

In animals, the mevalonic acid pathway supplies isopentenyl diphosphate (IPP, 9) and dimethylallyl diphosphate (DMAPP, 10) for isoprenoid biosynthesis. While in green algae, eubacteria, and plant plastids, IPP and DMAPP are provided by the deoxyxylulose phosphate (DXP) pathway.[1] Methyl-d-erythritol-2,4-cyclodiphosphate (MEcPP, 7, Scheme 1) was the first DXP-pathway intermediate to be identified, indeed it was isolated almost a decade before the DXP pathway was discovered. Starting from MEcPP, two reductive deoxygenation reactions, catalyzed by IspG and IspH (Scheme 1), lead to the production of both IPP and DMAPP.[2] Preliminary biochemical studies indicate that both IspG and IspH are unique iron-site-containing [4Fe–4S] proteins.[2d, 3] Here we report a study of the reversible cleavage of the MEcPP C–O bond at the C2 position (Scheme 1) and discuss its mechanistic implications.
Scheme 1.
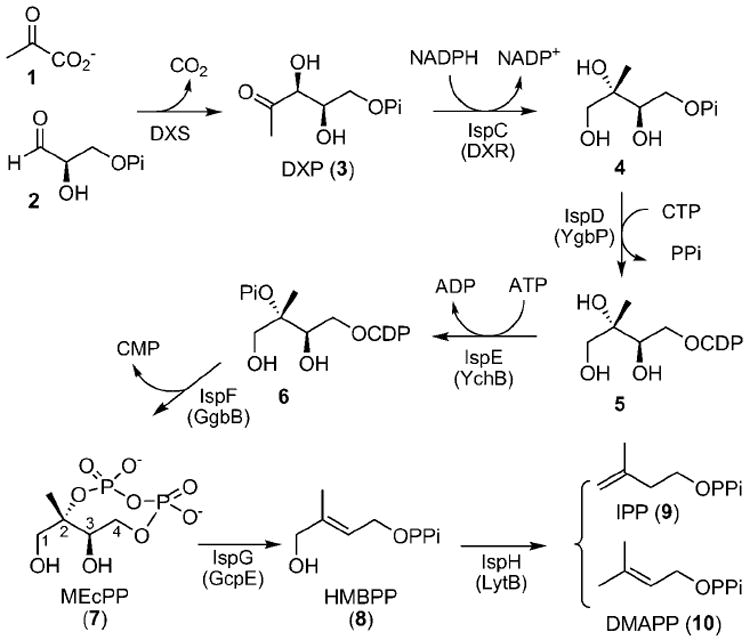
Deoxyxylulose phosphate (DXP) biosynthetic pathway.
Several mechanistic models have been proposed to account for the IspG-catalyzed reductive deoxygenation of MEcPP to 4-hydroxy-3-methyl-butenyl diphosphate (HMBPP, 8), including the vitamin K epoxyquinone reductase model,[4] the anaerobic ribonucleotide reductase (RNR) model,[4-5] and the deoxygenation model utilized in ascarylose biosynthesis.[6] Rohdich and Seemann have argued against the RNR-like mechanism based on the presumption that all three conserved Cys in IspG are [4Fe–4S] cluster ligands;[3b, 7] as there are no remaining Cys residues to act as the thiyl radical carrier. Further, isolated IspG does not contain a pyridoxamine cofactor, which argues against the deoxygenation mechanism utilized in ascarylose biosynthesis.
Several years ago, Rohdich et al. proposed an epoxide deoxygenation model (Scheme 2A),[7] although the proposed epoxy-HMBPP (11, Scheme 2) intermediate has not yet been directly observed in IspG-catalyzed reductive deoxygenation. Several lines of evidence suggest the plausibility of this model. First, it is known that synthetic [4Fe–4S] clusters catalyze epoxide-deoxygenation reactions to generate alkenes.[8] Second, we have synthesized epoxy-HMBPP (11), and our initial characterization revealed that IspG catalyzes reductive deoxygenation of epoxy-HMBPP to HMBPP with an efficiency comparable to that of MEcPP.[9] In addition, we have demonstrated that, in the absence of reductants, IspG can catalyze the production of MEcPP from epoxy-HMBPP.[10] Following our initial report on epoxy-HMBPP studies,[9] a report by Oldfield et al. suggested, on the basis of EPR analysis, that a common organometallic transient species en route to HMBPP is generated from both MEcPP and epoxy-HMBPP.[11] Another proposed mechanism (Scheme 2 B) is the cation model.[3b, 12] According to the cation model, heterolytic cleavage of the C–O bond at the C2 position (the first step) results in the formation of a tertiary cation intermediate. Brandt, et al. proposed, on the basis of computational studies, that the formation of a cation intermediate is energetically feasible.[13]
Scheme 2.
