

Abstract
The three members of the human neurexin gene family, neurexin 1 (NRXN1), neurexin 2 (NRXN2), and neurexin 3 (NRXN3), encode neuronal adhesion proteins that have important roles in synapse development and function. In autism spectrum disorder (ASD), as well as in other neurodevelopmental conditions, rare exonic copy-number variants and/or point mutations have been identified in the NRXN1 and NRXN2 loci. We present clinical characterization of four index cases who have been diagnosed with ASD and who possess rare inherited or de novo microdeletions at 14q24.3–31.1, a region that overlaps exons of the alpha and/or beta isoforms of NRXN3. NRXN3 deletions were found in one father with subclinical autism and in a carrier mother and father without formal ASD diagnoses, indicating issues of penetrance and expressivity at this locus. Notwithstanding these clinical complexities, this report on ASD-affected individuals who harbor NRXN3 exonic deletions advances the understanding of the genetic etiology of autism, further enabling molecular diagnoses.
Main Text
Incremental progress has been made in elucidating genetic factors involved in autism spectrum disorder (ASD [MIM 209850]). Most notably, researchers have identified both rare inherited and de novo copy-number variants (CNVs) and have subsequently performed sequence-based validation.1–9 NRXN1,1 NLGN3,10 NLGN4,10 SHANK3,11,12 and SHANK213 and genomic regions at 1q21.1,1 16p11.2,3,14 DDX53-PTCHD1,3,5,15 and others9 are all bona fide ASD risk loci.
Rare exonic deletions within NRXN1 (MIM 600565) at 2p16.3 are among the most consistently observed findings from CNV investigations of ASD. After the initial report of 300 kb hemizygous de novo lesions that eliminated several NRXN1 exons in two female siblings with ASD,1 several other studies have also detected deletions at this locus in ASD.4–7,16–20 Exonic NRXN1 deletions, which are extremely rare in controls,19 have also been reported in cases with schizophrenia,17,21–26 intellectual disability,17,19,27 bipolar disorder,28 attention deficit hyperactivity disorder (ADHD),29 Tourette syndrome,30 and autosomal-recessive Pitt-Hopkins syndrome.31 These reports, taken together with findings of cognitive impairments, aberrant synaptic electrophysiology, and behavioral changes in Nrxn1-knockout mice,32 provide strong evidence for the involvement of NRXN1 in the etiology of ASD and other neurodevelopmental disorders. There is also a report of a truncating mutation in NRXN2 (MIM 600566) in an ASD case.33
The human neurexin gene family includes three unlinked genes (NRXN1, NRXN2, and NRXN3 [MIM 600567]), each of which encodes alpha and beta isoforms.34 The neurexins are highly expressed in presynaptic terminals and have been shown to have important roles in synaptic cell adhesion and neurotransmitter secretion.35 Here, we describe the pedigrees (Figure 1), detailed phenotypes (Table 1), and genotypes (Figure 2 and Tables S1, S2, and S4–S7, available online) of ASD cases with rare deletions at 14q24.3-31.1, which overlaps NRXN3.
Figure 1.
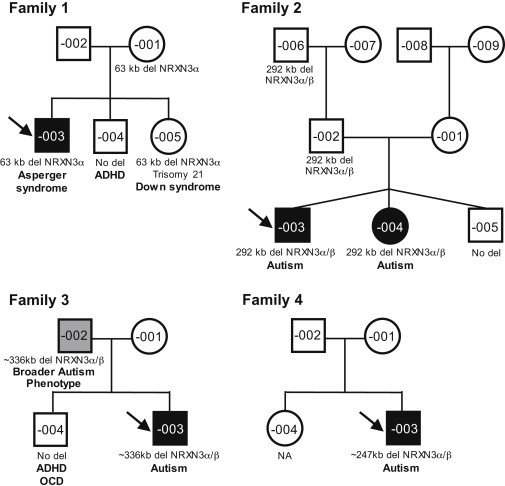
Pedigrees of Four Unrelated Families with Deletions at the NRXN3 Locus
Black-filled symbols represent ASD-affected individuals, gray-filled symbols represent broader autism phenotype (BAP)-affected individuals, and unfilled symbols represent apparently unaffected individuals. Probands are marked with an arrow. Clinical diagnosis and segregation of the NRXN3 deletions are shown. Children are placed left to right in birth order from eldest to youngest. Other CNVs and genetic variants are found in Tables S1, S2, and S4–S7. Abbreviations are as follows: ADHD, attention deficit hyperactivity disorder; OCD, obsessive compulsive disorder; and NA, not assessed.
