

Abstract
Kabuki syndrome (KS) is a rare genetic disease that causes developmental delay and congenital anomalies. Since the identification of MLL2 mutations as the primary cause of KS, such mutations have been identified in 56%–76% of affected individuals, suggesting that there may be additional genes associated with KS. Here, we describe three KS individuals with de novo partial or complete deletions of an X chromosome gene, KDM6A, that encodes a histone demethylase that interacts with MLL2. Although KDM6A escapes X inactivation, we found a skewed X inactivation pattern, in which the deleted X chromosome was inactivated in the majority of the cells. This study identifies KDM6A mutations as another cause of KS and highlights the growing role of histone methylases and histone demethylases in multiple-congenital-anomaly and intellectual-disability syndromes.
Main Text
Kabuki syndrome (KS; MIM 147920) was first described in 1981 by Niikawa and Kuroki,1, 2 and more than 400 cases have been reported in the literature. The main clinical characteristics are distinctive facial features, developmental delay, mild to moderate intellectual disability, post-natal growth retardation, and additional features including skeletal anomalies, hypodontia, and persistent fetal fingertip pads. Comparative genomic hybridization (CGH) microarray analysis failed to detect a recurrent anomaly in 72 KS individuals.3, 4, 5, 6, 7, 8 Use of the exome-sequencing strategy recently led to the identification of MLL2 (MIM 602113) mutations as a major cause of KS.9 In five recently published series, mutations in MLL2 were found in 56%–76% of KS patients.9, 10, 11, 12, 13
Because a significant proportion of patients do not have a detectable MLL2 mutation, we postulated the existence of additional genes associated with KS. In the quest for a another KS-causing genetic mutation, ten genes interacting with MLL2 were screened in 15 MLL2-mutation-negative KS individuals, and no pathogenic mutations were identified.11 Another gene coding for an MLL2-interacting protein, KDM6A (previously known as UTX; MIM 300128), was screened in 22 MLL2-mutation-negative KS individuals, and again, no causative mutations were detected.13
By using array CGH analysis (Agilent platform 244K), we identified de novo Xp11.3 microdeletions in two Belgian MLL2-mutation-negative KS girls (patients 1 and 2). Because both deletions were de novo, they are probably pathogenic. Both deletions included either a portion of or all of KDM6A. Moreover, there were no KDM6A deletions in a cohort of 411 normal controls in a previous study.14 The deletion in patient 1 included KDM6A exons 21–29, which code for the terminal part of the catalytic domain of KDM6A, and CXorf36, a gene recently implicated in X-linked autism.15 In patient 2, KDM6A, CXorf36, DUSP21 (MIM 300678), and FUNDC1 (Figure 1) were removed completely. The functions of DUSP21 and FUNDC1 remain unknown.
Figure 1.
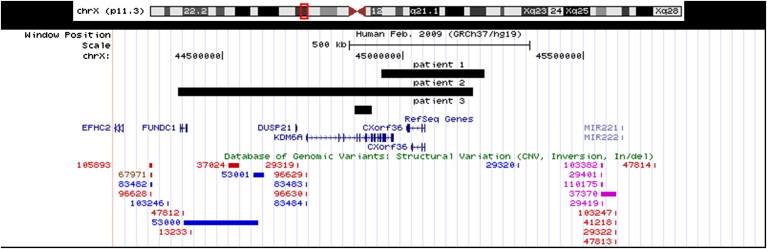
Region Xp11.3 Showing the Patients' Deletions
Region Xp11.3 shows the deletions drawn from the UCSC Genome Browser (GRCh37/hg19) for patients 1, 2, and 3. The black full tracks represent each patient's deletion, and each patient's number is above his or her respective track. The deletion in patient 1 spans 283.5 kb from base 44,941,324 to base 45,224,829, patient 2's deletion spans 815.7 kb from base 44,377,858 to base 45,193,629, and patient 3's deletion spans 45.4 kb from base 44,866,302 to base 44,912,718. The genes in the area are noted below the deletion tracks. CNVs in the Database of Genomic Variants are shown on the bottom lines. There are no previous reports of KDM6A copy-number changes.
