

Summary
Background
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of motor neurons that results in progressive weakness and death from respiratory failure, commonly within about 3 years. Previous studies have shown association of a locus on chromosome 9p with ALS and linkage with ALS–frontotemporal dementia. We aimed to test whether this genomic region is also associated with ALS in an independent set of UK samples, and to identify risk factors associated with ALS in a further genome-wide association study that combined data from the independent analysis with those from other countries.
Methods
We collected samples from patients with sporadic ALS from 20 UK hospitals and obtained UK control samples from the control groups of the Depression Case Control study, the Bipolar Affective Case Control Study, and the British 1958 birth cohort DNA collection. Genotyping of DNA in this independent analysis was done with Illumina HumanHap550 BeadChips. We then undertook a joint genome-wide analysis that combined data from the independent set with published data from the UK, USA, Netherlands, Ireland, Italy, France, Sweden, and Belgium. The threshold for significance was p=0·05 in the independent analysis, because we were interested in replicating a small number of previously reported associations, whereas the Bonferroni-corrected threshold for significance in the joint analysis was p=2·20×10−7
Findings
After quality control, samples were available from 599 patients and 4144 control individuals in the independent set. In this analysis, two single nucleotide polymorphisms in a locus on chromosome 9p21.2 were associated with ALS: rs3849942 (p=2·22×10−6; odds ratio [OR] 1·39, 95% CI 1·21–1·59) and rs2814707 (p=3·32×10−6; 1·38, 1·20–1·58). In the joint analysis, which included samples from 4312 patients with ALS and 8425 control individuals, rs3849942 (p=4·64×10−10; OR 1·22, 95% CI 1·15–1·30) and rs2814707 (p=4·72×10−10; 1·22, 1·15–1·30) were associated with ALS.
Interpretation
We have found strong evidence of a genetic association of two single nucleotide polymorphisms on chromosome 9 with sporadic ALS, in line with findings from previous independent GWAS of ALS and linkage studies of ALS–frontotemporal dementia. Our findings together with these earlier findings suggest that genetic variation at this locus on chromosome 9 causes sporadic ALS and familial ALS–frontotemporal dementia. Resequencing studies and then functional analysis should be done to identify the defective gene.
Funding
ALS Therapy Alliance, the Angel Fund, the Medical Research Council, the Motor Neurone Disease Association of Great Britain and Northern Ireland, the Wellcome Trust, and the National Institute for Health Research Dementias and Neurodegenerative Diseases Research Network (DeNDRoN).
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of motor neurons that results in progressive weakness and death from respiratory failure, commonly within about 3 years. About one in 300 people1 develops ALS but its prevalence is low—five per 100 000 people—because of its poor prognosis. About 5% of patients have a family history of ALS, and occasionally frontotemporal dementia. ALS and frontotemporal dementia can coexist in the same family or individual.
Several studies have reported linkage of ALS–frontotemporal dementia with a locus on chromosome 9p, and specifically with a shared region that spans about 3·6 Mb.2, 3, 4, 5, 6, 7 A genome-wide association study (GWAS) reported that sporadic ALS was associated with single nucleotide polymorphisms (SNPs) in the same region of chromosome 9p.8 Therefore, this locus is the focus of research into the genetic basis of familial ALS, the relation between ALS and frontotemporal dementia, and the mechanisms underlying sporadic ALS and frontotemporal dementia.
Previous GWAS in ALS have reported association results that have been replicated within the same study but rarely in independent studies.9 For example, variation in the ITPR2 gene was associated with ALS in a GWAS and was replicated within the study10 but has not been found by other investigators.8 Variation in DPP6 was reported to be associated with ALS and was replicated within the same study,11 and a second study found that the same SNPs were the most strongly associated, although they did not achieve genome-wide significance.12 A subsequent study failed to confirm the findings.13 Variation in the UNC13A gene was strongly associated with ALS and was replicated within the same study,8 but no successful replication of the association with this locus has been reported.
We therefore aimed to examine previously reported genetic associations for ALS in a GWAS in an independent set of UK samples. We also aimed to combine these data with existing genotypic data from seven other countries to generate a large GWAS in ALS.
Methods
Samples
Adults attending one of 20 UK hospitals and who had been diagnosed with ALS by two consultant neurologists were invited to participate in the UK National DNA Bank for Motor Neuron Disease Research (MND DNA bank), a national collection of ALS DNA samples derived from whole blood. Collection began in 2004, and enrolment is ongoing. Patients had no family history of ALS at the time of sampling, were of self-reported white European ancestry, and had onset of weakness on or after January, 2002. Patients provided ethically approved written consent for blood to be drawn. Control samples for the independent analysis were obtained from two sources: the control groups of the Depression Case Control (DeCC) study14 and the Bipolar Affective Case Control Study (BACCS)15 and data deposited by Panos Deloukas from the Wellcome Trust Sanger Institute (Cambridge, UK) and published online from the British 1958 birth cohort DNA collection.
