

Abstract
Collagen, type IV, alpha 1 (COL4A1) and alpha 2 (COL4A2) form heterotrimers and are abundant components of basement membranes, including those of the cerebral vasculature. COL4A1 mutations are an increasingly recognized cause of multisystem disorders, including highly penetrant cerebrovascular disease and intracerebral hemorrhage (ICH). Because COL4A1 and COL4A2 are structurally and functionally associated, we hypothesized that variants in COL4A2 would also cause ICH. We sequence COL4A2 in 96 patients with ICH and identify three rare, nonsynonymous coding variants in four patients that are not present in a cohort of 144 ICH-free individuals. All three variants change evolutionarily conserved amino acids. Using a cellular assay, we show that these putative mutations cause intracellular accumulation of COL4A1 and COL4A2 at the expense of their secretion, which supports their pathogenecity. Furthermore, we show that Col4a2 mutant mice also have completely penetrant ICH and that mutations in mouse and human lead to retention of COL4A1 and COL4A2 within the endoplasmic reticulum (ER). Importantly, two of the three putative mutations found in patients trigger ER stress and activate the unfolded protein response. The identification of putative COL4A2 mutations that might contribute to ICH in human patients provides insight into the pathogenic mechanisms of this disease. Our data suggest that COL4A2 mutations impair COL4A1 and COL4A2 secretion and can also result in cytotoxicity. Finally, our findings suggest that, collectively, mutations in COL4A1 and COL4A2 contribute to sporadic cases of ICH.
Main Text
Strokes are common and devastating neurological events with poor clinical outcomes for which effective treatment is limited. This is especially true for intracerebral hemorrhages (ICHs), which are associated with the highest rate of mortality despite only accounting for 10–15% of all strokes.1 Up to 50% of individuals die within the first year following ICH, and the majority of survivors suffer life-long disability.2 Prevention is therefore of central importance for reducing the personal and societal burden of ICH. Determining the genetic factors leading to cerebrovascular diseases allows identification of individuals who are at greater risk of developing ICH and for whom preventative interventions might be efficacious. Additionally, identifying causative or predisposing genetic risk factors facilitates understanding of the biological mechanisms underlying disease. Although sporadic ICH is often associated with cerebral amyloid angiopathy (CAA) or hypertensive vasculopathy, the precipitating events leading to hemorrhage are poorly defined.
Using genetic screens in mice and candidate gene approaches in humans, we showed that mutations in the gene coding for collagen, type IV, alpha 1 (COL4A1 [MIM 120130]) cause porencephaly (MIM 175780) and multisystem small vessel disease (MIM 607595), including nephropathy and ICH.3, 4 Col4a1 mutant mice had a broad spectrum of cerebrovascular diseases, including pre- and perinatal ICH, porencephalic cavities, progressive, multifocal, and recurrent ICH, and, occasionally, subarachnoid hemorrhages.3, 4 Dominant COL4A1 mutations are being increasingly recognized as an important cause of highly penetrant cerebrovascular diseases, including porencephaly and ICH.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 COL4A1 mutations in mice and humans are pleiotropic and can affect multiple organ systems with different levels of severity. Using controlled genetic approaches in mice, we have shown that at least some of the variability in the penetrance and severity of Col4a1-induced pathology can be attributed to environmental influences and to genetic context.3, 16, 17 Accumulating evidence suggests that allelic heterogeneity might also contribute to variable expressivity of COL4A1 mutations. A subset of individuals with COL4A1 mutations are reported to have a distinct clinical phenotype referred to as hereditary angiopathy, nephropathy, aneurysms, and cramps syndrome (HANAC [MIM 611773]), and the mutations in these individuals cluster within a 31 amino acid region of the COL4A1 protein that encompasses integrin binding sites.8, 12, 18, 19 These data suggest that alterations of specific functional domains could contribute to the phenotypic variability associated with COL4A1 mutations.
COL4A1 forms heterotrimers with COL4A2 (MIM 120090) and, together, they are the most abundant and prevalent proteins in basement membranes, including those of the cerebral vasculature. Heterotrimers composed of one COL4A2 and two COL4A1 peptides are assembled and modified within the endoplasmic reticulum (ER) before trafficking to the Golgi where they are packaged into vesicles for secretion into the vascular basement membranes.20, 21, 22 In the extracellular space, heterotrimers polymerize into flexible sheets, which not only provide strength to basement membranes but also participate in dynamic biological processes through interactions with growth factors and cell surface receptors, including integrins.23, 24, 25, 26
At the carboxy terminus of COL4A1 and COL4A2 is a globular noncollagenous (NC1) domain responsible for initiating heterotrimer formation within the ER.27 A 7S domain that is involved in lateral intertrimer associations in the extracellular matrix is present at the amino terminus of both proteins. Flanked by these terminal domains are the large triple-helix-forming domains that constitute more than 80% of both the COL4A1 and COL4A2 proteins. The triple-helix-forming domain is characteristic of collagens and is composed of repeating Gly-Xaa-Yaa amino acid residues where there is a requirement for a glycine every third amino acid and where the X and Y amino acids are often proline or lysine residues that undergo posttranslational processing in order to crosslink the constituent peptides of a heterotrimer.28, 29 The triple-helix-forming domains of type IV collagens also contain frequent and positionally conserved interruptions within the Gly-Xaa-Yaa repeats, which are thought to confer molecular flexibility to type IV collagens. Pathogenic mutations are reported in the NC1 domain, however, the vast majority of disease-causing mutations reported to date for various types of collagens, including type IV collagens, have been shown to interfere with triple-helix formation or stability.30
Because of the intimate structural and functional association between COL4A1 and COL4A2 and because of the presumed mechanisms by which COL4A1 mutations lead to disease, we expect that COL4A2 mutations will cause pathologies similar to those resulting from COL4A1 mutations. In support of this hypothesis, mutations in COL4A2 orthologs phenocopy mutations in COL4A1 orthologs in mice and in Caenorhabditis elegans.31, 32, 33, 34 However, to date, pathogenic COL4A2 mutations have not been reported in humans. To test the hypothesis that COL4A2 mutations cause ICH, we sequenced COL4A2 in a large cohort of unrelated individuals diagnosed with sporadic hemorrhages not related to arteriovenous malformations, tumors, or impaired coagulation. We identified putative pathogenic variants in four out of 96 people, showed that COL4A2 proteins are sequestered within the ER, and, in some cases, caused activation of the unfolded protein response (UPR).