We then sequenced KDM6A by Sanger sequencing and looked for intragenic deletions or duplications with a targeted custom Agilent array CGH in a cohort of 22 MLL2-mutation-negative KS individuals (8 females, 14 males). In accordance with the ethical standards of the Institut de Pathologie et de Génétique ethics committee, parental consent was obtained for DNA analysis of all the participants in this study and for the publication of photographs. The CGH microarray data (supplemental data, available online) discussed in this publication have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO)16 and are accessible under accession GSE32567 (see Accession Numbers section).
No point mutations were detected, but we identified a de novo intragenic deletion (exons 5–9) in one Italian, male KS individual (patient 3). We also sequenced UTY (MIM 400009), the Y chromosome paralog of KDM6A (see below), and looked for intragenic deletions or duplications as stated above, but we did not detect any mutations.
Patients 1 and 3 had a typical KS phenotype, including long palpebral fissures, lateral eversion of the lower eyelid, and moderate to severe intellectual disability (Table 1 and Figure 2). Although the facial features of patient 2 were not as classical, she displayed many features of this disorder, including lateral sparseness of the eyebrows, long eyelashes, strabismus, long palpebral fissures, large and prominent ears, persistent fetal fingertip pads, aortic coarctation, areolar fullness in infancy, and hirsutism. She presented with a mild developmental delay and had a normal verbal intelligence quotient (IQ) score, a poor performance IQ score, and hyperactive behavior (Table 1 and Figure 2). We noted that patients 1 and 2 had long halluces (Figure 3).
Table 1.
Clinical Features of Patients
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| General Characteristics | |||
| Gender | female | female | male |
| Maternal age at birth (yr) | 36 | 36 | 25 |
| Paternal age at birth (yr) | 39 | 29 | 27 |
| Age at examination (yr) | 13 | 10 | 2 |
| Weight <P3 | - | + | + |
| Length <P3 | + | + | + |
| OFC <P3 | + | + | + |
| NN hypoglycemia | + | - | + |
| Areolar fullness in infancy | + | + | + |
| Persistent finger pads | + | + | + |
| Brachydactyly | - | - | + |
| Hyperlaxity | + | + | + |
| Hirsutism | + | + | + |
| Feeding difficulties in infancy | + | - | + |
| CHD | ASD | AoC | - |
| Renal malformation | ND | - | - |
| Cancer | - | - | - |
| Developmental delay | severe | mild | moderate |
| IQ | Tot: 41a | V: 87, P:74b | Tot: 54c |
| Hypotonia | + | - | + |
| Behavior trouble | + | + | - |
| Facial Characteristics | |||
| Arched eyebrow | + | - | + |
| Lateral sparse of the eyebrow | - | + | + |
| Long palpebral fissure | + | + | + |
| Long eyelashes | + | + | + |
| Eversion of lateral third of the inferior eyelid | + | - | + |
| Broad tip | + | + | + |
| Depressed tip | + | - | + |
| Short columella | + | - | + |
| Strabismus | + | + | - |
| High arched palate | + | - | + |
| Neonatal teeth | - | - | - |
| Dental malocclusion | + | - | + |
| Ear Characteristics | |||
| Prominent | - | + | + |
| Cupped | - | - | + |
| Large auricle | + | + | - |
| Hearing loss | - | - | - |
Abbreviations are as follows: OFC, occipitofrontal circumference; NN, neonatal; CHD, congenital heart disease; ASD, atrial septal defect; AoC, aortic coarctation; ND, not determined; Tot, total; V, verbal; and P, performance.
IQ examined at 12 years of age with the Wechsler Intelligence Scale (WISC III).
IQ examined at 5 years of age with the Wechsler Intelligence Scale (WPPSI-R).
IQ examined at 14 months of age with the Griffith Mental Developmental Scale.
Figure 2.
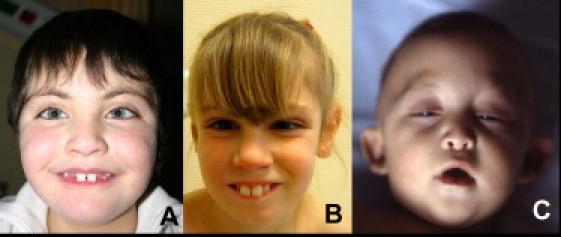
Facial Appearance in Affected Individuals
(A) Patient 1.
