

Abstract
The autosomal-recessive form of popliteal pterygium syndrome, also known as Bartsocas-Papas syndrome, is a rare, but frequently lethal disorder characterized by marked popliteal pterygium associated with multiple congenital malformations. Using Affymetrix 250K SNP array genotyping and homozygosity mapping, we mapped this malformation syndrome to chromosomal region 21q22.3. Direct sequencing of RIPK4 (receptor-interacting serine/threonine kinase protein 4) showed a homozygous transversion (c.362T>A) that causes substitution of a conserved isoleucine with asparagine at amino acid position 121 (p.Ile121Asn) in the serine/threonine kinase domain of the protein. Additional pathogenic mutations—a homozygous transition (c.551C>T) that leads to a missense substitution (p.Thr184Ile) at a conserved position and a homozygous one base-pair insertion mutation (c.777_778insA) predicted to lead to a premature stop codon (p.Arg260ThrfsX14) within the kinase domain—were observed in two families. Molecular modeling of the kinase domain showed that both the Ile121 and Thr184 positions are critical for the protein's stability and kinase activity. Luciferase reporter assays also demonstrated that these mutations are critical for the catalytic activity of RIPK4. RIPK4 mediates activation of the nuclear factor-κB (NF-κB) signaling pathway and is required for keratinocyte differentiation and craniofacial and limb development. The phenotype of Ripk4−/− mice is consistent with the human phenotype presented herein. Additionally, the spectrum of malformations observed in the presented families is similar, but less severe than the conserved helix-loop-helix ubiquitous kinase (CHUK)-deficient human fetus phenotype; known as Cocoon syndrome; this similarity indicates that RIPK4 and CHUK might function via closely related pathways to promote keratinocyte differentiation and epithelial growth.
Main Text
Limb pterygium syndromes (LPSs) are a clinically and genetically heterogeneous group of multiple-congenital-anomaly disorders characterized by multiple webbing on the neck, elbows, and knees that limit joint mobility and a wide range of other ectodermal structural abnormalities.1, 2 A well-known component of the LPS spectrum is popliteal pterygium syndrome (PPS) [MIM 119500]—an autosomal-dominant disease characterized by cleft lip and/or palate, lip pits, and digital and genital abnormalities, in addition to popliteal pterygium.1, 3, 4 Heterozygous mutations in IRF6, which encodes interferon regulatory factor 6 [MIM 607199], cause the autosomal-dominant form of PPS, although there is evidence for genetic heterogeneity.5, 6 An autosomal-recessive form of PPS, described as lethal-type popliteal pterygium syndrome (LPPS) [MIM 263650] and also known as Bartsocas-Papas syndrome, is characterized by a more severe phenotype than that associated with the autosomal-dominant form.7 The primary characteristics of Bartsocas-Papas syndrome are as follows: (1) marked popliteal pterygium leading to severe arthrogryposis, synostosis of the carpal/tarsal and phalangeal bones in the hands and feet, digital hypoplasia/aplasia, and complete soft-tissue syndactyly; (2) lack of nails; (3) ankyloblepharon filiforme adnatum; (4) lack of scalp hair, eyebrows and eyelashes, blepharophimosis, eyelid coloboma, and microphthalmia; (5) cleft lip and/or palate, facial cleft, filiform bands between the jaws, and hypoplastic nasal tip; (6) hypoplastic external genitalia; and (7) fetal and neonatal morbidity.1, 2, 3, 4, 7 Although Bartsocas-Papas syndrome was initially considered as a lethal disorder, there are reported cases at 2, 8, and 18 years of age,8, 9 supporting extensive clinical variability even for life span in LPPS. To date no gene has been associated with the autosomal-recessive form of PPS.
We studied a consanguineous Turkish family (BPS1) (Figures 1A–D and 2B), originally reported by Aslan et al.10 Marked popliteal pterygium associated with multiple congenital anomalies was observed in six members of the family, four of whom died within a week after birth. At the time this report was written, affected individuals V-12 and VI-3 (Figure 2B) were alive and 13 and 1.5 years old, respectively. Common shared findings in both were alopecia, ankyloblepharon filiforme adnatum, hypoplastic oral cavity and filiform bands between the jaws, limb abnormalities (including digital hypoplasia, syndactyly, nail hypoplasia, arthrogryposis, clubfeet, and popliteal, axillary, and inguinal pterygia), and hypoplastic labia major (Table 1). In addition to classical Bartsocas-Papas syndrome findings,11, 12, 13, 14 individual V-12 had additional abnormalities, including medial canthal webbing, hypoplasia of the iliac wing and scapulae, short stature, anal stenosis, and a rectal polyp (Table 1). Therefore, the initial diagnosis of individual V-12 was considered as variant of autosomal-recessive multiple pterygium syndrome (Aslan type) [MIM 605203].10 DNA samples were obtained from family members after receiving written informed consent from the adults and parents of children. The study protocol was approved by the Karadeniz Technical University, Faculty of Medicine Ethics Committee (approval number: 2010/5).
Figure 1.
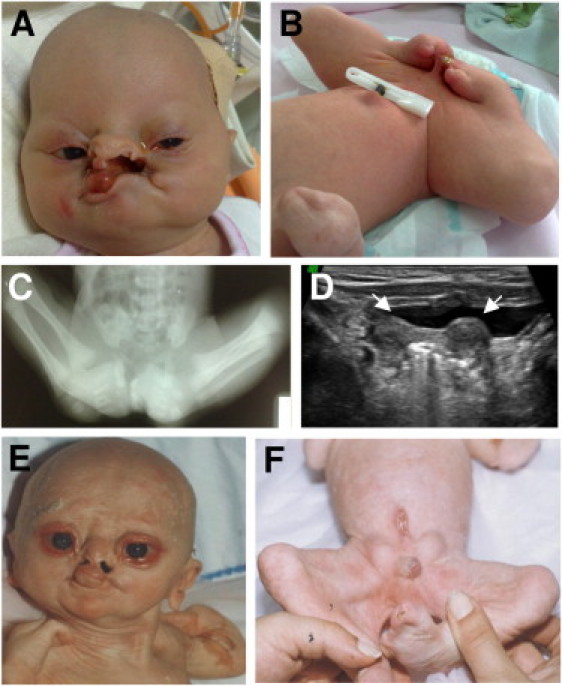
Clinical Characterization of the Affected Individuals from the BPS1 and BPS2 Families
(A and B) Frontal view of individual VI-3 from family BPS1 showing alopecia, ectropion, upslanting palpebral fissures, anteverted nostrils, oral clefts more prominent on the left side, synechia between lips, marked popliteal pterygium, total syndactyly of toes, and synechia of feet to genital region and fused digital mass of right hand.