For the affected cohort, we selected 96 individuals from among 800 consecutive participants with ICH presenting to Massachusetts General Hospital.35 Cranial computed tomography scans and/or magnetic resonance images of the head were completed for all individuals, and hemorrhages were classified according to the Boston Criteria.36 For the present study, 48 individuals with probable CAA-related ICH and 48 individuals with presumed hypertension-related deep ICH were chosen according to the inclusion and exclusion criteria published previously36 (summarized in Table 1). Simultaneously, we collected DNA from ethnically and age-matched individuals who were free of a history of hemorrhagic stroke and were drawn from the primary care practices at Massachusetts General Hospital. All participants or their surrogates provided informed consent for study participation and the Massachusetts General Hospital institutional review board approved all study procedures.
Table 1.
Inclusion and Exclusion Criteria
| Patient | Control |
|---|---|
| Inclusion Criteria | |
| either gender | either gender |
| ability and willingness to consent | ability and willingness to consent |
| symptomatic intracerebral hemorrhage | |
| Exclusion Criteria | |
| antecedent head trauma | history of intracerebral hemorrhage |
| ischemic stroke during 2 weeks prior to intracerebral hemorrhage | intracerebral tumor |
| intracerebral tumor | arteriovenous malformation, aneurysm, or central nervous system vasculitis |
| arteriovenous malformation, aneurysm, or central nervous system vasculitis | primary coagulopathy, blood dyscrasia, or active liver disorder |
| primary coagulopathy, blood dyscrasia, or active liver disorder | use of cocaine or sympathomimetic drug |
| use of cocaine or sympathomimetic drug | alcohol abuse |
| alcohol abuse | |
We performed direct sequence analysis (ABI BigDye v3.1) of the entire coding sequence of COL4A2 including the flanking intronic regions (more than 50 nucleotides for most introns and never less than 20 nucleotides) by using 42 pairs of primers (Table S1, available online). Using Sequencher software (Gene Codes Corporation), we identified 168 sequence variants of which 137 were intronic (Table S2). Because we expected that pathogenic mutations having a strong effect will be rare, nonsynonymous variants, we focused our attention on these. It is possible that intronic or synonymous variants are pathogenic, in which case our estimation of the role of COL4A2 in ICH would be understated. Of the 31 coding variants, ten were nonsynonymous (Table 2). Four nonsynonymous variants, c.1550G>A, c.2048G>C, c.2152C>T, and c.4195G>A, were found at high frequency and considered unlikely to be causative mutations (p.Arg517Lys, p.Gly683Ala, p.Pro718Ser, and p.Val1399Ile, hereafter referred to as COL4A2R517K, COL4A2G683A, COL4A2P718S and COL4A2V1399I, respectively). Notably, one of these variants changes a glycine residue, and glycine missense mutations within the triple-helix domain constitute the prototypic pathogenic mutation for several types of collagens. However, this variant, COL4A2G683A, is highly polymorphic (17 out of 92 affected individuals were heterozygous and 4 out of 92 were homozygous for the minor allele) and impacts a glycine that is part of a repeat interruption and not part of the Gly-Xaa-Yaa tripeptide repeat pattern necessary for triple-helix formation. Thus, out of a cohort of 96 individuals, we identified six nonsynonymous rare variants that we deemed to be potentially pathogenic (Figure 1A).
Table 2.
Coding SNPs Identified
|
Variant |
Patients |
Controls |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon | DNA | Protein | Homozygous Major | Heterozygous | Homozygous Minor | Total | Homozygous Major | Heterozygous | Homozygous Minor | Total | |
| Nonsynonymous | |||||||||||
| 9 | c.574G>T | p.Val192Phe | V192F | G/G (91) | G/T (1) | T/T (0) | 92 | G/G (144) | G/T (0) | T/T (0) | 144 |
| 22 | c.1550G>A | p.Arg517Lys | R517K | A/A (33) | A/G (45) | G/G (15) | 93 | ||||
| 27 | c.2048G>C | p.Gly683Ala | G683A | G/G (71) | G/C (17) | C/C (4) | 92 | ||||
| 28 | c.2102A>G | p.Lys701Arg | K701R | A/A (94) | A/G (1) | G/G (0) | 95 | A/A (137) | A/G (0) | G/G (0) | 137 |
| 28 | c.2152C>T | p.Pro718Ser | P718S | C/C (57) | C/T (27) | T/T (11) | 95 | C/C (92) | C/T (41) | T/T (4) | 137 |
| 36 | c.3326G>A | p.Arg1109Gln | R1109Q | G/G (92) | G/A (1) | A/A (1) | 94 | G/G (132) | G/A (1) | A/A (0) | 133 |
| 37 | c.3368A>G | p.Glu1123Gly | E1123G | A/A (91) | A/G (2) | G/G (0) | 93 | A/A (142) | A/G (0) | G/G (0) | 142 |
| 37 | c.3448C>A | p.Gln1150Lys | Q1150K | C/C (92) | C/A (1) | A/A (0) | 93 | C/C (142) | C/A (0) | A/A (0) | 142 |