Case and control samples for the joint analysis were from the UK, USA, Netherlands, Belgium, France, Italy, Ireland, and Sweden.8, 11, 16, 17 Demographic, ascertainment, and quality control details of these samples have been published previously.8, 11, 16, 17 All participating individuals gave written informed consent, and patients were selected for sporadic ALS. Samples were matched for geographical origin and ethnic origin only.
This research was approved by the research ethics committees of each relevant institution for all cohorts.
Procedures
DNA was extracted by use of standard methods at three centres within 1 week of the blood being drawn (usually on the same day) and was stored centrally at the UK DNA banking network in Manchester. We used a barcode-based sample tracking system to minimise the risk of clerical error.
Genotyping of DNA from samples held with the UK MND DNA bank was done with Illumina HumanHap550 BeadChips (Illumina, CA, USA) by UCL Genomics, London, UK. All DNA samples went through stringent quality control, and processing was done under full laboratory information management system control. The raw data were analysed with GenomeStudio (Illumina) and extracted for statistical analysis.
Quality control procedures were applied to individual and SNP data (webappendix pp 1–3). Individuals were excluded if more than 1% of their genotypic data was missing; if they had abnormal heterozygosity, poor cluster separation, or a sex assignment that conflicted with phenotypic data; if they shared more than 5% of alleles identical by descent with any other study member; or if they were of non-European ancestry. SNPs with minor allele frequency less than 1%, showing non-random missingness between genotypes or cases and controls, or showing departure from Hardy-Weinberg equilibrium (p<1×10−6) were excluded.
For the joint analysis, raw genotypes were obtained from public databases or directly from research groups and merged into a single file for analysis, with a covariate file listing the site of origin. Genotyping and quality control measures for the studies in the joint analysis have been published previously.8, 11, 16, 17
All data were deposited at the ALS online genetics database18 and the European genome-phenome archive.
Statistical analysis
We did power analysis with the Genetic Power Calculator.19 For the independent analysis there was 80% power to detect a typical previously reported association with an odds ratio (OR) of 1·2 and minor allele frequency of 0·25 at p=0·05. We report power at p=0·05 in the independent analysis because, although we have done a GWAS, we are interested in replicating association at five specified loci (FGGY, DPP6, ITPR2, UNC13A, and chromosome 9p21.2). For the joint analysis, we investigated the possibility of new associations within the genome and therefore we corrected for multiple testing. We used Bonferroni correction, which assumes the tests of association at each of the 227 475 SNPs are independent; the corrected threshold is therefore 2·20×10−7 (0·05/227 475), and there was 83·5% power to detect a similar variant at the threshold of p=2·20×10−7. Our aim was to use all of the available samples.
For the joint analysis, population stratification was controlled for by principal component analysis in EIGENSTRAT.20 We used the Tracy-Widom distribution to calculate the number of significant principal components needed as covariates in subsequent analyses, using the twstats program of the Eigensoft package.21
We tested association of genotypes with ALS by logistic regression with 30 principal components as covariates in the program PLINK (version 1.07).22 We used the expectation-maximisation algorithm—as used in PLINK—for haplotype estimation. Haplotypic association with ALS was tested with a sliding window of varying sizes, including a window sufficient to include all SNPs in the haplotype block that contained the most strongly associated SNPs.
We imputed genotypes by use of Impute2,23 with data from the 1000 Genomes Project and the International HapMap as templates.24 Genotype imputation allows the prediction of genotypes at untyped SNPs by the correlation between SNP genotypes in different populations. Association analysis can then be done on the predicted genotypes. Association analysis was done with SNPTEST,25 which accounts for uncertainty in the imputed genotypes, using the first ten principal components as covariates (the maximum possible). The correction for population stratification in the imputed data will have been weakened by the use of fewer principal components; however, the use of Impute2 increased our ability to deal with genotype uncertainty. We did this to identify variants for follow-up genotyping in a focused region, and thus the trade-off was deemed worthwhile.
Population attributable risk was estimated as:
where q is the risk allele frequency, p=1–q, and genotype relative risk is denoted by g, with the subscript showing heterozygote or homozygote status. Penetrance for each genotype was estimated as:
where K is the lifetime prevalence and f is the genotype frequency.
Role of the funding source
The sponsor of the study had no role in study design, data analysis, data interpretation, writing of the report, or the decision to submit the paper for publication. The Motor Neurone Disease Association is the sponsor of the UK National MND DNA bank, and therefore had a role in data collection. The corresponding author had full access to all of the data in the study and had final responsibility for the decision to submit the paper for publication.
Results
For the independent analysis, genotypes were obtained from 663 individuals in the UK MND DNA bank, 1589 control samples from the DeCC study and BACCS, and 2930 control samples from the 1958 birth cohort. After quality control measures, samples were available from 599 people with ALS and 4144 control individuals. Table 1 summarises clinical features of the ALS cohort in the independent analysis.
Table 1.
Clinical features of patients with amyotrophic lateral sclerosis in the independent analysis
| Case cohort (n=599) | ||
|---|---|---|
| Presentation* | ||
| Bulbar symptoms | 165 (28%) | |
| Limb weakness | 375 (63%) | |
| Respiratory | 11 (2%) | |
| Mixed† | 42 (7%) | |
| Age of onset‡ (years) | 60·7 (61·7, 22·9–86·5) | |
| Diagnostic delay§ (months) | 21·5 (18·3, 2·0–80·1) | |
Data are number (%) or mean (median, range).