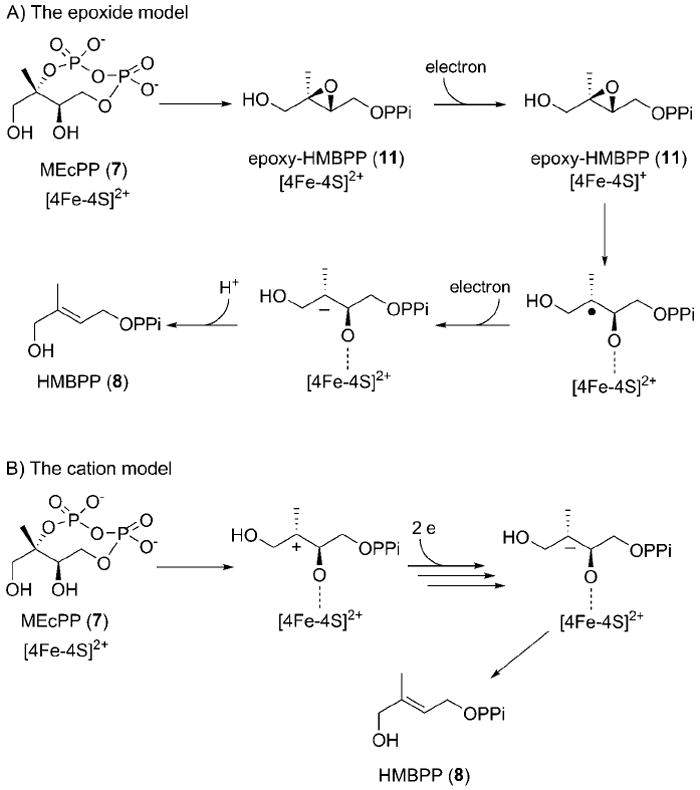
The epoxide and cation mechanistic models of IspG-catalyzed reductive deoxygenation of MEcPP to HMBPP.
In the above two models, the first step, conversion of MEcPP to epoxy-HMBPP or the production of a carbocation, does not involve redox chemistry. Previous studies in our laboratory did not detect any accumulation of either of the proposed intermediates during catalytic turnover. Interestingly, in the absence of reductants, the conversion of epoxy-HMBPP to MEcPP was observed.[10] Based on these features of the two mechanistic models in Scheme 2, we hypothesized that we might be able to study the C2 position reactivity by blocking the subsequent steps; this can be achieved by excluding the reduction system (e.g., NADPH-flavodoxin reductase or dithionite/methyl viologen). When these steps are blocked, the intermediate formed (epoxy-HMBPP or the carbocation) might have a high probability of reforming the substrate, MEcPP. Such an important mechanistic feature can be studied by measuring positional isotopic exchange, which is the focus of this study.
We synthesized [2-13C,18O]-MEcPP (7b) enzymatically. DXS, IspC (DXR), IspD, IspE, and IspF proteins were expressed as His6-tagged proteins and purified aerobically by Ni-NTA affinity chromatography. IspG was expressed as a Strep-tagged protein and purified anaerobically by using Strep-tactin resin as we reported recently.[3a] [2-13C,18O]-MEcPP was then synthesized by using these enzymes according to the route outlined in Scheme 3. [4-13C]-DXP (3b) was generated by DXP synthase (DXS) from [2-13C]-pyruvate (1b) and glyceraldehyde 3-phosphate (2), which was produced in situ by aldolase and triose phosphate isomerase (TPI) from fructose-1,6-biphosphate (12).[14] After the reaction was complete, enzymes were removed in a spin column with a molecular cut-off of 10 kDa. The filtrate containing product was then lyophilized, and the resulting [4-13C]-DXP (3b)-containing powder was incubated in H218O to produce [4-13C,18O]-DXP (3c).[15] After the 18O-exchange was complete, DXR protein was added along with a NADPH-regeneration system (glucose, glucose dehydrogenase, and NADP+) to convert 3c to [3-13C,18O]-MEP (4b). Compound 4b was purified by anion-exchange chromatography on AG 1-X2 resin (Bio-Rad). The overall yield of 4b from 1b was 92%. Once pure 4b was obtained, it was converted according to reported procedures.[16] The resulting 7b was purified in 77% yield from 4b.[16-17] MEcPP 7b was synthesized and purified in 62% yield over seven steps. By following the procedure outlined in Scheme 3, but excluding the 18O-exchange step, [2-13C]-MEcPP (7d) was also obtained in a similar yield. Syntheses and characterizations of compounds 7b and 7d are included in the Supporting Information.