(C) X-ray scan of individual VI-3 from family BPS1 presenting synostosis of phalangeal bones of the toes and missing metatarsals.
(D) Pelvic ultrasonography of individual VI-3 from family BPS1 showing a bicornuate uterus. Arrows indicate the two horns of the bicornuate uterus.
(E and F) Frontal view of individual IV-1 from family BPS2 presenting alopecia, scaly skin, ectropion, cloudy cornea, anteverted nostrils, oral clefts, synechia between lips, and syndactlylous missing fingers of both hands, severe popliteal pterygia, syndactyly of toes, and ambiguous genitalia (phallus and incomplete fusion of genital folds, and scrotal folds), and an inferiorly placed umbilicus.
Figure 2.
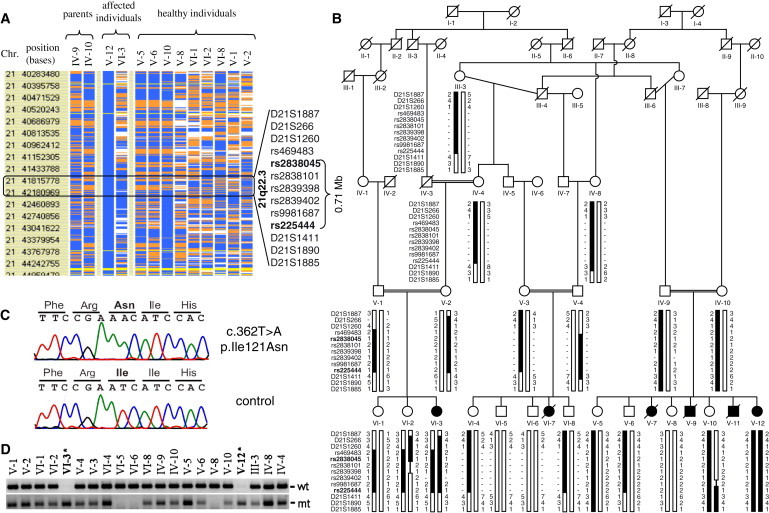
Homozygosity Mapping and Mutation Analysis in Family BPS1
(A) Schematic representation of the genome-wide homozygosity mapping data. Genotype files (CHP files) were generated with Affymetrix GTYPE software and were transferred to the VIGENOS (Visual Genome Studio) program (Hemosoft, Ankara), which facilitates visualization of a large quantity of genomic data in comprehensible visual screens. Homozygous genotypes identical to the genotype data obtained from the affected individual V-12 are shown in blue, whereas contrasting homozygote genotypes are shown in white and heterozygous SNPs appear in orange. Noninformative SNPs appear in yellow, and gray indicates no call. A single homozygote segment approximately 0.71Mb in size was observed and is marked in the rectangle. Microsatellite DNA markers selected from the interval are shown on the right side of the image.
(B) Pedigree and haplotype analysis of family BPS1 linked to chromosomal region 21q22.3. Genotyping data and haplotype bars for 21q markers are shown below the symbol for each individual. The order of DNA markers was obtained from the UCSC Human Genome Working Draft (build 36.1) and is listed on the left side of each genotype. Black bars denote the disease-associated region. Thin bars represent noninformative genotypes. SNP markers harboring the critical region are marked as bold.
(C) Sequence chromatograms showing the identified c.362T>A mutation in RIPK4 in family BPS1. The upper chromatogram shows the homozygous mutant sequence, and the lower chromatogram shows the wild-type sequences.
(D) Genotypes of the c.362T>A mutation in 21 members of family BPS1 were analyzed with the amplification refractory mutation system. The upper panel shows the wild-type allele (wt) and the lower panel shows the mutant allele (mt). Asterisks (∗) indicate the affected family members.
Table 1.
Clinical Findings in Patients with the Autosomal-Recessive Form of Popliteal Pterygium Syndrome
| Patient | VI-3 (BPS1) | V-12 (BPS1) | IV-1 (BPS2) | IV-1 (BPS3) |
|---|---|---|---|---|
| Consanguinity | + | + | + | + |
| Sex | female | female | male | female |
| Life span | 18 months old (alive) | 13 years old (alive) | 57 days (deceased) | 3.5 months (deceased) |
| Birth weight (at term) | 2350 g | 3.050 g | 2200 g | NA |
| Height at birth (due to pterygium) | NA | 37.4 cm | 35 cm | NA |
| Alopecia | + | + | + | + |
| Ectropion/lagophthalmos/keratitis | +/−/+ | +/−/+ | +/+/+ | −/−/NA |
| Corneal ulcerations | + | + | + | + |
| Colobomas of lower eyelids | + | − | NA | + |
| Ankyloblepharon/blepharophimosis | +/+ | +/+ | −/− | +/+ |
| Absence of eyebrows/eyelashes | +/+ | +/+ | +/+ | +/+ |
| Hypoplastic alae nasi and nose | + | + | + | + |
| Mouth border | irregular | irregular | irregular | absent |
| Hypoplastic oral/nasopharyngeal cavities | +/+ | +/+ | NA | NA |
| Orolabial synechia (filiform bands between jaws) | + | + | + | no mouth opening |
| Cleft lip/palate | + (unilateral)/+ | −/− | + (bilateral)/+ | no lip/no palate |
| Low set ears | + | + | + | − |
| Expressionless face | + | + | + | + |
| Upward slanting of palpebral fissures | + | + | + | + |
| Short neck | + | + | − | + |
| Micrognathia | + | + | NA | + |
| Limb Findings | ||||
| Short, bowed limbs | + | + | + | + |
| Digital hypoplasia/thumb aplasia | +/+ | +/+ | +/+ | +/+ |
| Syndactyly of fingers/toes | +/+ | +/+ | +/+ | +/+ |
| Hypoplasia of nails | + | + | + | + |
| Absence of palmar/plantar lines | + | + | + | NA |
| Arthrogryposis | + | + | NA | NA |
| Clubfoot deformity | + | + | + | NA |
| Flexion contractures | NA | + | + | + |
| Coetaneous and Musculoskeletal Findings | ||||
| Popliteal/axillary/inguinal pterygia | +/−/+ | +/−/+ | +/+/+ | +/+/+ |
| Filiform bands between feet/feet and pelvic region | +/+ | +/− | −/+ | NA |
| Anal stenosis/atresia/rectal polyps or skin tags | −/−/− | +/−/+ | −/−/− | +/−/+ |
| Genitourinary System | ||||
| Bicornuate-bicollis uterus | + | NA | male | NA |
| Hypoplastic labia major | + | + | male | + |
| Micropenis/hypoplastic scrotum\ | female | female | +/+ | female |