| 44 | c.4195G>A | p.Val1399Ile | V1399I | G/G (86) | G/A (8) | A/A (0) | 94 | ||||
| 48 | c.5068G>A | p.Ala1690Thr | A1690T | G/G (92) | G/A (1) | A/A (0) | 93 | G/G (143) | G/A (0) | A/A (0) | 143 |
| Synonymous | |||||||||||
| 3 | c.49C>T | p.Leu17Leu | L17L | C/C (91) | C/T (1) | T/T (0) | 92 | ||||
| 5 | c.297A>G | p.Thr99Thr | T99T | A/A (91) | A/G (2) | G/G (0) | 93 | ||||
| 10 | c.594C>T | p.Pro198Pro | P198P | C/C (92) | C/T (1) | T/T (0) | 93 | ||||
| 17 | c.1008C>T | p.Pro336Pro | P336P | C/C (26) | C/T (48) | T/T (17) | 91 | C/C (30) | C/T (68) | T/T (38) | 136 |
| 17 | c.1011G>A | p.Lys337Lys | K337K | G/G (92) | G/A (1) | A/A (0) | 93 | G/G (135) | G/A (1) | A/A (0) | 136 |
| 19 | c.1095G>A | p.Pro365Pro | P365P | G/G (86) | G/A (0) | A/A (8) | 94 | ||||
| 19 | c.1179C>T | p.Ile393Ile | I393I | C/C (82) | C/T (2) | T/T (10) | 94 | ||||
| 20 | c.1308C>T | p.Pro436Pro | P436P | C/C (92) | C/T (1) | T/T (0) | 93 | ||||
| 22 | c.1488G>A | p.Pro496Pro | P496P | A/A (33) | A/G (44) | G/G (16) | 93 | ||||
| 34 | c.3204C>T | p.Ser1068Ser | S1068S | C/C (91) | C/T (1) | T/T (0) | 92 | ||||
| 41 | c.3804A>T | p.Pro1268Pro | P1268P | A/A (88) | A/T (4) | T/T (0) | 92 | ||||
| 41 | c.3807T>C | p.Gly1269Gly | G1269G | T/T (88) | T/C (4) | C/C (0) | 92 | ||||
| 41 | c.3876C>T | p.Leu1292Leu | L1292L | C/C (91) | C/T (1) | T/T (0) | 92 | ||||
| 43 | c.4083T>C | p.Thr1361Thr | T1361T | T/T (89) | T/C (3) | C/C (0) | 92 | ||||
| 43 | c.4089G>A | p.Ala1363Ala | A1363A | G/G (61) | G/A (25) | A/A (6) | 92 | ||||
| 45 | c.4292T>C | p.Phe1430Phe | F1430F | T/T (90) | T/C (4) | C/C (0) | 94 | ||||
| 46 | c.4428G>A | p.Pro1476Pro | P1476P | G/G (92) | G/A (1) | A/A (0) | 93 | ||||
| 46 | c.4515A>G | p.Pro1505Pro | P1505P | G/G (63) | G/A (28) | A/A (2) | 93 | ||||
| 47 | c.4617G>A | p.Ala1539Ala | A1539A | G/G (78) | G/A (15) | A/A (0) | 93 | ||||
| 47 | c.4737C>T | p.Ala1579Ala | A1579A | C/C (92) | C/T (1) | T/T (0) | 93 | ||||
| 48 | c.4929G>A | p.Pro1643Pro | P1643P | G/G (92) | G/A (1) | A/A (0) | 93 | G/G (141) | G/A (2) | A/A (0) | 143 |
Figure 1.
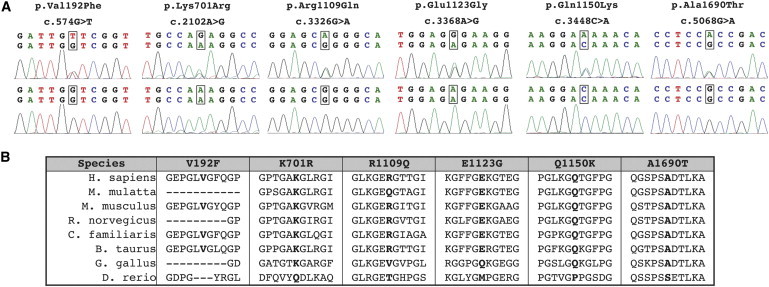
Six Nonsynonymous Rare Variants Identified in Patients Result in Missense Changes in Amino Acids that Are Highly Conserved across Species
(A) Electropherograms of genomic DNA from patients (top panels) or control individuals (lower panels) showing coding variants in patients.
(B) Multispecies alignment of COL4A2 orthologs shows that most of the rare coding variants identified in patients change highly conserved amino acids.
We resequenced at least 264 control chromosomes for each exon in which one of the six nonsynonymous variants was found (Table 2). One variant, c.3326G>A ([p.Arg1109Gln] hereafter referred to as COL4A2R1109Q), was identified in two affected individuals (including one homozygote) and in a single control individual, suggesting that this variant might not be pathogenic and other variants were present in dbSNP. However, because reduced penetrance is documented in COL4A1 mutations and because genetic context dependency has been demonstrated for Col4a1 mutations in mice, we chose to include these variants for further analysis.
We performed multispecies alignments to determine whether the affected amino acids were conserved and whether there was precedence for any of the variants in other species (Figure 1B). Five of the six variants were highly conserved across species and perfectly conserved among mammals. COL4A2R1109Q was less conserved and, in fact, Rhesus monkey (Macaca mulatta) has a glutamine (Q) at this position of its COL4A2 ortholog, further suggesting that this variant might not be pathogenic. We then used commonly referenced algorithms to help predict the potential impact of each variant. The automated tools SIFT (Sorting Intolerant from Tolerant, version 2.0) and PolyPhen (Polymorphism Phenotyping-2) lead to different and even contradictory results. The evolutionary-based approach, SIFT, predicted only one damaging variant (c.574G>T [p.Val192Phe], hereafter referred to as COL4A2V192F), whereas the structure- and evolutionary-based approach, PolyPhen, predicted two different variants to be possibly damaging (c.3448C>A [p.Gln1150Lys] and c.5068G>A [ p.Ala1690Thr]; hereafter referred to as COL4A2Q1150K and COL4A2A1690T) and one different variant to be probably damaging (c.3368A>G [p.Glu1123Gly]; hereafter referred to as COL4A2E1123G). However, PolyPhen also predicted the COL4A2P718S variant to be possibly damaging despite this variant being highly polymorphic and found in both cohorts at high frequencies and therefore highly unlikely to be pathogenic. Based on these results, we contend that in silico approaches are currently unlikely to accurately predict functional consequences of COL4A2 variants.
