivin G.P., Cardell E.L., Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hogan B.L. Bone morphogenetic proteins in development. Curr. Opin. Genet. Dev. 1996;6:432–438. doi: 10.1016/s0959-437x(96)80064-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H., Bradley A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. 1996;122:2977–2986. doi: 10.1242/dev.122.10.2977. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki N., Labosky P.A., Furuta Y., Hargett L., Dunn R., Fogo A.B., Takahara K., Peters D.M., Greenspan D.S., Hogan B.L. Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development. 1996;122:3587–3595. doi: 10.1242/dev.122.11.3587. [DOI] [PubMed] [Google Scholar]
- 37.Katagiri T., Boorla S., Frendo J.L., Hogan B.L., Karsenty G. Skeletal abnormalities in doubly heterozygous Bmp4 and Bmp7 mice. Dev. Genet. 1998;22:340–348. doi: 10.1002/(SICI)1520-6408(1998)22:4<340::AID-DVG4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Howe J.R., Roth S., Ringold J.C., Summers R.W., Järvinen H.J., Sistonen P., Tomlinson I.P., Houlston R.S., Bevan S., Mitros F.A., et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 39.Gallione C.J., Repetto G.M., Legius E., Rustgi A.K., Schelley S.L., Tejpar S., Mitchell G., Drouin E., Westermann C.J., Marchuk D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 40.Zbuk K.M., Eng C. Hamartomatous polyposis syndromes. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007;4:492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 41.Govani F.S., Shovlin C.L. Hereditary haemorrhagic telangiectasia: A clinical and scientific review. Eur. J. Hum. Genet. 2009;17:860–871. doi: 10.1038/ejhg.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallione C., Aylsworth A.S., Beis J., Berk T., Bernhardt B., Clark R.D., Clericuzio C., Danesino C., Drautz J., Fahl J., et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am. J. Med. Genet. A. 2010;152A:333–339. doi: 10.1002/ajmg.a.33206. [DOI] [PubMed] [Google Scholar]
- 43.Teer J.K., Mullikin J.C. Exome sequencing: The sweet spot before whole genomes. Hum. Mol. Genet. 2010;19(R2):R145–R151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilissen C., Hoischen A., Brunner H.G., Veltman J.A. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228. doi: 10.1186/gb-2011-12-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Titomanlio L., Marzano M.G., Rossi E., D'Armiento M., De Brasi D., Vega G.R., Andreucci M.V., Orsini A.V., Santoro L., Sebastio G. Case of Myhre syndrome with autism and peculiar skin histological findings. Am. J. Med. Genet. 2001;103:163–165. doi: 10.1002/ajmg.1517. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.