Table 1.
Summary of Clinical Characteristics for Individuals with NRXN3 Deletions
| Patient ID and Karyotype | NRXN3 Deletion Coordinatesaand Deleted Exon(s) |
Clinical Details |
||
|---|---|---|---|---|
| Dx | Ax | Other | ||
| F1-003 (proband) 46,XY |
14q24.3(77,933,816–77,996,755) exon 1 (alpha) |
Asperger syndrome (ADI-R and clinical Dx) | Age: 16 yr, 7 months; IQ: Leiter-R IQ= 119 (92%); Language: OWLS: RL = 121 (92%) and EL = DTC; Adaptive Behavior: VABS-I: ABC = 75(5%), COM = 96 (39%), DLS = 94 (34%), and SOC = 52 (<1%) | aggression, anger, anxiety, transition and stimulation (photo-, phono-, and osmophobia) difficulties, sleeplessness, depression, and headaches |
| F1-005 (sister) 47,XX,+21 |
14q24.3(77,933,816–77,996,755) exon 1 (alpha) |
Down syndrome (karyotype and clinical Dx) | First Ax. Age: 11 yr, 4 months: ADOS-2 = ASD; ADI-R = Non-ASD (except behavior); did not meet clinical criteria (by developmental pediatrician); no ASD Dx; IQ: Leiter-R IQ = 46 (<1%); Language: OWLS: TL = 40 (<1%), RL = < 40 (<1%), and EL = 43 (<1%); Adaptive Behavior: VABS-I: ABC = 49 (<1%), DLS = 46 (<1%), and SOC = 64 (1%) | born at 34 weeks, heart defect, sleep apnea, ear infections, fatigue, fear and anxiety, obsessive behavior, impulsive behavior, and difficulties with transitions, attention, and emotional regulation |
| Second Ax. Age: 16 yr: ADOS-3 and ADI-R, met criteria for BEV and COM, but not for SD; did not meet clinical criteria (by developmental pediatrician and psychologist); no ASD Dx; IQ: WASI: FSIQ = 51 (<1%), VIQ = 55 (<1%), PIQ = 54 (<1%); Language: OWLS: TL = 40 (<1%), RL = 40 (<1%), EL = 40 (<1%) | ||||
| F1-001 (mother) 46,XX |
14q24.3(77,933,816–77,996,755) exon 1 (alpha) |
none | high energy, social difficulties, anxiety, and auditory and language-processing difficulties | |
| F2-003 (proband) 46,XY |
14q31.1(79,194,918–79,486,635) exons 14-17 (alpha) and 3–7 (beta) |
autism (ADI-R and ADOS-1) | Age: 3 yr, 5 months; IQ: Leiter-R IQ = INC; Language: OWLS: INC; PPVT-4: SS = 67 (<1%); Adaptive Behavior: VABS-II: ABC = 61 (<1%), COM = 63 (1%), DLS = 60 (<1%), SOC = 59 (<1%), and MOT = 72 (3%) | born at 31 weeks, asthma, and juvenile arthritis |
| F2-004 (sister) 46,XX |
14q31.1(79,194,918–79,486,635) exons 14-17 (alpha) and 3–7 (beta) |
autism (ADI-R and ADOS-1) | Age: 3 yr, 9 months; IQ: Leiter-R IQ = 82 (12%); Language: OWLS: INC; PPVT: INC; Adaptive Behavior: VABS-II: ABC = 60 (<1%), COM = 54 (<1%), DLS = 64 (1%), SOC = 57 (<1%), and MOT = 75 (5%) | born at 31 weeks, asthma, and nonabsent seizures |
| F2-001 (father) 46,XY |
14q31.1(79,194,918–79,486,635) exons 14-17 (alpha) and 3–7 (beta) | none | Alexithymia: TAS-20 = 41 (not alexithymic) | color blind |
| F3-003 (proband) 46,XY |
14q31.1(∼78,503,451–78,839,469) exons 10-12 (alpha) and 1 (beta) |
autism (ADI-R) | Age: 13 yr; IQ: WASI: FSIQ = 71 (3%) | aggression, self harm, obsessive behavior, suicidal and homicidal thoughts, delusional and persecutory ideas, sleep issues, and obesity |
| F3-001 (father) 46,XY |
14q31.1(∼78,503,451–78,839,469) exons 10-12 (alpha) and 1 (beta) |