| Low set umbilicus | + | + | + | + |
| Inguinal hernia | − | − | + | NA |
| Radiological Findings | ||||
| Radius aplasia | NA | NA | + (unilateral left) | −/− |
| Bowing of ulnae | NA | NA | + | −/− |
| Hypoplasia of metacarpals/phalanges | +/+ | +/+ | +/+ | +/+ |
| Hypoplasia of iliac wing/pelvic bones/scapula | −/+/NA | +/+//+ | +/NA/NA | NA |
| Cranial MRI | normal | normal | NA | NA |
| Abdominal and Renal USG | normal | normal | ectopic kidney | NA |
| Others | ||||
| Dry, scaly skin | +/−/+ | − | + | + |
| Asymmetric/wide-set nipples | + | +/+ | −/+ | + |
| Cardiac murmur | patent foramen ovale | − | − | NA |
| Echocardiogram | mitral regurgitation | normal | NA | NA |
| Pulmonary hypoplasia | − | − | − | NA |
| Mental retardation | NA | − | NA | NA |
| Molecular Basis | ||||
| RIPK4 mutation | c.362T>A (p.Ile121Asn) | c.362T>A (p.Ile121Asn) | c.551C>T (p.Thr184Ile) | c.777_778insA (p.Arg260ThrfsX14) |
NA is an abbreviation for not available or unknown.
Genome-wide homozygosity mapping of the two affected (V-12 and VI-3) and ten unaffected members (V-1, V-2, VI-1, VI-2, IV-9, IV-10, V-5, V-6, V-8, and V-10) of the family was performed with Affymetrix 250K SNP arrays. A single homozygous segment on chromosomal region 21q22 was observed. Within this region, haplotypes residing between SNP markers rs2838045 and rs225444 were identical in both affected individuals, which indicated homozygosity by descent (Figure 2A). Subsequent microsatellite-marker genotyping that included all family members confirmed the 21q22.3 locus (Figure 2B). Critical recombination events observed in two healthy family members (VI-2 and V-10) positioned the mutant gene between SNP markers rs2838045 and rs225444, within an interval of approximately 0.71 Mb (Figure 2B). In this critical region there are seven protein-coding genes, TMPRSS2 [MIM 602060], RIPK4 [MIM 605706], PRDM15, C2CD2, ZNF295, UMODL1 [MIM 613859], and ABCG1 [MIM 603076] and three expressed sequence tags.
The human ortholog of the mouse receptor-interacting serine/threonine-protein kinase 4 gene (RIPK4) was a highly relevant candidate gene because homozygous Ripk4 knockout mice (Ripk4−/−) exhibit such severe ectoderm-originated organ anomalies as short limbs and tail partially fused to the body cavity; digital fusion; atresia of the oral cavity, esophagus, and all external orifices; reduced skin folds; and disrupted keratinocyte differentiation leading to expanded spinous and granular layers and an outermost layer of parakeratotic cells instead of enucleated squamous cells.15, 16 Homozygous Ripk4-deficient mice are not viable and die within several hours after birth because of suffocation caused by oral fusion. Heterozygous Ripk4-deficient mice (Ripk4+/−) are normal.15, 16
Sequencing of all 8 exons and exon-intron boundaries of RIPK4 (NM_020639) from the genomic DNA of affected individual V-12 showed a homozygous c.362T>A transversion in the second exon of the gene, which leads to substitution of asparagine with an evolutionarily highly conserved isoleucine at position 121 (p.Ile121Asn) in the serine/threonine kinase domain of the protein (Figure 2C). Primer3 web software was used to design primer sequences for amplification and the subsequent sequencing reaction (Table S1, available online).17 The detected c.362T>A mutation was tested in all available family members with the amplification refractory mutation system (ARMS) (Table S2). Both affected individuals (VI-3 and V-12) were homozygous, and the parents and individuals VI-1, VI-2, VI-4, VI-8, V-5, V-6, and V-10 were heterozygous for this mutation (Figure 2D). The c.362T>A substitution was not detected with the ARMS protocol in 336 healthy Turkish control samples.
We extended the study to an additional Turkish family (BPS2) with one affected male child that exhibited the primary characteristic features of Bartsocas-Papas syndrome (his parents are first cousins) (Figures 1E–1F and 3A and Table 1). Unfortunately, we could not obtain a DNA sample from the affected individual because he died prior to the study; however, DNA sequencing analysis of RIPK4 in parental DNA showed that both parents were heterozygous for a c.551C>T transition in the third exon, which leads to substitution of isoleucine with threonine at evolutionarily conserved position 184 (p.Thr184Ile) in the serine/threonine kinase domain of the protein (Figures 3B and 4A–4C). This change was not detected with the ARMS protocol with allele-specific primers (Table S2) in 336 healthy controls.
Figure 3.
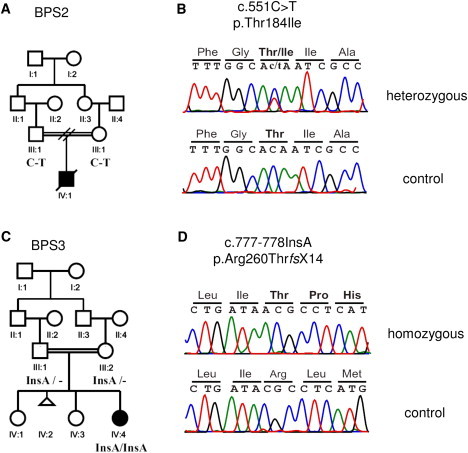
Mutation Analysis of RIPK4 in the BPS2 and BPS3 Families
(A) Pedigree of family BPS2 shows consanguinity and allele segregation. C and T below family members III-1 and III-2 indicate the wild-type and mutant alleles, respectively.