Data missing for six patients.
Simultaneously reported weakness of limbs and bulbar musculature.
Data missing for three patients.
Time between symptom onset and diagnosis. Data missing for eight patients.
For the joint analysis, genotypes were obtained from 4312 patients with ALS and 8425 control individuals. After quality control measures, samples were available from 4133 patients with ALS and 8130 control individuals, which were typed for 227 475 SNPs to generate 2 789 513 662 genotypes. Table 2 and the webappendix p 4 summarise demographic data.
Table 2.
Characteristics of the component populations for the joint analysis after quality control
| Cases | Controls | Total | Men | |
|---|---|---|---|---|
| Belgium8 | 286 | 307 | 593 | 313 |
| France16 | 230 | 700 | 930 | 805 |
| Netherlands8, 10, 12 | 977 | 988 | 1965 | 1152 |
| Ireland11 | 210 | 200 | 410 | 219 |
| Italy17 | 259 | 241 | 500 | 271 |
| Sweden8 | 422 | 441 | 863 | 481 |
| USA16 | 923 | 904 | 1827 | 1061 |
| UK16 | 227 | 205 | 432 | 264 |
| UK MND DNA bank | 599 | 0 | 599 | 382 |
| DeCC study and BACCS | 0 | 1505 | 1505 | 567 |
| 1958 birth cohort | 0 | 2639 | 2639 | 1357 |
| Total | 4133 | 8130 | 12 263 | 6872 |
The final three rows refer to samples used in the independent study. DeCC=Depression Case Control. BACCS=Bipolar Affective Case Control Study.
In the independent analysis, SNPs rs3849942 (p=2·22×10−6; OR 1·39, 95% CI 1·21–1·59) and rs2814707 (p=3·32×10−6; 1·38, 1·20–1·58) on chromosome 9 showed association with ALS (figure 1, table 3, webappendix pp 5–6). rs903603, which had strong linkage disequilibrium with the associated SNPs (D'=0·98), was the SNP most strongly associated with ALS in the independent set (p=8·92×10−8; OR 0·71, 95% CI 0·63–0·81). No other loci achieved genome-wide significance.
Figure 1.
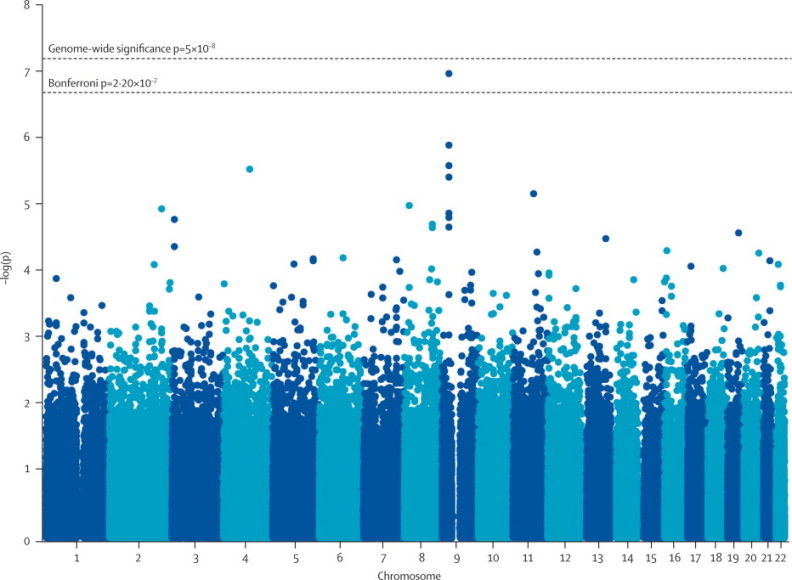
p values for association in the independent genome-wide association study
Table 3.
Associations for genes reported to be associated with amyotrophic lateral sclerosis in previous genome-wide association studies
| n for associated SNP in previous studies* | Associated SNP | Controls | p value in previous studies |
Independent analysis† |
Joint analysis‡ |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| p value |
Minor allele frequency |
p value |
Minor allele frequency |
|||||||
| Cases | Controls | Cases | Controls | |||||||
| FGGY9 | 1287 | rs6700125 | 1567 | 6·0×10−4 | 0·71 | 0·342 | 0·337 | 0·08 | 0·351 | 0·339 |
| DPP612 | 1767 | rs10260404 | 1916 | 5·4×10−8 | 0·89 | 0·397 | 0·395 | 0·08 | 0·391 | 0·380 |
| ITPR210 | 1337 | rs2306677 | 1356 | 3·28×10−6 | 0·98 | 0·100 | 0·100 | 0·97 | 0·100 | 0·096 |
| UNC13A8 | 4855 | rs12608932 | 14 953 | 2·53×10−14 | 0·42 | 0·366 | 0·354 | 5·14×10−4 | 0·382 | 0·344 |
| 9p21.28 | 4855 | rs2814707 | 14 953 | 7·45×10−9 | 3·32×10−6 | 0·305 | 0·243 | 4·72×10−10 | 0·272 | 0·236 |
| 9p21.28 | 4855 | rs3849942 | 14 953 | 1·01×10−8 | 2·22×10−6 | 0·304 | 0·241 | 4·64×10−10 | 0·271 | 0·235 |
| 9p21.28 | 4855 | rs903603 | 14 953 | .. | 8·92×10−8 | 0·411 | 0·493 | 1·37×10−7 | 0·456 | 0·492 |
Previously reported significant associations and the results we obtained for those genes in the two parts of this study are shown.8, 9, 10, 12
Total combined sample for the reported SNP, which includes a GWAS component and a non-GWAS follow-up study.