Therefore, to evaluate the functional consequences of putative COL4A2 mutations, we used a cell-culture-based assay that we have previously validated for COL4A1 mutations.17 Pathogenic mutations in genes coding for several types of collagen often disrupt normal heterotrimer formation leading to intracellular accumulation of misfolded proteins and decreased heterotrimer secretion. Thus, we used an in vitro secretion assay to test whether rare COL4A2 variants changed the ratio of extracellular to intracellular COL4A2 and COL4A1 proteins in cultured cells. As described previously,17 we transfected HT1080 cells (which endogenously express COL4A1 and COL4A2) with a control COL4A2 cDNA (NM_001846.2) or with COL4A2 cDNA that contained one of the six nonsynonymous rare sequence variants. At 80%–90% confluence, cells were serum deprived and treated with ascorbic acid (50 μg/ml), and after 24 hr the conditioned medium was collected and cells were lysed for subsequent immunoblot analysis with rat COL4A1 (H11) or rat COL4A2 (H22) monoclonal antibody (1:100 and 1:200, respectively; Shigei Medical Research Institute, Japan). Densitometric analysis was performed on low-exposure images with ImageJ software (National Institutes of Health) and comparison of values was carried out with Student's t test with p < 0.05 considered as statistically significant. The extracellular to intracellular ratios of COL4A2 and COL4A1 obtained from cells transfected with each of the variants were expressed relative to the ratio obtained from cells transfected with the control COL4A2. As controls for the functional assay, we also tested a pathogenic mutation (c.4165G>A [p.Gly1389Arg] referred to as COL4A2G1389R) that causes familial porencephaly and small-vessel disease (data not shown) and the highly polymorphic common variant (COL4A2P718S) that is expected to have no impact on COL4A2 biosynthesis. As predicted, cells expressing the validated mutation had significantly decreased extracellular to intracellular ratio of COL4A2, whereas cells expressing the common polymorphism did not differ significantly from cells transfected with the control cDNA (Figure 2A). Using the same assay, we found that three variants identified only in the affected cohort had significant reduction in the ratio of extracellular to intracellular COL4A2 and three did not (Figure 2A). Because COL4A1 and COL4A2 are secreted as heterotrimers, we also determined the extracellular to intracellular levels of COL4A1 and found that the results correlated perfectly in that the positive control and the same three rare variants all showed significant decreases in the extracellular to intracellular ratio (Figure 2B). COL4A2R1109Q, which was present in one control individual and is also present in M. mulatta, was among the three variants that did not have an effect on COL4A2 and COL4A1 secretion. These results strengthen the suggestion that COL4A2P718S and COL4A2R1109Q are not pathogenic and also suggest that COL4A2V192F and c.2102A>G ([p.Lys701Arg] referred to as COL4A2K701R) are not pathogenic. Importantly, our assay showed that COL4A2E1123G, COL4A2Q1150K, and COL4A2A1690T variants have functional consequences as they interfere with proper secretion of COL4A2 and COL4A1 and therefore constitute putative pathogenic mutations.
Figure 2.
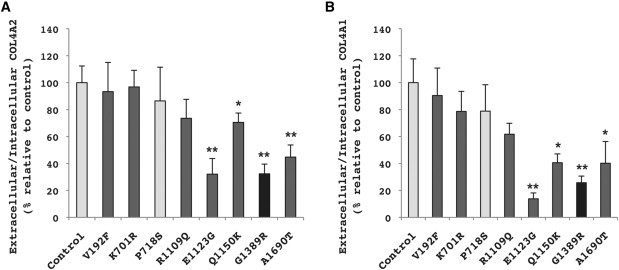
Functional Assay Reveals that COL4A2E1123G, COL4A2Q1150K, and COL4A2A1690T Impair COL4A2 and COL4A1 Secretion
Ratios of extracellular to intracellular COL4A2 (A) or COL4A1 (B) from cells expressing different COL4A2 alleles were determined by immunoblot analysis and are expressed as a percent relative to control (NM_001846.2) COL4A2 cDNA (mean of five independent experiments ± standard error of the mean [SEM]). The highly polymorphic common variant COL4A2P718S was used as a presumptive negative control, and the pathogenic mutation COL4A2G1389R was used as a positive control of the functional assay. The variants COL4A2V192F, COL4A2K701R, or COL4A2R1109Q have no functional consequences on the biosynthesis of COL4A1/COL4A2 heterotrimers. The COL4A2E1123G, COL4A2Q1150K, and COL4A2A1690T variants significantly reduce the extracellular to intracellular ratio of COL4A2 and COL4A1, suggesting that they are pathogenic mutations (∗p < 0.05, ∗∗p < 0.01). The control cDNA and a common polymorphism are shown in light gray, the known pathogenic variant is shown in black, and the rare variants identified in the patient cohort are shown in dark gray.
The COL4A2E1123G variant was identified in two affected individuals and was not found in 142 ethnically matched control individuals. The first individual was a 72-year-old white non-Hispanic man with probable CAA-related ICH in the right occipital lobe. He has no family history of ICH and was not taking aspirin or an anticoagulant at the time of his hemorrhage. The second was a 72-year-old white non-Hispanic woman with a history of hypertension who developed hemorrhage in the left basal ganglia while taking aspirin. There was no family history of ICH. These two individuals had adenine to guanine transitions (c.3368A>G) in exon 37, leading to glutamate to glycine substitutions in the X-position amino acid of a Gly-Xaa-Yaa repeat within the triple-helix-forming domain of the COL4A2 protein.
The COL4A2Q1150K variant was found in one affected individual but not in the control cohort. The affected individual was a 45-year-old white Hispanic man with a history of hypertension who developed hemorrhage in the left basal ganglia. He was not taking aspirin or anticoagulant at the time of his hemorrhage. Notably, two sisters of this individual had ICH. At the time of subject enrollment, both sisters were over age 40 and had no history of dementia; however, the family members were not available for follow-up. The variant was a cytosine to adenine transversion (c.3448C>A) in exon 37, leading to a glutamine to lysine replacement in the X-position of a Gly-Xaa-Yaa repeat.
The third variant, COL4A2A1690T, was identified in one individual but none of the controls and was not a known variant. The individual was a 69-year-old African American man with probable CAA-related ICH in the right occipital lobe. He was not taking aspirin or anticoagulant at the time of his hemorrhage. There was no family history of ICH. He had a guanine to adenine transition (c.5068G>A) in exon 48 leading to an alanine to threonine substitution. Unlike the previous mutations, which occur in the triple-helix-forming domain, amino acid 1690 is close to the C terminus and is located in the globular NC1 domain of the protein (which does not participate in triple-helix formation).