(B) Sequence chromatogram shows the identified c.551C>T mutation in RIPK4 observed in family BPS2. The upper chromatogram shows the heterozygous mutant sequences from the parents of affected individual IV-1, and the lower chromatogram shows the homozygous wild-type sequences from a healthy control.
(C) The pedigree of family BPS3 shows consanguinity and allele segregation. The mutant allele is shown below family members III-1, III-2, and IV-4.
(D) Sequence chromatogram shows a one base-pair insertion mutation (c.777_778insA) in RIPK4 in family BPS3. The upper chromatogram shows the homozygous mutant sequence in individual IV-1, and the lower chromatogram shows the homozygous wild-type sequences from a healthy control.
Figure 4.
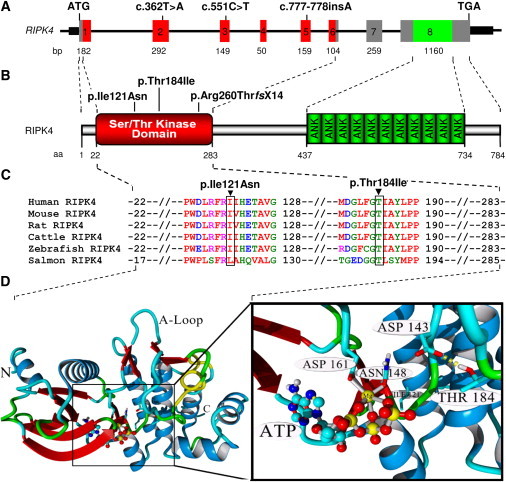
Diagrammatic Representation of RIPK4 and In-Silico Analysis of the RIPK4 Serine/Threonine Kinase Domain
(A) Diagram of RIPK4 and localization of the identified mutations (c.362T>A, c.551C>T, and c.777_778insA) through the exons. The length of each exon is indicated below the diagram.
(B) Diagram of the known functional domains of human RIPK4 and localization of the three identified mutations in the serine/threonine kinase domain. The numbers of the first and last amino acids of the each domain are shown below the diagram.
(C) Multiple alignments of all the partial amino acid sequences of the known RIPK4 serine/threonine kinase domain in a variety of species. Amino acid positions are indicated on the right side of the alignments, and the missense mutations (p.Ile121Asn and p.Thr445Ile) are shown above the alignment.
(D) Molecular model of the RIPK4 kinase domain showing the active site. Residues Asn148 and Asp161 are predicted to bind to ATP via an Mg2+ ion. The catalytic residue Asp143 is stabilized by a hydrogen bond to Thr184 (which is lost upon mutation to isoleucine), whereas the second residue of interest (Ile121) is located in the background, forming part of the hydrophobic core. Molecular graphics were created with YASARA and POVRay software. The following abbreviations are used: N, Amino terminus of the kinase domain; C, carboxyl terminus of the kinase domain; A-loop: activation loop.
Recently, a consanguineous Egyptian family (BPS3) with one affected and two healthy siblings was referred to us (Figure 3C and Table 1).18 The 3.5-month-old affected individual had total absence of body hair and multiple pterygia in almost all joints of the body, including shoulders, elbows, wrists, hips, knees, and ankles. A very large and thick popliteal pterygium and severe distortion of both lower limbs was observed. There were multiple eye anomalies and a bilateral orofacial cleft that did not follow the embryological fusion line. No oral stoma or lips were observed. The mandible and maxilla were severely hypoplastic. The hands and feet had complete syndactyly, absent thumbs, and variable degrees of phalangeal hypoplasia. The affected individual had abnormal external genitalia and the anal orifice was stenotic. Antenatal history was normal except for weak fetal movement. Sequencing of RIPK4 in the genomic DNA of the affected individual showed a homozygous one base-pair insertion in exon 5 (c.777_778insA), which was heterozygous in the parents (Figures 3D and 4A). This insertion is predicted to cause a frameshift at arginine 260 and an altered sequence of 13 amino acids followed by a premature stop codon (p.Arg260ThrfsX14) (Figure 4B). RNA from the affected individual was not available for further investigation to determine whether this premature stop codon triggers nonsense-mediated decay of the mutant RNA or not. The truncated protein is predicted to lose the last eight amino acids from the carboxyl terminus of the kinase domain, intermediate domain, and all ankyrin repeats of the protein; therefore, it is expected to be nonfunctional.
RIPK4 is a member of the receptor-interacting protein (RIP) kinase family, which is comprised of critical regulators of the signal transduction mechanisms that mediate cell survival and death.19, 20 RIPK4 was initially reported to interact with an isoform of the protein kinase C (PKC) family and was referred to as PKC-delta-interacting protein kinase (DIK) [MIM 605706].21 Independently, the mouse ortholog of RIPK4 was described as a membrane-associated protein kinase that interacts with PKCβ on the basis of yeast two-hybrid screening and was referred to as protein kinase C-associated kinase (PKK).22 Afterward, because of the similarities between the kinase domain of DIK and PKK and that of the other RIP family members, DIK and PKK were subsequently referred to as RIPK4 in humans and mice, respectively.23
RIPK4 encodes a 784-amino acid protein (RIPK4) that comprises an N-terminal serine/threonine kinase domain followed by an intermediate region, and a C-terminal putative regulatory domain containing 11 ankyrin repeats.21, 22, 23 In the present study, the regions in which the RIPK4 mutations occurred—exons 2, 3, and 5—correspond to the kinase domain of RIPK4 (Figures 4A–B). In order to analyze the possible effects of the two amino acid substitutions (p.Ile121Asn and p.Thr184Ile), a homology model of the RIPK4 N-terminal kinase domain was built with YASARA molecular modeling software (Figure 4D).24 Models were built for the closest ten templates with the YASARA's standard protocol hm_build.25 The best model was obtained from the RCSB Protein Data Bank (PDB) 3PJC file (crystal structure of JAK3 at 2.2 Å resolution),26 which yielded an overall structure validation Z score of −0.86. The placement of ATP in the RIPK4 model was derived from template 3PLS (RON kinase), which contains an ATP analog.27 The homology model is available from the authors upon request. In this model, residue Ile121 is placed in helix 3 (Pro114-His133); it forms part of the hydrophobic core and is in contact with the beta hairpin that supports the magnesium-binding residues Asn148 and Asp161 in the ATP-binding site. A mutation to asparagine thus has obvious destabilizing effects, and the FoldX program28 predicts a reduction in protein stability of 11 kJ/mol.