599 cases and 4144 controls.
4133 cases and 8130 controls.
In the joint analysis, rs3849942 (p=4·64×10−10; OR 1·22, 95% CI 1·15–1·30), rs2814707 (p=4·72×10−10; 1·22, 1·15–1·30), and rs903603 (p=1·37×10−7; 0·86, 0·82–0·91) showed association with ALS (figure 2, table 3, webappendix pp 7–8). SNPs at other loci that were associated with ALS in previous studies did not reach significance in the joint analysis, except the previously associated SNP in UNC13A (rs12608932; table 3). The genotype relative risk for rs3849942 was 1·24 (heterozygote) and 1·49 (homozygote), with minor allele frequencies of 0·27 in patients with ALS and 0·24 in controls. The population attributable risk of rs3849942 was 0·10 and the penetrance of the risk allele was 0·004 (heterozygote) and 0·005 (homozygote).
Figure 2.
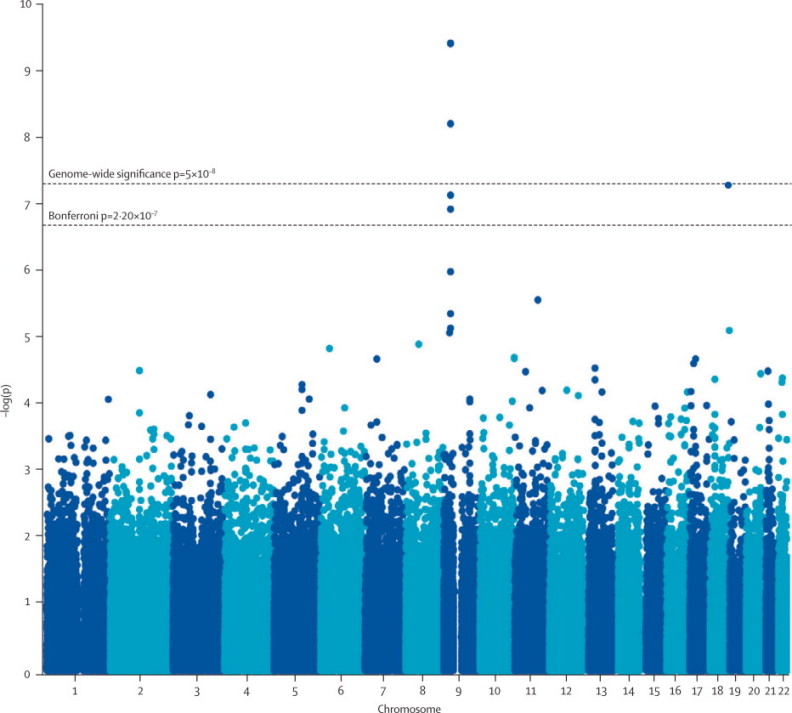
p values for association in the joint analysis
A haplotypic association of rs3849942 and rs10122902 (omnibus p=1·51×10−11) was identified in the joint analysis. Of the three haplotypes in the population (GA, AG, and GG), one had equal frequency in people with ALS and controls (GA: frequency cases 0·21, controls 0·20; p=0·123), one was associated with increased risk of ALS (AG: frequency cases 0·27, controls 0·23, p=7·76×10−10), and was one associated with reduced risk of ALS (GG: frequency cases 0·52, controls 0·57; p=4·83×10−11). Similar results were obtained for the independent study: rs3849942 and rs10122902 showed haplotypic association (omnibus p=1·01×10−7), and the same three haplotypes had similar control frequencies (GA: frequency cases 0·21, controls 0·19, p=0·074; AG: frequency cases 0·30, controls 0·24, p=3·18×10−6; GG: frequency cases 0·48, controls 0·57, p=4·24×10−8). Longer SNP haplotypes did not result in stronger association in either part of the study (data not shown). The sliding window showed that pairs of SNPs within a 106·5 kb block of linkage disequilibrium flanked by rs4879515 and rs702231 were nearly all associated with ALS at p<1×10−3, whereas outside this region there were no significant associations.
Imputation in the joint analysis in the region of interest (ie, the 3·6 Mb region identified by linkage) gave results for 21 899 imputed SNPs that were derived from 519 typed SNPs. Five SNPs were more strongly associated with ALS than was rs3849942; the most strongly associated SNP was a rare variant, chr9:27573575 (minor allele frequency 0·054; p=6·40×10−15). The most strongly associated variant with a minor allele frequency greater than 0·1 was rs13295103 (p=1·97×10−12; figure 3).