Scheme 3.
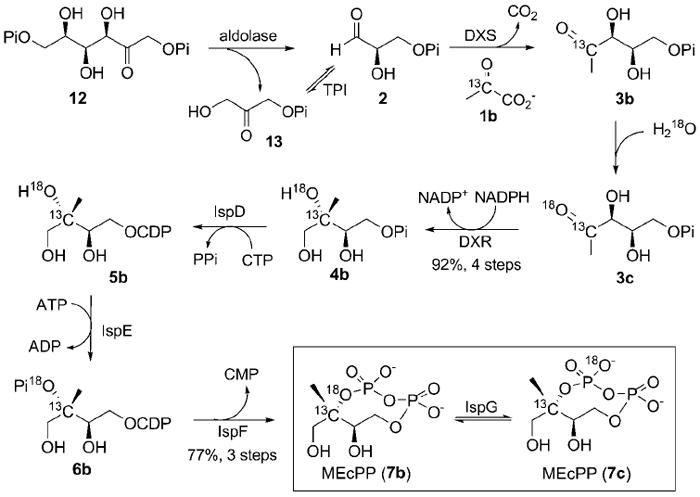
[2-13C,18O]-MEcPP (7b) synthesis and potential outcomes of IspG-catalyzed positional isotopic exchange in MEcPP.
Compounds 7b and 7d were characterized by 13C NMR spectroscopy (Figure 1 A–C). Their C2 13C NMR signals are observed as doublets with a coupling constant of 8.38 Hz as a result of the coupling between the C2 carbon and the neighboring 31P nucleus of the diphosphate linkage. A 1.0:1.1 mixture of 7b and 7d exhibits a 13C NMR spectrum consisting of two sets of doublets corresponding to the C2 carbons (Figure 1 C). The up-field shift of 0.037 ppm for 7b signals relative to those of 7d is due to the 18O isotope substitution effect.[18] These data confirmed the successful incorporation of 18O into the MEcPP bridging position. High-resolution mass spectrometry characterization of 7b and 7d further supported this conclusion (Supporting Information).
Figure 1.
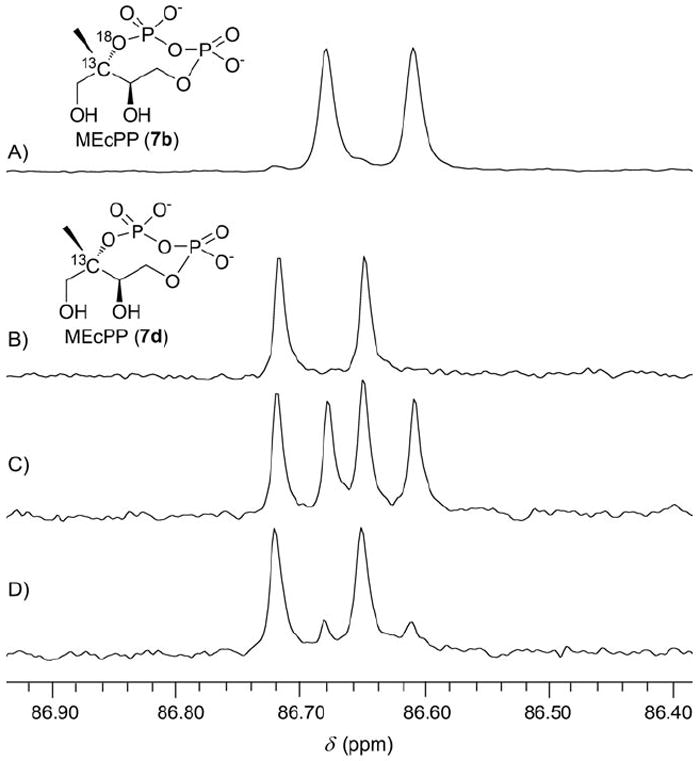
The C2 carbon 13C NMR signals of A) 7b, B) 7d, C) a 1.0:1.1 mixture of 7b and 7d; D) sample (C) was incubated with 10 μm of IspG at 37°C for 20 h in 100 mm Tris, pH 8.0.