In mice, Col4a2 mutations were reported to cause prenatal focal ICHs in six out of 11 embryos between embryonic day (E) E15 and E17, and so we sought to determine whether adult Col4a2 mutant mice also have ICH.31 We have previously shown that phenotypes in Col4a1 mutant mice are genetic-context dependent; therefore, we bred the mouse p.Gly646Asp (COL4A2G646D) mutation onto a uniform genetic background by iteratively crossing to C57BL/6J mice for at least seven generations. Col4a2+/+ and Col4a2+/G646D mice were aged to 7.5 to 9.5 months and then tested for the presence of ICH by staining with Prussian Blue as described previously.3 ICH were never observed in control mice (n = 7) however, we found that all mutant mice (n = 5) had multifocal ICHs in the subcortical region of the forebrain and in the cerebellum (Figure 3A). We quantified the extent of ICH by measuring the percentage of the cross sectional area that stained with Prussian blue and compared it to the extent of ICH in the brains of mice with a severe mutation in Col4a1.3, 4, 16 Compared to Col4a1+/Δex40 mutant mice, ICH in Col4a2+/G646D mice was consistently much less severe (Figure 3B). One interpretation is that Col4a2 mutations lead to a less severe phenotype than Col4a1 mutations. This explanation could be rationalized by the fact that COL4A1/A2 heterotrimers have a 2:1 ratio of COL4A1:COL4A2. Thus, in heterozygous Col4a1 mutants, 75% of the heterotrimers would have at least one mutant COL4A1 peptide, whereas in heterozygous Col4a2 mutants only 50% of the heterotrimers would be expected to contain a mutant COL4A2 peptide (assuming that normal and mutant peptides participate equally in heterotrimer formation). Although this interpretation is possible it is inconsistent with observations of mild ICH in mice with other Col4a1 mutant alleles, which can also be much milder than the phenotype of Col4a1+/Δex40 mutant mice (data not shown). These data support that both intergenic and intragenic allelic heterogeneity influence variable expressivity, however, their relative contributions remain to be determined.
Figure 3.
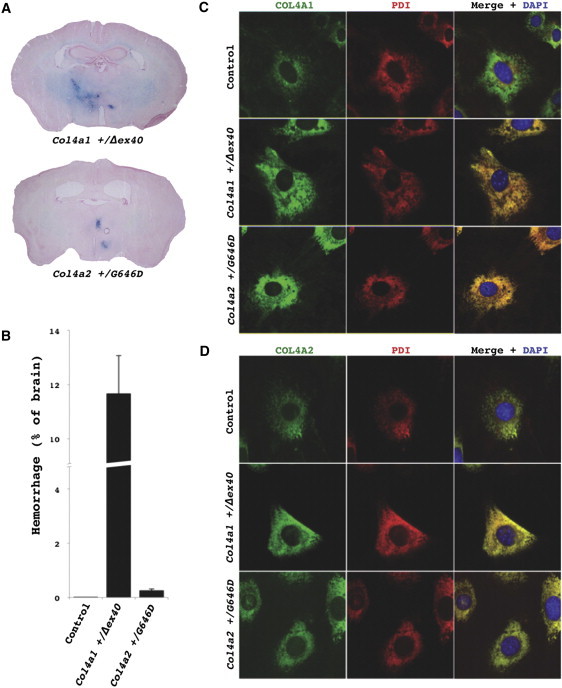
Col4a2 Mutant Mice Have Intracerebral Hemorrhages and Retention of COL4A1/A2 Heterotrimers within the ER
(A) Prussian Blue staining of brains from Col4a2+/G646D mutant mice revealed multifocal ICHs in all mutant mice (n = 5) and none in control mice (C57BL/6J Col4a1+/+; Col4a2+/+, n = 7, data not shown). The extent of the ICH appeared much less severe than ICH detected in Col4a1+/Δex40 mutant mice.
(B) To compare the severity of ICH between the two mutant lines, we measured the proportional area of ICH in brain sections at regularly spaced intervals (mean ± SEM). Control (C57BL/6J) mice never had detectable ICH and ICHs in Col4a2+/G646D mutant mice were much less severe than those observed in Col4a1+/Δex40 mutant mice.
(C and D) Immunofluorescent labeling of COL4A1 or COL4A2 (rat H11 or rat H22 [1:200], respectively, with AlexaFluor488 anti-rat IgG [1:500], Invitrogen; green) and the ER marker PDI (mouse ID3 [1:500], Stressgen with AlexaFluor594 anti-mouse IgG [1:500], Invitrogen; red) was performed in MEFs.38 MEFs were grown on glass coverslips, serum-deprived and treated with ascorbic acid (50 μg/ml) for 24 hr, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton in PBS. Cells were then mounted with Mowiol containing 4′, 6-diamidino-2-phenylindole (DAPI) to label nuclei. Colabeling with PDI and anti-COL4A1 (C) or anti-COL4A2 (D) in control (C57BL/6J Col4a1+/+; Col4a2+/+) cells showed considerable COL4A1/A2 labeling that did not colocalize with PDI and therefore was not within the ER. In contrast, in Col4a1+/Δex40 or Col4a2+/G646D mutant cells all of the detectable COL4A1 and COL4A2 labeling was colocalized with the ER marker, indicating ER retention of the COL4A1 and COL4A2 proteins. All animal experiments were done with the approval of the UCSF institutional animal care and use committee.
To test whether the mouse Col4a2+/G646D mutation also led to intracellular accumulation of COL4A1 and COL4A2 proteins at the expense of their secretion we isolated and cultured mouse embryonic fibroblasts (MEFs) from E14.5 embryos. Western analysis showed diminished secretion and increased intracellular accumulation (data not shown). Further, immunolabeling of cultured MEFs from control C57BL/6J mice with anti-COL4A1 or anti-COL4A2 revealed colocalization of COL4A1/A2 heterotrimers with protein disulphide isomerase (PDI; ER resident protein necessary for collagen biosynthesis) but also presence of COL4A1/A2 heterotrimers in secretory vesicles (Figures 3C and 3D). In contrast, in MEFs derived from Col4a2+/G646D embryos, there is less COL4A1 and COL4A2 undergoing secretion, and instead there is an increase in the intensity of COL4A1 and COL4A2 labeling in the ER. The results for Col4a2+/G646D, although less severe, are consistent with the results for Col4a1+/Δex40 mutant mice and suggest that ER retention of mutant proteins underlies diminished secretion.
Next, we sought to determine whether the reduction in extracellular to intracellular ratio of the three putative COL4A2 mutations that we found in the ICH cohort were also a consequence of increased retention in the ER. We transfected COL4A2 cDNA into HT1080 cells, HeLa cells or MEFs, and all cell types revealed similar results. In each case, cells expressing the control cDNA, the common polymorphism (COL4A2P718S) or the three likely nonpathogenic variants (COL4A2V192F, COL4A2K701R and COL4A2R1109Q) had COL4A2 immunolabeling in both the ER, where it colocalized with PDI, and in secretory vesicles being trafficked from the ER toward the plasma membrane (Figure 4A, arrow, and data not shown). In contrast, the expression of either the validated pathogenic variant (COL4A2G1389R) or the three putative mutations (COL4A2E1123G, COL4A2Q1150K, and COL4A2A1690T) resulted in increased colocalization with PDI and absence of COL4A2 in secretory vesicles and confirm that the reduction in extracellular to intracellular COL4A1 and COL4A2 ratios is the consequence of retention within the ER.