Figure 3.
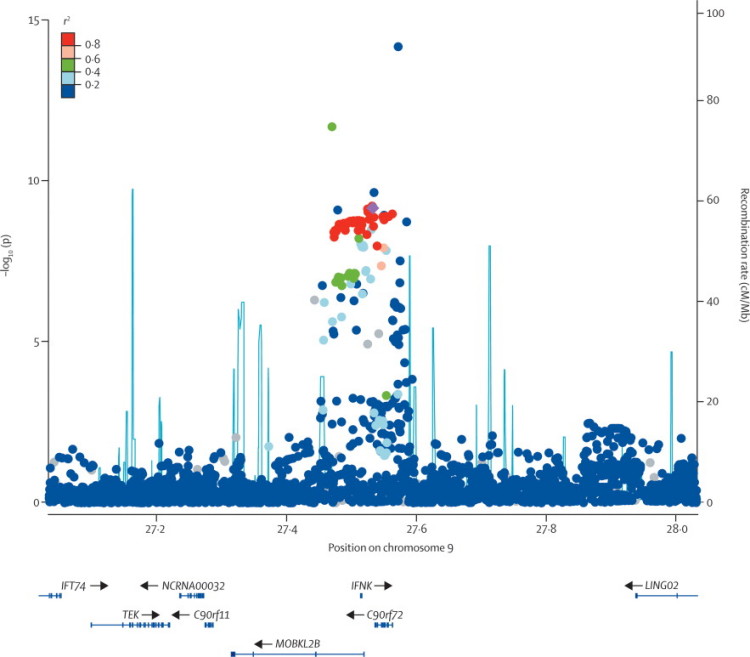
Genetic architecture of the associated region
Circles=−log10 of the p value of association with amyotrophic lateral sclerosis for typed and imputed single nucleotide polymorphisms in the joint analysis, coloured according to linkage disequilibrium with rs3849942 (shown as a purple diamond) as measured by r2. Blue lines=recombination rate across the locus based on the 1000 genomes project. Genes in the region are displayed below the graph. Arrows indicate the direction of transcription.
Discussion
In our independent study we have confirmed association of a chromosome 9 locus with ALS. We did not replicate any of the other previously reported associations, suggesting that our study gave a false-negative result or that they were population specific or false-positive results (panel, table 3).8, 9, 10, 11, 12 In our joint analysis, which is the largest GWAS of ALS to date, chromosome 9p21.2 and UNC13A were the only significantly associated loci. The association of UNC13A with ALS was weaker in the joint analysis than in the study that identified the association,8 which was well powered and replicated the results within the same study. Our independent samples do not show the association, possibly because the UNC13A variation is not a major cause of ALS in the UK, and so adding in these samples to the joint analysis we inevitably weaken the previously reported association. Given that the heritability of sporadic ALS is 0·61 (95% CI 0·38–0·78),26 our findings suggest that even larger GWAS are required to have sufficient power to detect genetic variations responsible for ALS. A potential limitation of this study is that not all control individuals were age and sex matched, although they were matched for geographical origin and ancestry. For a disease as rare as ALS, age and sex matching does not increase statistical power, whereas it would for a more common disorder.
Panel. Research in context.
Systematic review
To identify previously published genome-wide association studies (GWAS) in amyotrophic lateral sclerosis (ALS), we searched PubMed and the ALSOD database (http://www.alsod.iop.kcl.ac.uk), and contacted known ALS genetics research groups. Four GWAS reported findings that reached genome-wide significance;8, 9, 10, 11, 12 we designed the independent study to replicate these. Association studies were selected for the joint analysis if research diagnostic criteria for ALS were used, if microarrays of at least 300 000 single nucleotide polymorphisms were used, and if individual rather than pooled genotypes were available. Because the main finding of the independent and joint analyses was of an association to a region on chromosome 9 also known to be associated with familial ALS–frontotemporal dementia and sporadic frontotemporal dementia, we searched PubMed and contacted research groups and identified a further seven families with ALS–frontotemporal dementia2, 3, 4, 7 and a GWAS of frontotemporal dementia.29 These linkage data have been well summarised in a linkage paper.4
Interpretation
The ALS-associated locus on chromosome 9p was independently replicated, and no other loci were identified, suggesting that even larger genome-wide association studies are needed. We have shown the association signal on chromosome 9p lies in a well circumscribed block of linkage disequilibrium of about 106·5 Kb in size, which is flanked by markers rs4879515 and rs702231 and includes three annotated genes. This locus has also previously been implicated in sporadic frontotemporal dementia and in families with ALS–frontotemporal dementia. Because there is pathological and clinical overlap of ALS and frontotemporal dementia, the most logical interpretation is that dysfunction of a single gene product is responsible for some cases of ALS, frontotemporal dementia, and ALS–frontotemporal dementia.