BAP (clinical Dx) | social avoidance, depression, aggression, alcoholism, obsessive behavior, compulsive behavior, illiteracy, asthma, and emphysema | |
| F4-003 (proband) 46,XY |
14q31.1 (∼78,766,127–79,013,263) exon 13 (alpha) and 1–2 (beta) |
autism (clinical Dx, C-TRF, DSM) | Age: 3 yr, 6 months; IQ: Leiter-R IQ = INC | aggression, anger, anxiety, temper tantrums, social avoidance, sucking and biting of hands and fingers, sleep-onset disorder, upper-body hypotonia, thumb-flexion difficulty, oppositional defiance, previous head banging, and prior speech delay |
Abbreviations are as follows: Dx, diagnosis; Ax, assessment; COM, communication; SD, social deficit; TL, total language; RL, receptive language; EL, expressive language; DTC, declined to complete; ABC, adaptive behavior composite; DLS, daily living skills; SOC, socialization; INC, incomplete (test was attempted but subject failed to complete); MOT, motor skills; SS, standardized score; WASI, Wechsler Abbreviated Scale of Intelligence; FSIQ, full scale IQ; VIQ, verbal IQ; and PIQ, performance IQ. See text for remaining abbreviations.
NCBI 36 (hg18).
Figure 2.
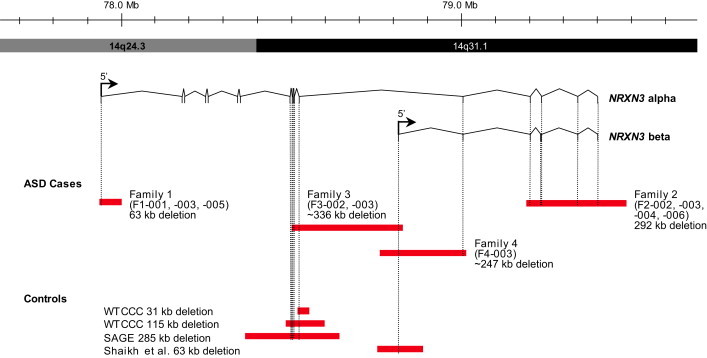
Extent of Genomic NRXN3 Deletions at Chromosomal Region 14q24.3-q31.1
Deletions are shown in red. CNVs detected in control populations (described in this paper) and from the Database of Genomic Variants55, 56 are also shown. Genomic coordinates and isoform information are from hg18. Control data are as described in this paper.
Initially, to assess the presence of CNVs in a research cohort of 1,158 Canadian individuals with ASD, we genotyped DNA with Affymetrix GeneChip SNP 6.0 (family 1 proband [F1-003] and family 2 sister [F2-004]), Illumina Infinium 1M single SNP (F1-003 and family 2 proband, F2-003), Illumina Omni 2.5M (family 1 sister, F1-005), and/or Agilent Human comparative genomic hybridization (CGH) 1M (F2-003) microarrays. These studies were approved by the Hospital for Sick Children (Toronto) research ethics board, and informed consent was obtained. We analyzed CNVs by using published methods,5,36,37 and we classified them as described.38 To ascertain the prevalence of NRXN3 CNVs, we examined the NRXN3 region in published data from 2,026 healthy individuals from the Children's Hospital of Philadelphia,39 from 2,493 controls genotyped at the University of Washington40 and in microarray data that our group analyzed from 10,603 population-based controls.3,5,15,36 This latter dataset included 1,123 controls from northern Germany,41 1,234 controls from Ottawa42 1,120 population controls from Ontario,43 1,056 HapMap samples,44 4,783 controls from the Wellcome Trust Case Control Consortium (WTCCC),45 and 1,287 controls recruited by the Study of Addiction: Genetics and Environment (SAGE) consortium.46 Control individuals were predominantly of European ancestry, which was comparable to the ancestry of the ASD subjects. Families 3 and 4 were found to have NRXN3 deletions through custom Agilent 44K microarray analyses, which were performed at the clinical genetics laboratories at Guy's Hospital and the Mayo Clinic, respectively. DNA from family 4 was also run on a 244K oligo Agilent array.