The threonine residue (Thr184) is located even closer to the active site in the A (Activation)-loop, which has been reported to regulate access to the ATP-binding site and to carry residues involved in autophosphorylation.28 As the A-loop is rather flexible and exhibits large variability between homologs; its predicted structure is not entirely reliable. Nevertheless and interestingly, Thr184 is found to form a hydrogen bond with Asp 143—the key active site residue that is assumed to abstract the proton from the hydroxyl group to be phosphorylated.28 A mutation to isoleucine disrupts this stabilizing interaction, and FoldX predicts a smaller energetic cost of 3.3 kJ/mol, which does not include the negative influence on the catalytic activity of Asp 143. However, replacement of Thr184 with an alanine or glutamate via site-directed mutagenesis disrupts the in vitro and in vivo kinase activity of the RIPK4 protein.29 Substitution of aspartic acid with an alanine or asparagine at position 143 also disrupts the in vitro and in vivo kinase activity of the RIPK4.29, 30 As such, amino acid substitutions in both the Ile121 and Thr184 positions have critical effects on the protein's stability and kinase activity, and thereby support their causative nature of these mutations.
A luciferase reporter assay was also performed in order to determine whether Ile121Asn, and Thr184Ile variants of RIPK4 have enough activity to evoke a potent nuclear factor-κB (NF-κB) response or not. To generate both RIPK4 expression vectors (which harbor Ile121Asn and Thr184Ile variants) previously described full-length N-terminal Flag-tagged RIPK4 cDNA22 was used. The two RIPK4 variants were recreated by site-directed mutagenesis as an N-terminal Flag-tagged RIPK4 cDNA in a pCMV-driven mammalian expression vector. The PCR products were digested with Dpn I restriction enzyme (Agilent) and transformed into One Shot TOP10 chemically competent E. coli cells (Invitrogen). Clones harboring the desired single nucleotide change were verified by DNA sequencing. Ile121Asn and Thr184Ile encoding vectors were introduced transiently into HEK293T cells, along with a NF-κB firefly luciferase reporter construct and a Renilla luciferase reporter construct. Comparisons were made with respect to wild-type RIPK4, the previously described The184Ala mutant,29 and Chk2 constructs because CHK2 is a kinase that does not activate NF-κB, and hence it was used as a negative control. The assays were performed with a dual luciferase reporter assay system (Promega). Whole-cell lysates were also subjected to SDS/8%PAGE for immunoblotting analysis with anti-Flag to confirm the expression levels of each of these proteins.
Luciferase reporter assays showed that both of the RIPK4 variants associated with the autosomal-recessive form of popliteal pterygium syndrome, Ile121Asn and Thr184Ile, were catalytically inactive in this study and failed to evoke a robust NF-κB response (Figures 5 A–C). These data provide further evidence that both Ile121 and Thr184 residues located in the kinase domain of the protein are critical for catalytic activity of RIPK4, and their substitution explains the etiopatogenesis of the disease evaluated in this study.
Figure 5.
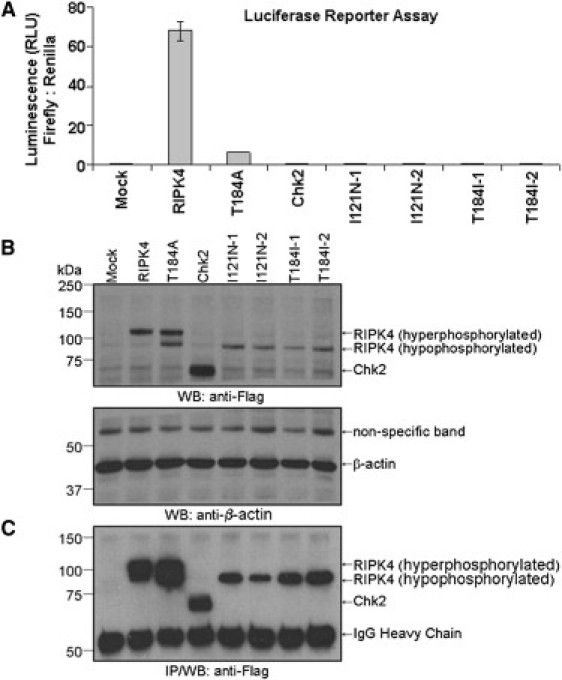
Functional Analysis RIPK4 Variants Described in This Study
RIPK4 and various mutants described in the text were cotransfected with pBIIxLuc NF-κB firefly luciferase reporter and pRL-TK Renilla luciferase reporter into HEK293T cells. Two representative clones (denoted as Ile121Asn-1 and Ile121Asn-2, and Thr184Ile-1 and Thr184Ile-2) each harboring the desired disease-associated RIPK4 variation were analyzed by this method.
(A) This figure depicts a typical luciferase assay, which was performed in triplicate. The Renilla luciferase construct was used to normalize for transfection efficiency. Data shown are mean ± standard error of the mean.
(B) An aliquot from each of the cell lysates used above was loaded on to SDS/8%PAGE gel and subjected to an anti-Flag immunoblot and anti-β-actin immunoblot to determine protein expression.
(C) The remaining cell lysates were immunoprecipitated (IP) with anti-Flag to enrich for Flag-tagged proteins, and then subjected to immunoblotting analysis with anti-Flag to confirm the expression levels of each of these proteins.