The associated 9p21 locus is the focus of much interest for researchers of both ALS and frontotemporal dementia. Although frontotemporal dementia and ALS are independent diseases, there is some overlap. About 5% of people with ALS have overt frontotemporal dementia and up to 51% have subtle cognitive deficits that suggest frontal and temporal lobe dysfunction.27 Conversely, about 50% of people with frontotemporal dementia have features of motor neuron degeneration.28 A locus on chromosome 9 has been independently linked to ALS–frontotemporal dementia in seven families;2, 3, 4, 5, 6, 7 this 3·6 Mb locus is defined across studies by the flanking markers D9S169 and D9S251. The SNPs we have identified lie within this region, with the peak association at 106·5 Kb. A GWAS that used pathological subtyping of patients with frontotemporal dementia to increase homogeneity also identified the same SNPs as those most strongly associated with sporadic frontotemporal dementia (rs3849942 p=1·38×10−6 and rs2814707 p=5·72×10−6).29 Therefore, variation in the same gene is likely to be responsible for sporadic ALS, sporadic frontotemporal dementia, and familial ALS–frontotemporal dementia. Assuming that the association locus is the same as the linkage locus, the region of interest in families with ALS–frontotemporal dementia must now be thought to be much narrower than that defined by recombination; however, if a synthetic association of rare variants and ALS is being detected, the disease-causing variation could still lie a substantial distance from the association peak.30
Penetrance is about 20% in the family with the strongest association between chromosome 9 and ALS–frontotemporal dementia.3 Thus, familial variants might underlie at least some of the sporadic cases of ALS and frontotemporal dementia. The large signal seen in our independent study of UK samples is consistent with a founder effect in the UK population, which possibly extends out to other populations. In this respect, that a GWAS of a cohort of Finnish patients with ALS and controls also detected the chromosome 9p locus (rs3849942 p=9·11×10−11) is interesting.31 This association signal was driven mainly by people with familial ALS; the most strongly associated SNP in this subset of patients was rs2225389 (p=2·23×10−12). All the patients with familial ALS who were not homozygous for the D90A allele of the SOD1 gene, and almost 20% of the patients with sporadic ALS, shared a 42-SNP haplotype, which strongly suggests a founder effect for the chromosome 9p locus, at least in Finland. However, the penetrance of the associated risk allele of rs3849942 in sporadic ALS in our study is only slightly higher than the background risk of ALS (0·36–0·45%),1 whereas the penetrance of the familial variants is about 20%; thus, the likelihood that the same variation is responsible for both familial and sporadic ALS is questionable, which makes a founder effect less likely. The significant association of rs3849942 with sporadic ALS in our joint analysis including multiple populations suggests that a common genetic variation might be contributing to disease risk. The two-SNP haplotype that was most strongly associated with ALS in the joint analysis (rs3849942-rs10122902) mimics the behaviour of the APOE association with Alzheimer's disease; the three APOE alleles—ɛ2, ɛ3, and ɛ4—are in fact pairs of alleles at two SNPs forming a haplotype. When a haplotype is formed from alleles at two SNPs there are four possible haplotypes, although not all will necessarily exist in the population. For both the rs3849942-rs10122902 haplotype in ALS and the APOE haplotype in Alzheimer's disease, only three of the four possible haplotypes are present, and the fourth does not exist in the population. In both cases, all identified haplotypes are common, and one is associated with risk of the disease (APOE ɛ4 in Alzheimer's disease; AG in ALS), one is associated with protection from the disease (APOE ɛ2 in Alzheimer's disease; GG in ALS), and one is neutral (APOE ɛ3 in Alzheimer's disease; GA in ALS). Thus, common variation, as with APOE in Alzheimer's disease, rather than a founder effect, could be responsible for sporadic ALS. However, because the penetrance at a functional variant might be much higher than at an associated tag SNP, which would account for the discrepancy between the penetrance for familial ALS and sporadic ALS, a founder effect cannot be excluded. There must be multiple variants worldwide because families with chromosome 9p-linked ALS–frontotemporal dementia do not all share a common haplotype, and so multiple founders must exist. However, the chromosome 9p locus is important in ALS because the population attributable risk estimate shows that it is involved in 10% of all cases of sporadic ALS.
The disease-associated SNPs we have identified lie between two genes, C9orf72 and MOBKL2B, and are close to IFNK, which lies in an intron of MOBKL2B. The protein encoded by MOBKL2B is Mps one binder kinase activator-like 2B, which is involved in kinase regulation. On the opposite strand at the same location is IFNK, which encodes interferon κ precursor, which is important for immunity to viral infection. The SNPs comprising the most strongly associated haplotype (rs3849942 and rs10122902) and imputed SNP (chr9:27573575) overlap or lie within C9orf72, which encodes a protein that might be involved in cell development and possibly spermatogenesis.32
Although linkage of ALS–frontotemporal dementia to this region of chromosome 9 was first identified in 2006,2, 3 sequencing has not yet yielded a disease-causing variant such as a non-synonymous SNP or splice-site variation. If this remains the case, one explanation could be that the variation is tagged by SNPs but not easily detected by exome capture and high throughput sequencing or Sanger sequencing. The disease-causing variant might be in an unknown exon or gene and has therefore not been sequenced, or it might be a trinucleotide repeat expansion in an intron, although such repeats are not seen in the surrounding region. Copy number variation is not necessarily easily detected, depending on the sequencing method used, but three microarray-based studies in ALS have not detected ALS-associated copy number variation in this region of chromosome 9.33, 34, 35 Finally, a microRNA or non-annotated gene that has not been examined, or an inversion, which would be difficult to detect by sequencing, might be the disease-causing variant. Our results suggest genetic variation at this locus on chromosome 9 causes sporadic amyothophic lateral sclerosis and familial ALS–frontotemporal dementia. Resequencing studies and then functional analyses are needed to identify the defective gene.