Figure 4.
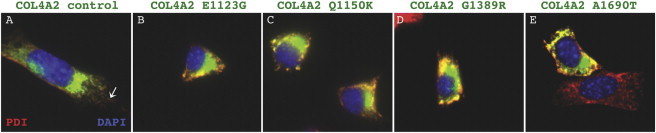
Putative Human Mutations, COL4A2E1123G, COL4A2Q1150K, and COL4A2A1690T Encode Proteins that Are Retained within the Endoplasmic Reticulum
Immunofluorescent labeling of COL4A2 (green; as described in Figure 3 legend) was performed in cells expressing control COL4A2 cDNA or different COL4A2 variants. (A) In cells transfected with control COL4A2 cDNA, COL4A2 (green) is detected in the ER where it colocalized with PDI (red) and in post-ER vesicles (arrow). (D) In cells transfected with the pathogenic mutation COL4A2G1389R, all of the COL4A2 labeling was perinuclear and colocalized with PDI consistent with ER retention. Similarly, in cells transfected with the three putative mutations (B, C, and E), COL4A2 colocalized with PDI in a tight perinuclear pattern. (E) A nontransfected cell (red) that does not express the COL4A2A1690T variant labeled with PDI and demonstrates a more disperse ER network compared to the transfected cell expressing the putative mutation (yellow).
Intracellular accumulation of mutant proteins can lead to ER stress and activation of the unfolded protein response (UPR) in Col4a1 mutant mice.16, 37 To investigate whether putative COL4A2 mutations might also lead to activation of these same pathways, we performed quantitative RT-PCR (qPCR) to determine the expression levels of multiple ER stress markers and UPR effectors in cells stably transfected with control COL4A2 cDNA or COL4A2 cDNA containing the various variants. We generated multiple clones of HT1080 cells that stably expressed control COL4A2 cDNA or variant COL4A2 cDNA and we selected four independent clones for each allele that had similar COL4A2 expression levels. We first confirmed that there were no significant differences in the level of COL4A1 expression or the level of expression of the collagen-binding chaperone HSP47 (Figure 5). To test for the presence of ER stress or for the activation of the UPR, we performed qPCR for the ER resident chaperone BiP, two ER-resident UPR transducers PERK and ATF6, a UPR-activated transcription factor ATF4, and the UPR target gene CHOP (see Table S3 for primers).
Figure 5.
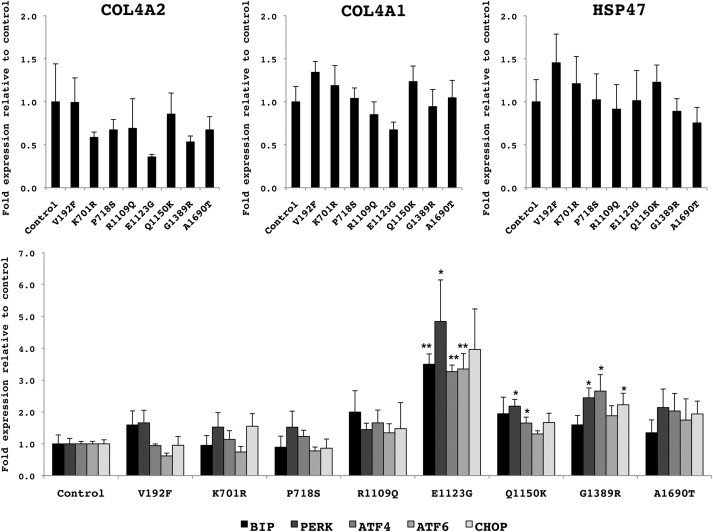
Putative Human Mutations, COL4A2E1123G and COL4A2Q1150K, Trigger ER Stress and Activation of the UPR
Quantitative RT-PCRs for ER stress and UPR markers were performed on cells stably transfected with control COL4A2 cDNA or different COL4A2 variants that expressed similar levels of COL4A2, COL4A1, and collagen-binding chaperone HSP47 (top panels). For analysis, cells were serum-deprived and treated with ascorbic acid (50 μg/ml) for 24 hr before being harvested. Total RNAs were isolated from each clone with RNeasy mini kit (QIAGEN) and used for the generation of cDNA with iScript Supermix (Bio-Rad). RT-qPCRs were performed with SsoFast EvaGreen supermix (Bio-Rad) on the equivalent of 20 ng of RNA. Presence of ER stress and UPR activation were investigated with the expression levels of the ER chaperone BIP, two ER-resident transducers of the UPR PERK and ATF6, the downstream transcription factor ATF4 and the UPR target gene CHOP (lower panel). The common variant COL4A2P718S and the pathogenic mutation COL4A2G1389R were used as controls. Levels of expression are expressed relative to control (mean of four independent clones ± SEM). ER stress and UPR activation were detected in cells expressing the putative mutant COL4A2E1123G and COL4A2Q1150K (∗p < 0.05, ∗∗p < 0.01).
Consistent with the absence of intracellular accumulation for the control COL4A2 cDNA, the common polymorphism, and the three likely nonpathogenic variants, we did not detect ER stress or UPR activation for these variants (Figure 5). In contrast, the pathogenic mutation and two of the three putative mutations (COL4A2E1123G and COL4A2Q1150K) triggered ER stress and UPR activation. One putative mutation, COL4A2A1690T, had no evidence of ER stress or UPR activation. This variant is within the NC1 domain, and these data support the possibility that mutations affecting the NC1 domain might have distinct intracellular consequences compared to mutations affecting the triple-helical domain. For example, mutations within the triple-helix-forming domain could result in misfolding of partially assembled heterotrimers, whereas mutation within the NC1 domain could interfere with the initiation of heterotrimer assembly.
The current study provides strong evidence for involvement of COL4A2 mutations in sporadic ICH. We resequenced all coding sequences and flanking intervening sequences for the entire COL4A2 gene, and we identified three putative pathogenic mutations in four individuals with ICH that were not present in at least 284 control chromosomes. The putative mutations COL4A2E1123G, COL4A2Q1150K, and COL4A2A1690T resulted in missense changes in amino acids that are highly conserved across species. Notably, none of these putative mutations are glycine missense variants, which in COL4A1 are well established to underlie severe, early-onset, and highly penetrant cerebrovascular diseases. This could reflect an ascertainment bias and perhaps nonglycine variants represent alleles that present with milder disease later in life. It is also possible that these variants become pathogenic later in life as a consequence of interactions with other factors that predispose to ICH. Although current in silico algorithms are not ideal for predicting functional consequences of COL4A2 variants, we showed that all three putative mutations lead to decreased ratios of extracellular to intracellular COL4A2 as a consequence of their retention in the ER. Similarly, a mutation in the mouse ortholog, Col4a2G646D, caused intracellular retention of COL4A1 and COL4A2 within the ER and ICH in the mutant animals. Finally, two of the three putative mutations that we identified also caused significant ER stress and UPR activation. Both of these mutations occurred within the triple-helix-forming domain and the sole putative mutation that did not lead to ER stress occurred within the NCI domain.