Ripk4 is expressed in most tissues of the developing mouse embryo—the highest level of expression occurs in the basal and suprabasal layers of the epidermis—and plays a role in the homeostasis of epidermal keratinocyte proliferation and differentiation.22, 31, 32 In Ripk4-deficient mice, increased epidermal thickness due to abnormal keratinocyte differentiation was reported even though the keratinocyte proliferation rate was normal.15 The phenotype of Ripk4−/− mice is consistent with the human phenotype observed in the presented affected individuals, both phenotypes exhibit organ anomalies of surface ectoderm origin, such as dry and scaly skin, corneal ulcerations, hypoplastic alae nasi and nose, hypoplastic oral and nasopharyngeal cavities, orolabial synechia (filiform bands between the jaws), cleft lip and/or palate, and hypoplastic labia major. In addition, elevated expression of Ripk4 was noted in hair follicle keratinocytes, whereas its deficiency leads to abnormal hair follicle development and hair loss,15, 16, 31 as in the presented individuals that had alopecia. Limb and digital malformations in both humans and mice might also be a consequence of abnormal development of the apical ectodermal ridge during embryogenesis.
The physiological function of RIPK4 and the role of its kinase activity are largely unknown; however, it has been reported that RIPK4 is critical for activation of NF-κB, which is involved in regulating keratinocyte differentiation. The kinase domain of RIPK4 alone appeared to be as efficient as full-length RIPK4 at activating NF-κB.16, 19, 30 NF-κB is a dimeric transcription factor generated from combinations of five structurally related proteins—NF-κB1 (p50), NF-κB2 (p52), RelA (p65), c-Rel, and RelB.33 NF-κB/Rel protein complexes are retained in the cytoplasm of resting cells because their nuclear localization signal is masked by IκB inhibitory proteins.34 In response to stimulation, IκB kinase (IKK), which consists of two catalytic subunits (IKKα also known as conserved helix-loop-helix ubiquitous kinase [CHUK] and IKKβ) and one regulatory subunit (IKKγ [NEMO]), phosphorylates the IκB proteins. After phosphorylation, IκB proteins undergo polyubiquitination and subsequent degradation. When the IκB function is abolished, NF-κB dimers translocate to the nucleus and activate the expression of target genes.35, 36 Several studies have reported that the ubiquitously expressed NF-κB is a key player in regulation of epidermal differentiation, apical ectodermal ridge formation, limb outgrowth, digit development, cellular proliferation, organ development, apoptosis, and immunity.37, 38, 39, 40 Mice lacking Chuk (Chuk−/− mice) exhibit developmental abnormalities such as short limbs and tails, rounded heads because of the shorter jaws and nasal bones, syndactyly in both the fore and hind paws, retarded hair follicle development, reduced skinfolds, and predominant parakeratosis in the epidermis instead of an enucleated cornified layer because of the defect of differentiation of epidermal keratinocytes, and they die within a few hours after birth.41, 42, 43, 44, 45 There are close similarities in the phenotypes of Chuk knockout mice and Ripk4−/− mice. CHUK is responsible for the noncanonical pathway of NF-κB activation,46 whereas RIPK4 requires IKKβ for the induction of the canonical NF-κB pathway.30 However, both canonical and noncanonical NF-κB signaling appear to be required for epidermal differentiation in mice.
Lahtela et al.47 reported two human fetuses with a lethal condition known as Cocoon syndrome (MIM 613630), which is characterized by severe abnormalities in brain and facial structure and apparently absent limbs that are bound to the trunk and encased under the skin; this syndrome is a result of a homozygous stop codon mutation in CHUK—the human ortholog of mouse Chuk. Fetuses with Cocoon syndrome exhibit a more severe form of Bartsocas-Papas syndrome. The present study's results clearly show that these syndromes are not allelic; however, the morphological defects and skin abnormalities seen in both syndromes are similar and consistent with phenotypes observed in mice lacking genes that encode Chuk and Ripk4, which suggests that these two genes might function in closely linked pathways to regulate epidermal development and differentiation. Transgenic experiments have shown that Ripk4 functions in the epidermis via PKC-dependent signaling pathways but Ripk4 cannot rescue the defect seen mice that lack Chuk.16
NF-κB signaling pathway is also critical for proper IRF6 function in keratinocyte development.48, 49 Transcription of IRF6 is activated by ΔNp63α, which is the major isoform of p63 encoded by TP63 (MIM 603273).50, 51 Mutations of TP63, disrupting the function of the isoform ΔNp63α, causes developmental disorders that are characterized by various degrees of limb abnormalities, ectodermal dysplasia, and facial clefts.52 Various isoforms of p63 such as TAp63α and ΔNp63α function as direct upstream elements of Chuk.53 ΔNp63 also interacts with structural components cRel or RelA of the NF-κB/Rel protein complex to form novel binding complexes on p63 or NF-κB/Rel sites of multitarget gene promoters.54, 55 Taken together, these data suggest that IRF6, CHUK, and RIPK4 are important components of closely related pathways to control epidermal development.
The present study's findings show that recessive mutations in RIPK4 cause Bartsocas-Papas syndrome and its variant, the autosomal-recessive form of multiple pterygium syndrome (Aslan type). All mutations corresponded to the serine/threonine kinase domain of the protein, indicating the functional relevance of this domain. Missense and nonsense mutations cause both the lethal and nonlethal recessive forms of multiple pterygium syndrome characterized by severe organ anomalies of ectoderm origin. The abnormalities observed in presented individuals were similar but less severe than those seen in CHUK-deficient human fetuses with Cocoon syndrome. The present study's findings point out that RIPK4 and CHUK might function in closely related pathways to promote keratinocyte differentiation and epithelial growth.
Acknowledgments
We thank all the family members that participated in this study and M. Livaoğlu, N. Karacal, A. Yıldırım, K. Baydar, and Ö. Aliyazıcıoğlu for their indispensible technical assistance. This study was supported by the Karadeniz Technical University Research Fund (grant 2009.114.001.05 to E.K.) and the Scientific and Technological Research Council of Turkey (TÜBİTAK) (108S420 to N.A.A. and 108S418 to H.K.) grants under the E-RARE network CRANIRARE consortium (project no. R07197KS).