In the 1.0:1.1 mixture of 7b (1.0 mm) and 7d (1.1 mm), the doublet corresponding to 7b diminished dramatically when IspG (10.0 μm) was introduced; eventually, an equilibrium ratio of ~5.3:1 between the two sets of doublets was reached (Figure 1D). In the absence of IspG, MEcPP in the reaction buffer is stable, and there is no sign of decomposition. Even in the presence of alkaline phosphatase, there is no sign of MEcPP degradation after a day of incubation (Figure S2 in the Supporting Information). Thus, the reduction of the 7b signal in Figure 1D cannot be due to decomposition. Moreover, the 1.0:1.1 mixture of 7b and 7d was characterized by mass spectrometry before and after IspG was introduced. In the mass spectra, the ratio of the two signals at 325.99 (7b) and 323.99 (7d) is the same before and after IspG introduction (Figures S1 and S2). The best explanation for the reduction in the 7b signal in Figure 1D is an IspG-catalyzed positional isotopic exchange, as predicted in Scheme 3, in which the bridging 18O is shifted to a terminal position.
The equilibrium ratio of ~5.3:1 observed in Figure 1D is consistent with reversible C–O bond cleavage at C2. Following cleavage of the C–O bond, rotation of the terminal phosphate will result in scrambling of the 18O/16O atoms. In the absence of a reduction system, an intermediate cleaved at C2 cannot proceed to form the product, HMBPP (8). The diphosphate moiety instead undergoes C–O bond formation at the C2 position to re-form MEcPP. Because the above experiments were carried out in the absence of reductants, the detection of positional isotopic exchange supports the view that C–O bond cleavage at C2 is indeed independent of the reduction steps in the reductive deoxygenation of MEcPP to HMBPP. Because C–O bond cleavage is reversible, one third of the 18O label will remain at the bridging position once equilibrium is reached (Figure S3). The equilibrium ratio of ~5.3:1 is due to the 1.0:1.1 mixture of 7b (1.0 mm) and 7d (1.1 mm) that was used in this study. The kinetics of positional isotope exchange were also measured by using pure [2-13C,18O]-MEcPP (7b) as the substrate (Figure S3). At saturating MEcPP concentration (2.0 mm), the kobs of positional exchange is estimated to be 0.38 min−1, which is 63 times lower than the kcat of reductive deoxygenation of MEcPP (7) to HMBPP (8). This kinetic result suggests that in the MEcPP active site, the terminal phosphate might not freely rotate following C–O bond cleavage at C2 position.
The observation of positional isotopic exchange provides evidence for a reversible C–O bond cleavage at the inactivated C2 position prior to subsequent reduction steps. Scheme 4 represents our current understanding of this intriguing system by integrating all the mechanistic information published in recent months by several groups. While this manuscript was in preparation, the crystal structure of IspG from a thermophile,Aquifex aeolicus, was published.[19] IspG is a homodimer, and each subunit has two domains, an N-terminal triosephosphate isomerase (TIM) barrel domain and a C-terminal iron–sulfur domain. Interestingly, the proposed substrate binding site, which is in the TIM barrel domain, is ~20 Å away from the iron–sulfur cluster. In addition, recent spectroscopic characterizations of IspG by two research groups (Oldfield[11] and Hoffman and Duin[20]) suggest that MEcPP not only directly interacts with the iron–sulfur cluster, but is also converted to an organometallic intermediate (Scheme 4).
Scheme 4.
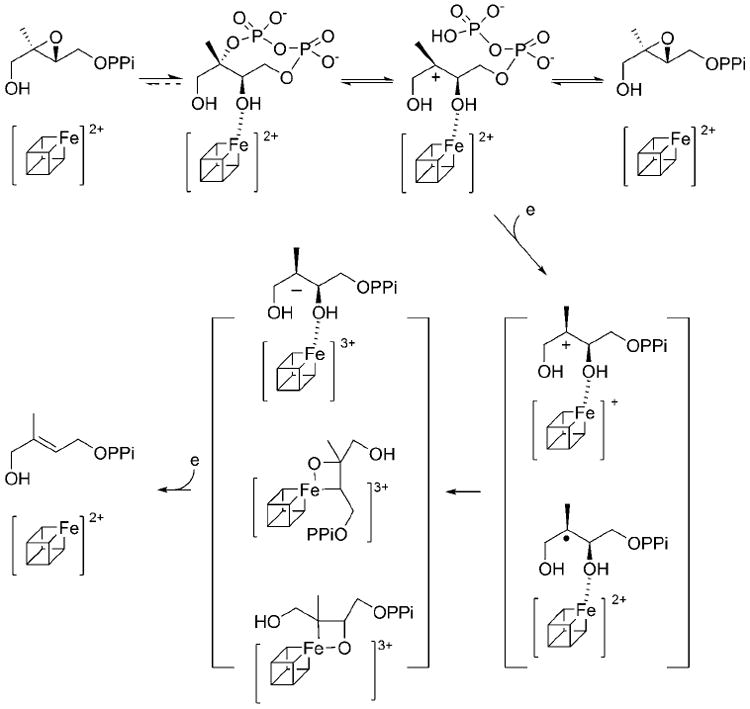
IspG mechanistic models.