ER stress has been proposed to be an important part of the pathophysiology of collagen mutations. However, because pathogenic COL4A1 and COL4A2 mutations most often result in intracellular accumulation at the expense of secretion, it is difficult to dissociate these two consequences with respect to their relative contributions to pathology. Although all three putative mutations showed increased ER retention of mutant proteins, the inability to detect ER stress in one of the mutations might suggest that UPR activation is not a necessary part of the cellular pathophysiology. It is possible that the extent of, and cellular response to, secretion differs in vivo compared to our observations in vitro. Regardless, the identification of putative COL4A2 mutations in four out of 96 individuals supports the hypothesis that COL4A2 mutations that perturb collagen biosynthesis can increase risk of sporadic ICH in humans. In addition to the potential cytotoxic effects of intracellular accumulation of mutant proteins, the molecular pathway(s) could involve extracellular insults (including a deficiency or the presence of abnormal COL4A1/COL4A2 heterotrimers in the basement membrane) that disrupt protein-protein interactions with other basement membrane components, growth factors,23, 25 and/or cell surface receptors. Evaluating these potential cellular mechanisms with Col4a2 mutant mice will inform treatment or intervention options to prevent ICH in patients with type IV collagen mutations.
Acknowledgments
This study was funded by a research grant from the American Heart Association (D.B.G.) and by National Institutes of Health (NIH)– National Institute of Neurological Disorders and Stroke grant R01NS059727 (S.M.G. and J.R.). Salary support was also provided by Research To Prevent Blindness (C.L.D.), and NIH-National Eye Institute (NEI) grant R01EY019887 (B.K.). Additional support was provided by a core grant from the NEI (EY02162) and a Research To Prevent Blindness Unrestricted Grant (UCSF Department of Ophthalmology).
Published online: December 29, 2011
Footnotes
Supplemental Data include three tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SIFT (Sorting Intolerant from Tolerant, version 2.0), http://sift.jcvi.org/
PolyPhen (Polymorphism Phenotyping-2), http://genetics.bwh.harvard.edu/pph/
Supplemental Data
References
- 1.Qureshi A.I., Tuhrim S., Broderick J.P., Batjer H.H., Hondo H., Hanley D.F. Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 2001;344:1450–1460. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 2.Rosand J., Eckman M.H., Knudsen K.A., Singer D.E., Greenberg S.M. The effect of warfarin and intensity of anticoagulation on outcome of intracerebral hemorrhage. Arch. Intern. Med. 2004;164:880–884. doi: 10.1001/archinte.164.8.880. [DOI] [PubMed] [Google Scholar]
- 3.Gould D.B., Phalan F.C., van Mil S.E., Sundberg J.P., Vahedi K., Massin P., Bousser M.G., Heutink P., Miner J.H., Tournier-Lasserve E., John S.W. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 4.Gould D.B., Phalan F.C., Breedveld G.J., van Mil S.E., Smith R.S., Schimenti J.C., Aguglia U., van der Knaap M.S., Heutink P., John S.W.M. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 5.Breedveld G., de Coo I.F., Lequin M.H., Arts W.F.M., Heutink P., Gould D.B., John S.W.M., Oostra B., Mancini G.M.S. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J. Med. Genet. 2006;43:490–495. doi: 10.1136/jmg.2005.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vahedi K., Kubis N., Boukobza M., Arnoult M., Massin P., Tournier-Lasserve E., Bousser M.-G. COL4A1 mutation in a patient with sporadic, recurrent intracerebral hemorrhage. Stroke. 2007;38:1461–1464. doi: 10.1161/STROKEAHA.106.475194. [DOI] [PubMed] [Google Scholar]
- 7.Sibon I., Coupry I., Menegon P., Bouchet J.-P., Gorry P., Burgelin I., Calvas P., Orignac I., Dousset V., Lacombe D., et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann. Neurol. 2007;62:177–184. doi: 10.1002/ana.21191. [DOI] [PubMed] [Google Scholar]
- 8.Plaisier E., Gribouval O., Alamowitch S., Mougenot B., Prost C., Verpont M.C., Marro B., Desmettre T., Cohen S.Y., Roullet E., et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 2007;357:2687–2695. doi: 10.1056/NEJMoa071906. [DOI] [PubMed] [Google Scholar]
- 9.de Vries L.S., Koopman C., Groenendaal F., Van Schooneveld M., Verheijen F.W., Verbeek E., Witkamp T.D., van der Worp H.B., Mancini G. COL4A1 mutation in two preterm siblings with antenatal onset of parenchymal hemorrhage. Ann. Neurol. 2009;65:12–18. doi: 10.1002/ana.21525. [DOI] [PubMed] [Google Scholar]
- 10.Shah S., Kumar Y., McLean B., Churchill A., Stoodley N., Rankin J., Rizzu P., van der Knaap M., Jardine P. A dominantly inherited mutation in collagen IV A1 (COL4A1) causing childhood onset stroke without porencephaly. Eur. J. Paediatr. Neurol. 2010;14:182–187. doi: 10.1016/j.ejpn.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Rouaud T., Labauge P., Tournier Lasserve E., Mine M., Coustans M., Deburghgraeve V., Edan G. Acute urinary retention due to a novel collagen COL4A1 mutation. Neurology. 2010;75:747–749. doi: 10.1212/WNL.0b013e3181eee440. [DOI] [PubMed] [Google Scholar]
- 12.Plaisier E., Chen Z., Gekeler F., Benhassine S., Dahan K., Marro B., Alamowitch S., Paques M., Ronco P. Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am. J. Med. Genet. A. 2010;152A:2550–2555. doi: 10.1002/ajmg.a.33659. [DOI] [PubMed] [Google Scholar]
- 13.Bilguvar K., DiLuna M.L., Bizzarro M.J., Bayri Y., Schneider K.C., Lifton R.P., Gunel M., Ment L.R., Pacifier and Breastfeeding Trial Group COL4A1 mutation in preterm intraventricular hemorrhage. J. Pediatr. 2009;155:743–745. doi: 10.1016/j.jpeds.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meuwissen M.E.C., de Vries L.S., Verbeek H.A., Lequin M.H., Govaert P.P., Schot R., Cowan F.M., Hennekam R., Rizzu P., Verheijen F.W., et al. Sporadic COL4A1 mutations with extensive prenatal porencephaly resembling hydranencephaly. Neurology. 2011;76:844–846. doi: 10.1212/WNL.0b013e31820e7751. [DOI] [PubMed] [Google Scholar]