Published online: December 22, 2011
Footnotes
Supplemental Data include two tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Human Genome Database, build hg18, March 2006, http://genome.ucsc.edu/
WHAT-IF server, http://swift.cmbi.ru.nl/servers/html/index.html
Yasara, http://www.yasara.org/
Supplemental Data
References
- 1.Hall J.G., Reed S.D., Rosenbaum K.N., Gershanik J., Chen H., Wilson K.M. Limb pterygium syndromes: A review and report of eleven patients. Am. J. Med. Genet. 1982;12:377–409. doi: 10.1002/ajmg.1320120404. [DOI] [PubMed] [Google Scholar]
- 2.Hall J.G. The lethal multiple pterygium syndromes. Am. J. Med. Genet. 1984;17:803–807. doi: 10.1002/ajmg.1320170410. [DOI] [PubMed] [Google Scholar]
- 3.Gorlin R.J., Sedano H.O., Cervenka J. Popliteal pterygium syndrome. A syndrome comprising cleft lip-palate, popliteal and intercrural pterygia, digital and genital anomalies. Pediatrics. 1968;41:503–509. [PubMed] [Google Scholar]
- 4.Escobar V., Weaver D. Popliteal pterygium syndrome: A phenotypic and genetic analysis. J. Med. Genet. 1978;15:35–42. doi: 10.1136/jmg.15.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo S., Schutte B.C., Richardson R.J., Bjork B.C., Knight A.S., Watanabe Y., Howard E., de Lima R.L., Daack-Hirsch S., Sander A., et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuzawa N., Kondo S., Shimozato K., Nagao T., Nakano M., Tsuda M., Hirano A., Niikawa N., Yoshiura K. Two missense mutations of the IRF6 gene in two Japanese families with popliteal pterygium syndrome. Am. J. Med. Genet. A. 2010;152A:2262–2267. doi: 10.1002/ajmg.a.33338. [DOI] [PubMed] [Google Scholar]
- 7.Bartsocas C.S., Papas C.V. Popliteal pterygium syndrome. Evidence for a severe autosomal recessive form. J. Med. Genet. 1972;9:222–226. doi: 10.1136/jmg.9.2.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolan S.M., Shanske A.L., Marion R.W., Gross S.J. First-trimester diagnosis of Bartsocas-Papas syndrome (BPS) by transvaginal ultrasound: Case report and review of the literature. Prenat. Diagn. 2003;23:138–142. doi: 10.1002/pd.560. [DOI] [PubMed] [Google Scholar]
- 9.Maganzini A.L., Rios A., Shanske A. Craniofacial characteristics evidenced in Bartsocas-Papas syndrome from birth to five years. Case report. N. Y. State Dent. J. 2006;72:34–37. [PubMed] [Google Scholar]
- 10.Aslan Y., Erduran E., Kutlu N. Autosomal recessive multiple pterygium syndrome: A new variant? Am. J. Med. Genet. 2000;93:194–197. doi: 10.1002/1096-8628(20000731)93:3<194::aid-ajmg6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 11.Martínez-Frías M.L., Frías J.L., Vazquez I., Fernández J. Bartsocas-Papas syndrome: Three familial cases from Spain. Am. J. Med. Genet. 1991;39:34–37. doi: 10.1002/ajmg.1320390109. [DOI] [PubMed] [Google Scholar]
- 12.Massoud A.A., Ammaari A.N., Khan A.S., ven Katraman B., Teebi A.S. Bartsocas-Papas syndrome in an Arab family with four affected sibs: Further characterization. Am. J. Med. Genet. 1998;79:16–21. [PubMed] [Google Scholar]
- 13.Veenstra-Knol H.E., Kleibeuker A., Timmer A., ten Kate L.P., van Essen A.J. Unreported manifestations in two Dutch families with Bartsocas-Papas syndrome. Am. J. Med. Genet. A. 2003;123A:243–248. doi: 10.1002/ajmg.a.20308. [DOI] [PubMed] [Google Scholar]
- 14.Kalender A.M., Dogan A., Sebik A., Gokalp M.A. “Ring leg deformity” in Bartsocas-Pappas syndrome. Eur. J. Med. Genet. 2009;52:269–270. doi: 10.1016/j.ejmg.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Holland P., Willis C., Kanaly S., Glaccum M., Warren A., Charrier K., Murison J., Derry J., Virca G., Bird T., Peschon J. RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr. Biol. 2002;12:1424–1428. doi: 10.1016/s0960-9822(02)01075-8. [DOI] [PubMed] [Google Scholar]
- 16.Rountree R.B., Willis C.R., Dinh H., Blumberg H., Bailey K., Dean C., Jr., Peschon J.J., Holland P.M. RIP4 regulates epidermal differentiation and cutaneous inflammation. J. Invest. Dermatol. 2010;130:102–112. doi: 10.1038/jid.2009.223. [DOI] [PubMed] [Google Scholar]
- 17.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 18.Abdalla E.M., Morsy H. Bartsocas-Papas Syndrome: Unusual Findings in the First Reported Egyptian Family. Case Reports in Genetics. 2011;2011 doi: 10.1155/2011/428714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meylan E., Tschopp J. The RIP kinases: Crucial integrators of cellular stress. Trends Biochem. Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Stanger B.Z., Leder P., Lee T.H., Kim E., Seed B. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 21.Bhr C., Rohwer A., Stempka L., Rincke G., Marks F., Gschwendt M. DIK, a novel protein kinase that interacts with protein kinase Cdelta. Cloning, characterization, and gene analysis. J. Biol. Chem. 2000;275:36350–36357. doi: 10.1074/jbc.M004771200. [DOI] [PubMed] [Google Scholar]
- 22.Chen L., Haider K., Ponda M., Cariappa A., Rowitch D., Pillai S. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J. Biol. Chem. 2001;276:21737–21744. doi: 10.1074/jbc.M008069200. [DOI] [PubMed] [Google Scholar]
- 23.Meylan E., Martinon F., Thome M., Gschwendt M., Tschopp J. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-kappa B and is processed during apoptosis. EMBO Rep. 2002;3:1201–1208. doi: 10.1093/embo-reports/kvf236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 25.Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thoma G., Nuninger F., Falchetto R., Hermes E., Tavares G.A., Vangrevelinghe E., Zerwes H.G. Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family. J. Med. Chem. 2011;54:284–288. doi: 10.1021/jm101157q. [DOI] [PubMed] [Google Scholar]
- 27.Wang J., Steinbacher S., Augustin M., Schreiner P., Epstein D., Mulvihill M.J., Crew A.P. The crystal structure of a constitutively active mutant RON kinase suggests an intramolecular autophosphorylation hypothesis. Biochemistry. 2010;49:7972–7974. doi: 10.1021/bi100409w. [DOI] [PubMed] [Google Scholar]