(B) Patient 2.
(C) Patient 3.
Note the long palpebral fissures in patients 1 and 2 and the arched eyebrows in patients 1 (mild) and 3.
Figure 3.
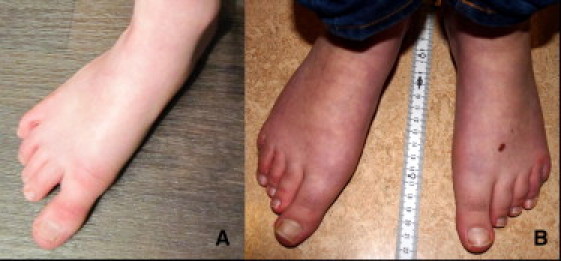
Appearance of Feet in Affected Individuals
(A) Patient 1.
(B) Patient 2.
Note the long halluces in both patients.
Thus, the KDM6A deletions in these three patients are associated with a broad phenotypic spectrum ranging from typical KS individuals (patients 1 and 3) to a milder clinical presentation (patient 2). This clinical variability is also a feature of patients with MLL2 mutations.13 However, the possible effects of CXorf36 deletion in patient 1 and of CXorf36, DUSP21, and FUNDC1 deletion in patient 2 on their respective phenotypes should also be considered.
KDM6A (29 exons) is one of the X chromosomal genes that largely escapes X inactivation.17 It encodes a 1,401 residue protein that contains two functional domains. The catalytic domain is a histone demethylase that specifically catalyzes demethylation of mono-, di-, and trimethylated lysine 27 on histone H3 (H3K27).18, 19 This demethylation mediates tissue-specific expression of various genes and is mostly involved in developmental processes and the cell cycle.19, 20, 21, 22 Interestingly, KDM6A and MLL2 act together in the epigenetic control of transcriptionally active chromatin by counteracting Polycomb-group (PcG) proteins.22 The other functional domain of KDM6A plays a role in chromatin remodeling by interacting with the switch/sucrose nonfermentable (SWI/SNF) remodeling complex that contains the transcription activator Brg1.23
Like MLL2, KDM6A plays a role in embryogenesis and development. Homozygous dUTX (Drosophila KDM6A ortholog) Drosophila mutants manifest rough eyes, dysmorphic wings, and modification of the sex combs.21 This phenotype resembles the trithorax phenotype, further supporting the notion that KDM6A counteracts PcG repression.21 Moreover, UTX-1 (C. elegans KDM6A ortholog) might affect developmental fate decisions in C. elegans vulval precursor cells via the transcription regulation of genes that encode the retinoblastoma (RB) protein complex, which has been conserved from worms to humans.20 Kdm6a1 is also important in the expression of posterior HOX genes in Zebrafish.19 In addition, KDM6A, together with MLL2, plays a major role in the regulation of muscle-specific genes during embryogenesis.22, 24 Finally, members of another important developmental gene family (T-box genes, which act in the mesoderm in the formation of the heart and vertebrae) recruit KDM6A to activate their target genes, again emphasizing the role of KDM6A in developmental processes.23 Interestingly, most of the pathogenic mutations for reported T-box genes in human diseases are located in the T-box domain that interacts with KDM6A.23
KDM6A escapes X inactivation,17 but it has been suggested that in mice, Kdm6a expression from the inactive X chromosome is significantly lower than that from the active X chromosome.25 Kdm6a expression is higher in the adult brain, the adult liver, and specific developing brain regions in females than in males.25 UTY is the paralog of KDM6A on the Y chromosome.17 UTY has 84% amino acid sequence similarity with KDM6A and might compensate for the increased expression of KDM6A in females, despite the fact that demethylase activity has yet to be demonstrated for this KDM6A paralog.14, 25
It is not surprising to find another gene associated with KS on the X chromosome because there have been reports of KS-like patients with small ring X (r[X]) chromosomes.26, 27, 28, 29, 30, 31, 32, 33, 34 In addition, there is a clear overlap between the congenital heart defects prevalent in male KS patients (aortic coarctation and other left-sided obstructions) and those found in patients with monosomy X and r(X) chromosomes.28, 35 Small r(X) chromosomes typically lead to Turner syndrome through the complete inactivation of the r(X). However, it is unclear why some individuals develop a KS-like phenotype. Incomplete X chromosome inactivation and the subsequent expression of usually repressed genes has been suggested as an explanation for the KS phenotype in some r(X) patients.32, 33, 34 KDM6A was deleted in the three patients who had both a KS-like phenotype and a r(X) in which the breakpoints had been mapped; this finding is consistent with the hypothesis that KDM6A deletion plays an important role in the KS-like phenotype observed in some patients with a r(X).30, 32, 34 We were unable to prove haploinsufficiency for KDM6A in patients 1 and 2 because KDM6A expression was very low in peripheral blood lymphocytes (data not shown). Nevertheless, studies have shown that two copies of Kdm6a are necessary for normal expression in female mouse embryos and adult mice,25 suggesting that haploinsufficiency might be the pathogenic mechanism. However, this explanation would not account for the fact that 45,X and other r(X) patients with a KDM6A deletion do not all develop the Kabuki phenotype.