A deletion overlapping exon 1 of the NRXN3 alpha isoform was detected in proband F1-003 (Figure 2). Sequencing across the breakpoints confirmed the deletion to be 62,939 bp (chr14: 77,933,816–77,996,755; NCBI Build 36 [hg18]). Microarray analysis of parental DNA detected the same deletion in the mother. Independent validation of the array CNV calls was performed with real-time quantitative PCR (qPCR) based on SYBR Green I, and two independent primer pairs each at NRXN3 and 7q31.1 (FOXP2) served as a negative (diploid) control.47 The microdeletion was found in the proband (F1-003), mother (F1-001), and Down-syndrome-affected sister (F1-005) (Figure S1 and Tables S1–S2). All other genic CNVs detected in the sample from proband F1-003 were previously detected in control samples, and those that were rare (without overlapping CNVs in the Database of Genomic Variants) occurred within regions devoid of any annotated genes. We also performed targeted exome resequencing of F1-001, -002, -003, -004, and -005 to search for variants in the nondeleted NRXN3 allele and to test for other potential genic variants that might also contribute to the ASD phenotype. The Agilent SureSelect 50 Mb human all exon capture kit was used, and the Life/AB SOLiD4 platform was subsequently used for sequencing (Table S3). Three potentially interesting missense variants in ASD candidate genes,9 namely contactin 3 (CNTN3 MIM [601325]), contactin-associated protein-like 3 (CNTNAP3 MIM [610517]), and E1A-binding protein p300 (EP300 MIM [602700]), were detected and validated with Sanger sequencing (Table S4). F1-003 had a normal karyotype and a normal FMR-1 repeat number.
In family 1, the male proband (F1-003) was conceived naturally by a 33-year-old mother (F1-001) and a 35-year-old father (F1-002; Figure 1). Table 1 summarizes the clinical details. The pregnancy was uncomplicated, although the mother contracted pneumonia at 7 months of gestation. The proband was delivered by spontaneous vaginal delivery at 38 weeks and had a nuchal cord and birth weight of 7 lbs, 3 oz. The parents first noted his advanced speech and memorization of detail, as well as social difficulties, at 12–24 months of age. At age 9, he was identified as “gifted” after being assessed for learning disabilities. When the boy was between the ages of 9 and 14, there were concerns of depression. When the proband was 10, a family stressor might have contributed to suicidal ideation. At 13, he was diagnosed with Asperger syndrome. He had difficulties with aggression, anger, transitions, and stimulation, including that from bright lights and small spaces. His nonverbal cognitive abilities were assessed at age 16 with the Leiter International Performance Scale-Revised (Leiter-R) measure. He performed above the expectations for his age compared to his same-aged peers (92nd percentile). In addition, he completed the Vineland Adaptive Behavior Scales (VABS) and had age-appropriate outcomes for communication (39th percentile) and daily living skills (34th percentile), but his socialization skills were severely delayed (below the first percentile). Oral and Written Language Scales (OWLS) testing indicated he had above average (93rd percentile) receptive language and listening comprehension, but he declined to complete measures of expressive language (Oral Expression Scale). The Autism Diagnostic Interview-Revised (ADI-R), combined with a clinical assessment, was consistent with a diagnosis of Asperger syndrome. The proband reported that his mind raced and that he had difficulty sleeping through the night. He had experienced recurrent episodes of aggressive outbursts and depression and subsequently received treatment with risperidone, which was found to have been ineffective. After he complained of recurrent headaches, he was diagnosed with bilateral chronic migraine headaches with episodic visual symptoms that were most likely due to migraine aura. At 16 years old, he attended university part-time while maintaining a part-time job.
F1-003 is one of three children of nonconsanguineous parents of Irish and English descent. The mother has self-reported high energy levels, social difficulties, and anxiety, as well as auditory and language-processing difficulties. She did not meet criteria for ASD or a BAP upon a clinical interview assessment. Within the extended maternal family, there is one third-degree relative with an ASD diagnosis, and there are several second- and third-degree relatives with suspected ASD characteristics. The father, who is apparently healthy, has a brother with Down syndrome and second- and third-degree relatives with suspected ASD characteristics.