- 28.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. The FoldX web server: An online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moran S.T., Haider K., Ow Y., Milton P., Chen L., Pillai S. Protein kinase C-associated kinase can activate NFkappaB in both a kinase-dependent and a kinase-independent manner. J. Biol. Chem. 2003;278:21526–21533. doi: 10.1074/jbc.M301575200. [DOI] [PubMed] [Google Scholar]
- 30.Muto A., Ruland J., McAllister-Lucas L.M., Lucas P.C., Yamaoka S., Chen F.F., Lin A., Mak T.W., Núñez G., Inohara N. Protein kinase C-associated kinase (PKK) mediates Bcl10-independent NF-kappa B activation induced by phorbol ester. J. Biol. Chem. 2002;277:31871–31876. doi: 10.1074/jbc.M202222200. [DOI] [PubMed] [Google Scholar]
- 31.Adams S., Pankow S., Werner S., Munz B. Regulation of NF-kappaB activity and keratinocyte differentiation by the RIP4 protein: Implications for cutaneous wound repair. J. Invest. Dermatol. 2007;127:538–544. doi: 10.1038/sj.jid.5700588. [DOI] [PubMed] [Google Scholar]
- 32.Adams S., Munz B. RIP4 is a target of multiple signal transduction pathways in keratinocytes: Implications for epidermal differentiation and cutaneous wound repair. Exp. Cell Res. 2010;316:126–137. doi: 10.1016/j.yexcr.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Baeuerle P.A., Baltimore D. NF-kappa B: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 34.Moynagh P.N. The NF-kappaB pathway. J. Cell Sci. 2005;118:4589–4592. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 35.Karin M., Delhase M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 36.Zandi E., Chen Y., Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: Discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 37.Verma I.M., Stevenson J.K., Schwarz E.M., Van Antwerp D., Miyamoto S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 38.Bushdid P.B., Brantley D.M., Yull F.E., Blaeuer G.L., Hoffman L.H., Niswander L., Kerr L.D. Inhibition of NF-kappaB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature. 1998;392:615–618. doi: 10.1038/33435. [DOI] [PubMed] [Google Scholar]
- 39.Kanegae Y., Tavares A.T., Izpisúa Belmonte J.C., Verma I.M. Role of Rel/NF-kappaB transcription factors during the outgrowth of the vertebrate limb. Nature. 1998;392:611–614. doi: 10.1038/33429. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Teran M., Ros M.A. The Apical Ectodermal Ridge: Morphological aspects and signaling pathways. Int. J. Dev. Biol. 2008;52:857–871. doi: 10.1387/ijdb.072416mf. [DOI] [PubMed] [Google Scholar]
- 41.Hu Y., Baud V., Delhase M., Zhang P., Deerinck T., Ellisman M., Johnson R., Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 42.Takeda K., Takeuchi O., Tsujimura T., Itami S., Adachi O., Kawai T., Sanjo H., Yoshikawa K., Terada N., Akira S. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 43.Li Q., Lu Q., Hwang J.Y., Büscher D., Lee K.F., Izpisua-Belmonte J.C., Verma I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Y., Baud V., Oga T., Kim K.I., Yoshida K., Karin M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 45.Sil A.K., Maeda S., Sano Y., Roop D.R., Karin M. IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. 2004;428:660–664. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 46.Oeckinghaus A., Hayden M.S., Ghosh S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 47.Lahtela J., Nousiainen H.O., Stefanovic V., Tallila J., Viskari H., Karikoski R., Gentile M., Saloranta C., Varilo T., Salonen R., Kestilä M. Mutant CHUK and severe fetal encasement malformation. N. Engl. J. Med. 2010;363:1631–1637. doi: 10.1056/NEJMoa0911698. [DOI] [PubMed] [Google Scholar]
- 48.Richardson R.J., Dixon J., Malhotra S., Hardman M.J., Knowles L., Boot-Handford R.P., Shore P., Whitmarsh A., Dixon M.J. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 2006;38:1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 49.Biggs L.C., Rhea L., Schutte B.C., Dunnwald M. Interferon Regulatory Factor 6 Is Necessary, but Not Sufficient, for Keratinocyte Differentiation. J. Invest. Dermatol. 2011 doi: 10.1038/jid.2011.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laurikkala J., Mikkola M.L., James M., Tummers M., Mills A.A., Thesleff I. p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development. 2006;133:1553–1563. doi: 10.1242/dev.02325. [DOI] [PubMed] [Google Scholar]
- 51.Thomason H.A., Dixon M.J., Dixon J. Facial clefting in Tp63 deficient mice results from altered Bmp4, Fgf8 and Shh signaling. Dev. Biol. 2008;321:273–282. doi: 10.1016/j.ydbio.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 52.van Bokhoven H., Brunner H.G. Splitting p63. Am. J. Hum. Genet. 2002;71:1–13. doi: 10.1086/341450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Candi E., Terrinoni A., Rufini A., Chikh A., Lena A.M., Suzuki Y., Sayan B.S., Knight R.A., Melino G. p63 is upstream of IKK alpha in epidermal development. J. Cell Sci. 2006;119:4617–4622. doi: 10.1242/jcs.03265. [DOI] [PubMed] [Google Scholar]
- 54.Yang X., Lu H., Yan B., Romano R.A., Bian Y., Friedman J., Duggal P., Allen C., Chuang R., Ehsanian R., et al. ΔNp63 versatilely regulates a Broad NF-κB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res. 2011;71:3688–3700. doi: 10.1158/0008-5472.CAN-10-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu H., Yang X., Duggal P., Allen C.T., Yan B., Cohen J., Nottingham L., Romano R.A., Sinha S., King K.E., et al. TNF-α Promotes c-REL/DeltaNp63α Interaction and TAp73 Dissociation from Key Genes That Mediate Growth Arrest and Apoptosis in Head and Neck Cancer. Cancer Res. 2011;71:6867–6877. doi: 10.1158/0008-5472.CAN-11-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.