In patients 1 and 2, the molecular X inactivation ratios are strongly skewed and are 89:11 and 97:3, respectively. The X inactivation profile was determined via PCR amplification of the CAG repeat in exon 1 of the androgen-receptor gene before and after DNA digestion with HpaII and CfoI. To determine whether the deleted copy of KDM6A was located on the active or inactive X chromosome, we performed fluorescence in situ hybridization (FISH) analysis with a KDM6A probe on the X chromosomes that had been differentially labeled by the incorporation of 5-bromo-2′-deoxyuridine (5-BrdU). The results showed that the deleted copy of KDM6A was located on the inactive X chromosome in all of the 70 mitoses analyzed in both patients (Figure 4). The fact that KDM6A escapes X-inactivation17 suggests that the phytohemagglutinin (PHA)-stimulated lymphocytes that have a deleted copy of KDM6A on the inactive X chromosome have a survival advantage over cell lines that have a deleted copy of KDM6A on the active X chromosome. This suggestion is in line with the hypothesis that although KDM6A escapes X inactivation, its expression is lower from the inactive X chromosome than from the active X chromosome.25
Figure 4.

Image of FISH Study on X Chromosomes Differentially Labeled with 5-BrdU
An image of a FISH study shows X chromosomes differentially labeled by the incorporation of 5-BrdU. The inactive X chromosome (in the white circle) appears brighter than the active X chromosome. Xqter subtelomeric probes hybridize to both X chromosomes (blue arrows). The KDM6A signal (white arrow) is absent from the inactive X chromosome and is present on the active X chromosome. For this experiment, peripheral blood lymphocytes from patients 1 and 2 were stimulated with phytohemagglutinin and were cultured for 72 hr. In order to identify the late-replicating inactive X chromosome, we treated the cells with 5-BrdU (30 μg/ml) 5 hr prior to harvesting. Colcemid was then added to a concentration of 0.2 μg/ml, and 1 hr later, metaphase preparations were produced via standard procedures involving swelling in 75 mM KCl and fixing in 3:1 methanol/acetic acid. We performed FISH analysis by simultaneously using a subtelomeric Xqter probe labeled with SpectrumOrange (Vysis) to indicate the X chromosomes (blue arrow) and RP11-435K1 labeled with SpectrumOrange (AmpliTech) to distinguish between the deleted and nondeleted X chromosomes (white arrow). To facilitate the detection of incorporated 5-BrdU, we denatured cellular DNA in 2N HCl for 30 min at 37°C. 5-BrdU was then labeled with a 5-BrdU-specific monoclonal antibody conjugated to fluorescein (Roche) (1 μg/ml). Finally, we counterstained the DNA by applying an antifade solution containing 0.1 μg/ml DAPI.