Acknowledgments
Acknowledgments
The independent study case samples were obtained from the UK DNA bank for Motor Neuron Disease Research, which was funded by the Motor Neurone Disease Association grant 3/3 and the Wellcome Trust grant 070122/A/02/Z, with additional support from the National Institute for Health Research Dementias and Neurodegenerative Diseases Research Network (DeNDRoN). Genotyping of the independent study case samples was funded by grants from the Angel Fund and ALS Therapy Alliance, with additional funding to RO and JH from the Motor Neurone Disease Association for genotyping (grant 10/57). We acknowledge use of genotype data from the British 1958 birth cohort DNA collection, which was funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02. Controls for the DeCC study and BACCS were genotyped under the Medical Research Council grant G0701420. This project has been supported by the Prinses Beatrix Fonds, VSB Fonds, The Kersten Foundation, The Netherlands ALS Foundation, and The Adessium Foundation (LHvdB). JHV is supported by grants from The Brain Foundation of the Netherlands. WR is supported by grants from the University of Leuven (Methusalem) and the Interuniversity Attraction Poles (IUAP) program P6/43 of the Belgian Federal Science Policy Office. VS is supported by the Italian Ministry of Health (Ricerca Finalizzata 2007 number 31). This work was supported in part by the Intramural Research Program of the National Institutes of Health, and the National Institute on Aging (Z01-AG000949-02). AC is supported by the Italian Ministry of Health (Ricerca Sanitaria Finalizzata, 2007, number 30), the Fondazione Vialli e Mauro per la SLA (grant 3/2008), and the Federazione Italiana Giuoco Calcio (Grant 2/2008). We thank the ALS Association, the ALS Society of Canada, MNDA Iceland, and the National Institute for Health Research Biomedical Research Centre for Mental Health for support, and Kuang Lin for programming advice.
Contributors
AS, KM, JH, RWO, and AAC did the genotyping and analyses. SN, MEW, BS, CV, LJ, OA, and JFP did the analyses. KM, JHV, MAvE, LHvdB, WR, OH, AEF, CML, AWB, PMA, IF, VS, AC, BJT, JM, VM, JEL, PM, JDG, HP, PNL, RHB, RWO, KEM, PJS, CES, and AAC contributed samples or genotypes or both. KM, PJS, CES, and AAC conceived the study. All authors contributed to the writing of the manuscript.
Contributing UK MND DNA bank investigators
Queen Elizabeth Hospital and University of Birmingham Hub J McConville, V Patterson (Belfast City Hospital); C Young (Walton Centre for Neurology and Neurosurgery); K Talbot (Radcliffe Hospitals NHS Trust, John Radcliffe Hospital); I Ormerod (North Bristol NHS Trust, Southmead Hospital). King's College Hospital and King's College London Hub R W Orrell (Royal Free Hospital and National Hospital for Neurology and Neurosurgery); C Hillier (Poole NHS Trust); J Frankel (Southampton University Hospitals NHS Trust, Southampton General Hospital); A Radunovic, A Malaspina (Barking, Havering and Redbridge University Hospitals NHS Trust); A Radunovic (Barts and the London NHS Trust); C Allen (Cambridge University Hospitals NHS Trust, Addenbrooks Hospital); J Zajicek (Derriford Hospital Plymouth). Royal Hallamshire Hospital and University of Sheffield Hub A J Wills (Nottingham University Hospitals NHS Trust, Queen's Medical Centre); R J Swingler (NHS Tayside, Ninewells Hospital); T L Williams (Newcastle Upon Tyne Hospitals NHS Foundation Trust, Royal Victoria Infirmary); D Mitchell (Lancashire Teaching Hospitals NHS Trust, Royal Preston Hospital); J Ealing (Greater Manchester Centre for Clinical Neuroscience and Hope Hospital Salford).
Conflicts of interest
WR has submitted a patent for a therapeutic molecule in neurodegeneration. OH has done consultancy work for Allergan, Merck, and Serono. VS has done consultancy work for Acceleron Pharma. AAC and PNL were funded by Trophos for a clinical trial of a therapeutic drug in ALS. PNL has done consultancy work for NeuroNova, Allon, and GlaxoSmithKline. RHB has done consultancy work for Biogen Idec and holds stock or stock options in Link Medicine and AviTx. PJS has submitted two patents for therapeutic molecules in ALS and has been a consultant to Sanofi-Aventis. All other authors declare no conflicts of interest.