The discrepancies between structural and spectroscopic results can be accounted for by a major conformational change upon MEcPP binding to IspG. It is known that reduction of the cluster in the absence of substrate is challenging,[2d, 3a, 20] and substrate binding might facilitate the subsequent reduction steps. The results presented in this study are consistent with this hypothesis. Oldfield and co-workers demonstrated that the same organometallic intermediate can be detected by using either MEcPP or epoxy-HMBPP as the substrate.[11] Recently, we have demonstrated the conversion of epoxy-HMBPP to MEcPP by IspG, consistent with the formation of the same organometallic intermediate from either substrate, although the conversion of epoxy-HMBPP to MEcPP was ten times slower than its conversion to HMBPP.[10] Hoffman and Duin also characterized such an intermediate and thoroughly discussed other possibilities for the identity of such an intermediate (Scheme 4).[20]
In the study presented here, all experiments were carried out in the absence of reductants. The detection of reversible C–O cleavage at the inactivated C2 position not only supports binding of MEcPP to IspG, but also implies that C–O bond cleavage at such a inactivated C2 position occurs prior to subsequent reduction chemistries. Once the activated intermediate is formed, its reduction can lead to the formation of organometallic intermediates. Several organometallic species have been proposed by Oldfield,[11] Hoffman and Duin[20] and co-workers as candidates for the downstream intermediates. Future studies are needed to differentiate among these options.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the NSF CAREER (CHE-0748504) and NIH (GM-093903) to P.L. The authors thank Dr. Norman Lee for his assistance with mass spectrometry characterization of the samples. John Bitok, Leighanne Brammer and Maria Hassis-LeBeau produced the IspC, D, E and F constructs.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201000716.
References
- 1.a) Eisenreich W, Bacher A, Arigoni D, Rohdich F. Cell Mol Life Sci. 2004;61:1401–1426. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qureshi N, Porter JW. In: Biosynthesis of Isoprenoid Compounds. Porter JW, Spurgeon SL, editors. Vol. 1. Wiley; New York: 1981. pp. 47–94. [Google Scholar]; c) FloresPérez Ú, Pérez-Gil J, Rodríguez-Villalón A, Gil MJ, Vera P, Rodríguez-Concepción M. Biochem Biophys Res Commun. 2008;371:510–514. doi: 10.1016/j.bbrc.2008.04.115. [DOI] [PubMed] [Google Scholar]; d) Rohdich F, Kis K, Bacher A, Eisenreich W. Curr Opin Chem Biol. 2001;5:535–540. doi: 10.1016/s1367-5931(00)00240-4. [DOI] [PubMed] [Google Scholar]
- 2.a) Campos N, Rodríguez-Concepción M, Seemann M, Rohmer M, Boronat A. FEBS Lett. 2001;488:170–173. doi: 10.1016/s0014-5793(00)02420-0. [DOI] [PubMed] [Google Scholar]; b) Querol J, Campos N, Imperial S, Boronat A, Rodríguez-Concepción M. FEBS Lett. 2002;514:343–346. doi: 10.1016/s0014-5793(02)02402-x. [DOI] [PubMed] [Google Scholar]; c) Okada K, Hase T. J Biol Chem. 2005;280:20672–20679. doi: 10.1074/jbc.M500865200. [DOI] [PubMed] [Google Scholar]; d) Adedeji D, Hernandez H, Wiesner J, Köhler U, Jomaa H, Duin EC. FEBS Lett. 2007;581:279–283. doi: 10.1016/j.febslet.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 3.a) Xiao Y, Zahariou G, Sanakis Y, Liu Y. Biochemistry. 