- 15.Vermeulen R.J., Peeters-Scholte C., Van Vugt J.J.M., Barkhof F., Rizzu P., van der Schoor S.R., van der Knaap M.S. Fetal origin of brain damage in 2 infants with a COL4A1 mutation: fetal and neonatal MRI. Neuropediatrics. 2011;42:1–3. doi: 10.1055/s-0031-1275343. [DOI] [PubMed] [Google Scholar]
- 16.Gould D.B., Marchant J.K., Savinova O.V., Smith R.S., John S.W.M. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum. Mol. Genet. 2007;16:798–807. doi: 10.1093/hmg/ddm024. [DOI] [PubMed] [Google Scholar]
- 17.Labelle-Dumais C., Dilworth D.J., Harrington E.P., de Leau M., Lyons D., Kabaeva Z., Manzini M.C., Dobyns W.B., Walsh C.A., Michele D.E., Gould D.B. COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 2011;7:e1002062. doi: 10.1371/journal.pgen.1002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plaisier E., Alamowitch S., Gribouval O., Mougenot B., Gaudric A., Antignac C., Roullet E., Ronco P. Autosomal-dominant familial hematuria with retinal arteriolar tortuosity and contractures: a novel syndrome. Kidney Int. 2005;67:2354–2360. doi: 10.1111/j.1523-1755.2005.00341.x. [DOI] [PubMed] [Google Scholar]
- 19.Alamowitch S., Plaisier E., Favrole P., Prost C., Chen Z., Van Agtmael T., Marro B., Ronco P. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology. 2009;73:1873–1882. doi: 10.1212/WNL.0b013e3181c3fd12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayne R., Wiedemann H., Irwin M.H., Sanderson R.D., Fitch J.M., Linsenmayer T.F., Kühn K. Monoclonal antibodies against chicken type IV and V collagens: electron microscopic mapping of the epitopes after rotary shadowing. J. Cell Biol. 1984;98:1637–1644. doi: 10.1083/jcb.98.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trüeb B., Gröbli B., Spiess M., Odermatt B.F., Winterhalter K.H. Basement membrane (type IV) collagen is a heteropolymer. J. Biol. Chem. 1982;257:5239–5245. [PubMed] [Google Scholar]
- 22.Fatemi S.H. The role of secretory granules in the transport of basement membrane components: radioautographic studies of rat parietal yolk sac employing 3H-proline as a precursor of type IV collagen. Connect. Tissue Res. 1987;16:1–14. doi: 10.3109/03008208709001990. [DOI] [PubMed] [Google Scholar]
- 23.Paralkar V.M., Vukicevic S., Reddi A.H. Transforming growth factor beta type 1 binds to collagen IV of basement membrane matrix: implications for development. Dev. Biol. 1991;143:303–308. doi: 10.1016/0012-1606(91)90081-d. [DOI] [PubMed] [Google Scholar]
- 24.Parkin J.D., San Antonio J.D., Pedchenko V., Hudson B., Jensen S.T., Savige J. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Hum. Mutat. 2011;32:127–143. doi: 10.1002/humu.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, X., Harris, R., Bayston, L., and Ashe, H. (2008). Type IV collagens regulate BMP signalling in Drosophila Nature 425, 72–77. [DOI] [PubMed]
- 26.Bunt S., Hooley C., Hu N., Scahill C., Weavers H., Skaer H. Hemocyte-secreted type IV collagen enhances BMP signaling to guide renal tubule morphogenesis in Drosophila. Dev. Cell. 2010;19:296–306. doi: 10.1016/j.devcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khoshnoodi J., Cartailler J.-P., Alvares K., Veis A., Hudson B.G. Molecular recognition in the assembly of collagens: terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J. Biol. Chem. 2006;281:38117–38121. doi: 10.1074/jbc.R600025200. [DOI] [PubMed] [Google Scholar]
- 28.Gorres K.L., Raines R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vranka J.A., Sakai L.Y., Bächinger H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004;279:23615–23621. doi: 10.1074/jbc.M312807200. [DOI] [PubMed] [Google Scholar]
- 30.Engel J., Prockop D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991;20:137–152. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 31.Favor J., Gloeckner C.J., Janik D., Klempt M., Neuhäuser-Klaus A., Pretsch W., Schmahl W., Quintanilla-Fend L. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–736. doi: 10.1534/genetics.106.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta M.C., Graham P.L., Kramer J.M. Characterization of alpha1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha1 and alpha2(IV) mutations on type IV collagen distribution. J. Cell Biol. 1997;137:1185–1196. doi: 10.1083/jcb.137.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo X.D., Johnson J.J., Kramer J.M. Embryonic lethality caused by mutations in basement membrane collagen of C. elegans. Nature. 1991;349:707–709. doi: 10.1038/349707a0. [DOI] [PubMed] [Google Scholar]
- 34.Sibley M.H., Graham P.L., von Mende N., Kramer J.M. Mutations in the alpha 2(IV) basement membrane collagen gene of Caenorhabditis elegans produce phenotypes of differing severities. EMBO J. 1994;13:3278–3285. doi: 10.1002/j.1460-2075.1994.tb06629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biffi A., Halpin A., Towfighi A., Gilson A., Busl K., Rost N., Smith E.E., Greenberg M.S., Rosand J., Viswanathan A. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology. 2010;75:693–698. doi: 10.1212/WNL.0b013e3181eee40f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knudsen K.A., Rosand J., Karluk D., Greenberg S.M. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/wnl.56.4.537. [DOI] [PubMed] [Google Scholar]
- 37.Firtina Z., Danysh B.P., Bai X., Gould D.B., Kobayashi T., Duncan M.K. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J. Biol. Chem. 2009;284:35872–35884. doi: 10.1074/jbc.M109.060384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson E.J. Oxford University Press; USA: 1987. Teratocarcinomas and embryonic stem cells. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.