The role of UTY is largely unknown, and it has no in vitro demethylase activity. The finding of a male KS individual (patient 3) with an intragenic deletion of KDM6A and with clinical severity similar to that of patient 1 suggests that UTY could partially compensate for the loss of KDM6A in male individuals, as previously suggested in the literature.25
As for MLL2, somatic homozygous and hemizygous mutations in KDM6A have been identified in various cancer types (multiple myeloma, esophageal squamous cell carcinoma, renal cell carcinoma, myeloid leukemia, breast cancer, colorectal cancer, and glioblastoma), suggesting that KDM6A is a tumor-suppressing gene.14 Nevertheless, cancer is not a key feature of KS, given that this complication has been reported in only seven KS patients36, 37 (acute lymphoblastic leukemia, Burkitt lymphoma, fibromyxoid sarcoma, synovial sarcoma, and hepatoblastoma were reported once, and neuroblastoma was reported in two KS individuals).35, 36 If loss of both alleles is sufficient enough to cause cancer, one would expect cancer to occur more frequently in KS, such as in retinoblastoma (MIM 180200) or Wilms tumor (MIM 194070). This suggests either that loss of the second allele in MLL2, KDM6A, or UTY is necessary but not sufficient enough for cancer development or that this complication is underrecognized, demonstrating the importance of natural-history studies in rare syndromes.37
The following histone methylases and histone demethylases have been implicated in other multiple-anomaly syndromes: EHMT1 (MIM 607001; Kleefstra syndrome [MIM 610253]), SETBP1 (MIM 611060; Schinzel-Giedion syndrome [MIM 269150]), JARID1C (MIM 314690; Claes-Jensen-type, X-linked mental-retardation syndrome [MIM 300534]), PHF8 (MIM 300560; Siderius-type, X-linked mental-retardation syndrome [MIM 300263]), and MLL2 in KS. The identification of KDM6A pathogenic mutations in KS patients expands the role of histone-modification factors in intellectual disability and congenital malformation.
In conclusion, we report KDM6A mutations as a cause of KS in one male and two females. Our findings confirm both KS genetic heterogeneity and a locus on the X chromosome, as has been suggested previously. Because some KS patients were negative for MLL2, KDM6A, and UTY sequencing and did not show deletions or duplications during screening, it is likely that other KS-associated genes remain to be discovered.
Acknowledgments
We thank the families for their participation, the “Fonds Marguerite-Marie Delacroix” for the research grant provided to D.L, and Isabelle Leroy for the neurologic evaluation of the patients. Our work was supported by the Institut de Recherche Scientifique en Pathologie et Génétique. We thank Judith Goodship for her critical reading of the article.
Published online: December 22, 2011
Footnotes
Supplemental Data include a PDF with the CGH procedure and a description of the .txt files and 11 .txt files with all the CGH microarray data.
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32567
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
CGH microarray data are available at the GEO repository under accession GSE32567.
Supplemental Data
References
- 1.Niikawa N., Matsuura N., Fukushima Y., Ohsawa T., Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 1981;99:565–569. doi: 10.1016/s0022-3476(81)80255-7. [DOI] [PubMed] [Google Scholar]
- 2.Kuroki Y., Suzuki Y., Chyo H., Hata A., Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 1981;99:570–573. doi: 10.1016/s0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman J.D., Zhang Y., Greshock J., Ciprero K.L., Emanuel B.S., Zackai E.H., Weber B.L., Ming J.E. Array based CGH and FISH fail to confirm duplication of 8p22-p23.1 in association with Kabuki syndrome. J. Med. Genet. 2005;42:49–53. doi: 10.1136/jmg.2004.024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoumans J., Nordgren A., Ruivenkamp C., Brøndum-Nielsen K., Teh B.T., Annéren G., Holmberg E., Nordenskjöld M., Anderlid B.M. Genome-wide screening using array-CGH does not reveal microdeletions/microduplications in children with Kabuki syndrome. Eur. J. Hum. Genet. 2005;13:260–263. doi: 10.1038/sj.ejhg.5201309. [DOI] [PubMed] [Google Scholar]