2009;48:10483–10485. doi: 10.1021/bi901519q. [DOI] [PubMed] [Google Scholar]; b) Seemann M, Bui BTS, Wolff M, Tritsch D, Campos N, Boronat A, Marquet A, Rohmer M. Angew Chem. 2002;114:4513–4515. doi: 10.1002/1521-3773(20021115)41:22<4337::AID-ANIE4337>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:4337–4339. doi: 10.1002/1521-3773(20021115)41:22<4337::AID-ANIE4337>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; c) Seemann M, Janthawornpong K, Schweizer J, Bottger LH, Janoschka A, Ahrens-Botzong A, Tambou MN, Rotthaus O, Trautwein AX, Rohmer M, Schunemann V. J Am Chem Soc. 2009;131:13184–13185. doi: 10.1021/ja9012408. [DOI] [PubMed] [Google Scholar]; d) Xiao Y, Chu L, Sanakis Y, Liu P. J Am Chem Soc. 2009;131:9931–9933. doi: 10.1021/ja903778d. [DOI] [PubMed] [Google Scholar]
- 4.Hecht S, Eisenreich W, Adam P, Amslinger S, Kis K, Bacher A, Arigoni D, Rohdich F. Proc Natl Acad Sci USA. 2001;98:14837–14842. doi: 10.1073/pnas.201399298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolff M, Seemann M, Grosdemange-Billiard C, Tritsch D, Campos N, Rodríguez-Concepción M, Boronat A, Rohmer M. Tetrahedron Lett. 2002;43:2555–2559. [Google Scholar]
- 6.He XM, Liu HW. Annu Rev Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 7.Rohdich F, Zepeck F, Adam P, Hecht S, Kaiser J, Laupitz R, Grawert T, Amslinger S, Eisenreich W, Bacher A, Arigoni D. Proc Natl Acad Sci USA. 2003;100:1586–1591. doi: 10.1073/pnas.0337742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh T, Nagano T, Sato M, Hirobe M. Tetrahedron Lett. 1989;30:6387–6388. [Google Scholar]
- 9.Nyland RL, II, Xiao Y, Liu P, Meyers CLF. J Am Chem Soc. 2009;131:17734–17735. doi: 10.1021/ja907470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao Y, Nyland RL, II, Freel Meyers CL, Liu P. Chem Commun. 2010;46:7220–7222. doi: 10.1039/c0cc02594a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Li J, Wang K, Huang C, Zhang Y, Oldfield E. Proc Natl Acad Sci USA. 2010;107:11189–11193. doi: 10.1073/pnas.1000264107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kollas AK, Duin EC, Eberl M, Altincicek B, Hintz M, Reichenberg A, Henschker D, Henne A, Steinbrecher I, Ostrovsky DN, Hedderich R, Beck E, Jomaa H, Wiesner J. FEBS Lett. 2002;532:432–436. doi: 10.1016/s0014-5793(02)03725-0. [DOI] [PubMed] [Google Scholar]
- 13.Brandt W, Dessoy MA, Fulhorst M, Gao W, Zenk MH, Wessjohann LA. ChemBioChem. 2004;5:311–323. doi: 10.1002/cbic.200300743. [DOI] [PubMed] [Google Scholar]
- 14.Taylor SV, Vu LD, Begley TP, Schorken U, Grolle S, Sprenger GA, Bringer-Meyer S, Sahm H. J Org Chem. 1998;63:2375–2377. [Google Scholar]
- 15.Dorrestein PC, Zhai H, Taylor SV, McLafferty FW, Begley TP. J Am Chem Soc. 2004;126:3091–3096. doi: 10.1021/ja039616p. [DOI] [PubMed] [Google Scholar]
- 16.Illarionova V, Kaiser J, Ostrozhenkova E, Bacher A, Fischer M, Eisenreich W, Rohdich F. J Org Chem. 2006;71:8824–8834. doi: 10.1021/jo061466o. [DOI] [PubMed] [Google Scholar]
- 17.Kuzuyama T, Takahashi S, Watanabe H, Seto H. Tetrahedron Lett. 1998;39:4509–4512. [Google Scholar]
- 18.Risley JM, Van Etten RL. J Am Chem Soc. 1979;101:252–253. [Google Scholar]
- 19.Lee M, Grawert T, Quitterer F, Rohdich F, Eppinger J, Eisenreich W, Bacher A, Groll M. J Mol Biol. 2010;404:600–610. doi: 10.1016/j.jmb.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 20.Xu W, Lees NS, Adedeji D, Wiesner J, Jomaa H, Hoffman BM, Duin EC. J Am Chem Soc. 2010;132:14509–14520. doi: 10.1021/ja101764w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.