Web Extra Material
References
- 1.Johnston CA, Stanton BR, Turner MR. Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol. 2006;253:1642–1643. doi: 10.1007/s00415-006-0195-y. [DOI] [PubMed] [Google Scholar]
- 2.Morita M, Al-Chalabi A, Andersen PM. A locus on chromosome 9p confers susceptibility to amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 3.Vance C, Al-Chalabi A, Ruddy D. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 4.Boxer AL, Mackenzie IR, Boeve BF. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2010 doi: 10.1136/jnnp.2009.204081. published online June 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valdmanis PN, Dupre N, Bouchard JP. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64:240–245. doi: 10.1001/archneur.64.2.240. [DOI] [PubMed] [Google Scholar]
- 6.Gijselinck I, Engelborghs S, Maes G. Identification of 2 loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–616. doi: 10.1001/archneurol.2010.82. [DOI] [PubMed] [Google Scholar]
- 7.Benajiba L, Le Ber I, Camuzat A. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- 8.van Es MA, Veldink JH, Saris CG. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- 9.Dunckley T, Huentelman MJ, Craig DW. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med. 2007;357:775–788. doi: 10.1056/NEJMoa070174. [DOI] [PubMed] [Google Scholar]
- 10.van Es MA, Van Vught PW, Blauw HM. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 2007;6:869–877. doi: 10.1016/S1474-4422(07)70222-3. [DOI] [PubMed] [Google Scholar]
- 11.Cronin S, Berger S, Ding J. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet. 2008;17:768–774. doi: 10.1093/hmg/ddm361. [DOI] [PubMed] [Google Scholar]
- 12.van Es MA, van Vught PW, Blauw HM. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2008;40:29–31. doi: 10.1038/ng.2007.52. [DOI] [PubMed] [Google Scholar]
- 13.Fogh I, D'Alfonso S, Gellera C. No association of DPP6 with amyotrophic lateral sclerosis in an Italian population. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.05.014. published online June 12. [DOI] [PubMed] [Google Scholar]
- 14.Lewis CM, Ng MY, Butler AW. Genome-wide association study of major recurrent depression in the UK population. Am J Psychiatry. 2010;167:949–957. doi: 10.1176/appi.ajp.2010.09091380. [DOI] [PubMed] [Google Scholar]
- 15.Gaysina D, Cohen-Woods S, Chow PC. Association of the dystrobrevin binding protein 1 gene (DTNBP1) in a bipolar case-control study (BACCS) Am J Med Genet B Neuropsychiatr Genet. 2009;150B:836–844. doi: 10.1002/ajmg.b.30906. [DOI] [PubMed] [Google Scholar]
- 16.Landers JE, Melki J, Meininger V. Reduced expression of the kinesin-associated protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2009;106:9004–9009. doi: 10.1073/pnas.0812937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schymick JC, Scholz SW, Fung HC. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2007;6:322–328. doi: 10.1016/S1474-4422(07)70037-6. [DOI] [PubMed] [Google Scholar]
- 18.Wroe R, Wai-Ling Butler A, Andersen PM, Powell JF, Al-Chalabi A. ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph Lateral Scler. 2008;9:249–250. doi: 10.1080/17482960802146106. [DOI] [PubMed] [Google Scholar]
- 19.Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 20.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 21.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International HapMap Consortium The international HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 25.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 26.Al-Chalabi A, Fang F, Hanby MF, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry (in press). [DOI] [PMC free article] [PubMed]
- 27.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 28.Lomen-Hoerth C. Characterization of amyotrophic lateral sclerosis and frontotemporal dementia. Dement Geriatr Cogn Disord. 2004;17:337–341. doi: 10.1159/000077167. [DOI] [PubMed] [Google Scholar]
- 29.Van Deerlin VM, Sleiman PM, Martinez-Lage M. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laaksovirta H, Peuralinna T, Schymick JC. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010 doi: 10.1016/S1474-4422(10)70184-8. published online Aug 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beaver JE, Tasan M, Gibbons FD, Tian W, Hughes TR, Roth FP. FuncBase: a resource for quantitative gene function annotation. Bioinformatics. 2010;26:1806–1807. doi: 10.1093/bioinformatics/btq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wain LV, Pedroso I, Landers JE. The role of copy number variation in susceptibility to amyotrophic lateral sclerosis: genome-wide association study and comparison with published loci. PLoS ONE. 2009;4:e8175. doi: 10.1371/journal.pone.0008175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cronin S, Blauw HM, Veldink JH. Analysis of genome-wide copy number variation in Irish and Dutch amyotrophic lateral sclerosis populations. Hum Mol Genet. 2008;17:3392–3398. doi: 10.1093/hmg/ddn233. [DOI] [PubMed] [Google Scholar]
- 35.Blauw HM, Veldink JH, van Es MA. Copy-number variation in sporadic amyotrophic lateral sclerosis: a genome-wide screen. Lancet Neurol. 2008;7:319–326. doi: 10.1016/S1474-4422(08)70048-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.