- 5.Miyake N., Shimokawa O., Harada N., Sosonkina N., Okubo A., Kawara H., Okamoto N., Ohashi H., Kurosawa K., Naritomi K., et al. No detectable genomic aberrations by BAC array CGH in Kabuki make-up syndrome patients. Am. J. Med. Genet. A. 2006;140:291–293. doi: 10.1002/ajmg.a.31012. [DOI] [PubMed] [Google Scholar]
- 6.Cuscó I., del Campo M., Vilardell M., González E., Gener B., Galán E., Toledo L., Pérez-Jurado L.A. Array-CGH in patients with Kabuki-like phenotype: identification of two patients with complex rearrangements including 2q37 deletions and no other recurrent aberration. BMC Med. Genet. 2008;9:27. doi: 10.1186/1471-2350-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuniba H., Yoshiura K., Kondoh T., Ohashi H., Kurosawa K., Tonoki H., Nagai T., Okamoto N., Kato M., Fukushima Y., et al. Molecular karyotyping in 17 patients and mutation screening in 41 patients with Kabuki syndrome. J. Hum. Genet. 2009;54:304–309. doi: 10.1038/jhg.2009.30. [DOI] [PubMed] [Google Scholar]
- 8.Maas N.M., Van de Putte T., Melotte C., Francis A., Schrander-Stumpel C.T., Sanlaville D., Genevieve D., Lyonnet S., Dimitrov B., Devriendt K., et al. The C20orf133 gene is disrupted in a patient with Kabuki syndrome. J. Med. Genet. 2007;44:562–569. doi: 10.1136/jmg.2007.049510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C., et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulussen A.D., Stegmann A.P., Blok M.J., Tserpelis D., Posma-Velter C., Detisch Y., Smeets E.E., Wagemans A., Schrander J.J., van den Boogaard M.J., et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011;32:E2018–E2025. doi: 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- 11.Li Y., Bögershausen N., Alanay Y., Simsek Kiper P.O., Plume N., Keupp K., Pohl E., Pawlik B., Rachwalski M., Milz E., et al. A mutation screen in patients with Kabuki syndrome. Hum. Genet. 2011;130:715–724. doi: 10.1007/s00439-011-1004-y. [DOI] [PubMed] [Google Scholar]
- 12.Micale L., Augello B., Fusco C., Selicorni A., Loviglio M.N., Silengo M.C., Reymond A., Gumiero B., Zucchetti F., D'Addetta E.V., et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J. Rare Dis. 2011;6:38. doi: 10.1186/1750-1172-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannibal M.C., Buckingham K.J., Ng S.B., Ming J.E., Beck A.E., McMillin M.J., Gildersleeve H.I., Bigham A.W., Tabor H.K., Mefford H.C., et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A. 2011;155A:1511–1516. doi: 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Haaften G., Dalgliesh G.L., Davies H., Chen L., Bignell G., Greenman C., Edkins S., Hardy C., O'Meara S., Teague J., et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009;41:521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aziz A., Harrop S.P., Bishop N.E. DIA1R is an X-linked gene related to Deleted In Autism-1. PLoS ONE. 2011;6:e14534. doi: 10.1371/journal.pone.0014534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenfield A., Carrel L., Pennisi D., Philippe C., Quaderi N., Siggers P., Steiner K., Tam P.P., Monaco A.P., Willard H.F., Koopman P. The UTX gene escapes X inactivation in mice and humans. Hum. Mol. Genet. 1998;7:737–742. doi: 10.1093/hmg/7.4.737. [DOI] [PubMed] [Google Scholar]
- 18.Hong S., Cho Y.W., Yu L.R., Yu H., Veenstra T.D., Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA. 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan F., Bayliss P.E., Rinn J.L., Whetstine J.R., Wang J.K., Chen S., Iwase S., Alpatov R., Issaeva I., Canaani E., et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 20.Wang J.K., Tsai M.C., Poulin G., Adler A.S., Chen S., Liu H., Shi Y., Chang H.Y. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 2010;24:327–332. doi: 10.1101/gad.1882610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herz H.M., Madden L.D., Chen Z., Bolduc C., Buff E., Gupta R., Davuluri R., Shilatifard A., Hariharan I.K., Bergmann A. The H3K27me3 demethylase dUTX is a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol. Cell. Biol. 2010;30:2485–2497. doi: 10.1128/MCB.01633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aziz A., Liu Q.C., Dilworth F.J. Regulating a master regulator: establishing tissue-specific gene expression in skeletal muscle. Epigenetics. 2010;5:691–695. doi: 10.4161/epi.5.8.13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller S.A., Mohn S.E., Weinmann A.S. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol. Cell. 2010;40:594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seenundun S., Rampalli S., Liu Q.C., Aziz A., Palii C., Hong S., Blais A., Brand M., Ge K., Dilworth F.J. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010;29:1401–1411. doi: 10.1038/emboj.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J., Deng X., Watkins R., Disteche C.M. Sex-specific differences in expression of histone demethylases Utx and Uty in mouse brain and neurons. J. Neurosci. 2008;28:4521–4527. doi: 10.1523/JNEUROSCI.5382-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niikawa N., Kuroki Y., Kajii T., Matsuura N., Ishikiriyama S., Tonoki H., Ishikawa N., Yamada Y., Fujita M., Umemoto H., et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am. J. Med. Genet. 1988;31:565–589. doi: 10.1002/ajmg.1320310312. [DOI] [PubMed] [Google Scholar]
- 27.Hughes H.E., Davies S.J. Coarctation of the aorta in Kabuki syndrome. Arch. Dis. Child. 1994;70:512–514. doi: 10.1136/adc.70.6.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Digilio M.C., Marino B., Toscano A., Giannotti A., Dallapiccola B. Congenital heart defects in Kabuki syndrome. Am. J. Med. Genet. 2001;100:269–274. doi: 10.1002/ajmg.1265. [DOI] [PubMed] [Google Scholar]
- 29.Dennis N.R., Collins A.L., Crolla J.A., Cockwell A.E., Fisher A.M., Jacobs P.A. Three patients with ring (X) chromosomes and a severe phenotype. J. Med. Genet. 1993;30:482–486. doi: 10.1136/jmg.30.6.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGinniss M.J., Brown D.H., Burke L.W., Mascarello J.T., Jones M.C. Ring chromosome X in a child with manifestations of Kabuki syndrome. Am. J. Med. Genet. 1997;70:37–42. [PubMed] [Google Scholar]
- 31.Abd S.E., Wilson L., Howlin P., Patton M.A., Wintgens A.M., Wilson R. Agenesis of the corpus callosum in Turner syndrome with ring X. Dev. Med. Child Neurol. 1997;39:119–124. doi: 10.1111/j.1469-8749.1997.tb07394.x. [DOI] [PubMed] [Google Scholar]
- 32.Stankiewicz P., Thiele H., Giannakudis I., Schlicker M., Baldermann C., Krüger A., Dörr S., Starke H., Hansmann I. Kabuki syndrome-like features associated with a small ring chromosome X and XIST gene expression. Am. J. Med. Genet. 2001;102:286–292. doi: 10.1002/ajmg.1462. [DOI] [PubMed] [Google Scholar]
- 33.Su P.H., Kuo P.L., Chen S.J., Chen J.Y., Yu J.S., Liu Y.L., Kao I.W. Kabuki make-up (Niikawa-Kuroki) syndrome with mosaicism ring chromosome X and incomplete XIST gene expression. Acta Paediatr. Taiwan. 2007;48:28–31. [PubMed] [Google Scholar]
- 34.Rodríguez L., Diego-Alvarez D., Lorda-Sanchez I., Gallardo F.L., Martínez-Fernández M.L., Arroyo-Muñoz M.E., Martínez-Frías M.L. A small and active ring X chromosome in a female with features of Kabuki syndrome. Am. J. Med. Genet. A. 2008;146A:2816–2821. doi: 10.1002/ajmg.a.32521. [DOI] [PubMed] [Google Scholar]
- 35.Digilio M.C., Baban A., Marino B., Dallapiccola B. Hypoplastic left heart syndrome in patients with Kabuki syndrome. Pediatr. Cardiol. 2010;31:1111–1113. doi: 10.1007/s00246-010-9773-y. [DOI] [PubMed] [Google Scholar]
- 36.Tumino M., Licciardello M., Sorge G., Cutrupi M.C., Di Benedetto F., Amoroso L., Catania R., Pennisi M., D'Amico S., Di Cataldo A. Kabuki syndrome and cancer in two patients. Am. J. Med. Genet. A. 2010;152A:1536–1539. doi: 10.1002/ajmg.a.33405. [DOI] [PubMed] [Google Scholar]
- 37.Casanova M., Selicorni A., Ferrari A. Cancer predisposition in children with Kabuki syndrome. Am. J. Med. Genet. A. 2011;155A:1504. doi: 10.1002/ajmg.a.33711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.