The proband's deletion-negative brother (F1-004) has a normal 46,XY karyotype and a normal FMR-1 repeat. He was diagnosed with ADHD at age 12; testing with the Autism Diagnostic Observation Schedule (ADOS: module 4) at age 14 showed that he did not meet formal diagnostic criteria for ASD. He performed as expected for his age on the Leiter-R test (63rd percentile). His receptive- and expressive-language skills exceeded expectations for his age (OWLS, 88th percentile), and his adaptive behavior was adequate in measures of communication, daily living skills, and socialization as assessed by VABS.
The proband's sister (F1-005) was diagnosed with Down syndrome and possesses a 47,XX,+21 karyotype and a normal FMR-1 repeat. A research assessment at age 11 revealed that she was below the first percentile for her age on the Leiter-R. Her receptive- and expressive-language skills also fell below the first percentile on the OWLS assessment. She showed mild to moderate deficits across her adaptive-behavior profile for her age. She met criteria for ASD on the ADOS module 2 and the ADI-R behavior dimension only, but she did not meet clinical criteria when assessed by a developmental pediatrician. Her behavior was consistent with her trisomy 21 status and complicated the diagnostic process. At age 16, heightened anxiety as well as increased obsessive and impulsive behaviors prompted a second clinical assessment (Table 1). The obsessive and repetitive components of her behavior were the most challenging and interfered with her adaptive functioning, but an overall diagnosis of non-ASD was given.
For family 2, analysis of the male proband's (F2-003's) DNA identified a ∼292 kb deletion overlapping several terminal exons of both the alpha and beta isoforms of NRXN3 (Figure 2). Microarray analysis of parental DNA detected the same deletion in the father (F2-002). Sequencing across the breakpoints showed the deletion to be 291,717 bp extending from chr14: 79,194,918–79,486,635 (hg18). Further qPCR validation (Figure S1) determined the presence of the NRXN3 deletion in the proband and the father, as well as in the sister (F2-004), who had an ASD diagnosis, and in the paternal grandfather (F2-006). Of the other CNVs identified in F2-003, all but one were previously detected in normal control samples (Table S5). CNVs identified in F2-004 are listed in Table S6. Subsequent exome resequencing of F2-001, -002, -003, -004, and -005 identified five missense variants in ASD candidate genes9 (Tables S3 and S4), contactin-associated protein-like 5 (CNTNAP5 MIM [610519]), F-box protein 40 (FBXO40 [MIM 609107]), and phosphodiesterase 1C, calmodulin-dependent 70 kDa (PDE1C [MIM 602987]), as well as the neuronally expressed prune homolog 2 (PRUNE2 [MIM 610691]; Drosophila) and aryl hydrocarbon receptor nuclear translocator 2 (ARNT2 [MIM 606036]).
Family 2 includes trizygotic triplets; the proband (F2-003) and an affected sister (F2-004) inherited the NRXN3 deletion from their father, whereas their unaffected brother (F2-005; Figure 1) did not inherit the deletion (Table 1). The triplets were conceived naturally by a 23-year-old mother (F2-001) and a 30-year-old father (F2-002) and were delivered at 31 weeks gestation. The weights of the affected proband and sister were 3 lbs, 12 oz. and 3 lbs, 4 oz., respectively. Multiple medical interventions were undertaken at birth as a result of the premature delivery. The proband was born with a right-sided cranial hemorrhage, and the affected sister was also suspected to have suffered a cranial hemorrhage at birth. All of the siblings have continuing difficulty with asthma, the proband suffers from juvenile arthritis, and the affected sister has had multiple ear and respiratory infections.
The proband was diagnosed with ASD after a clinical assessment at 3 years, 5 months of age. During testing for a research protocol when he was 5 years, 8 months of age, he met the criteria for an autism diagnosis based on the ADI-R and the ADOS Module 1. Nonverbal-communication ability and receptive- and expressive-language skills could not be assessed because the proband was unable to complete the intelligence quotient (IQ) (Leiter-R) and language (OWLS) tests. Testing with the Peabody Picture Vocabulary Test (PPVT-4) indicated that the proband had extremely poor receptive-vocabulary skills and adaptive behavior (VABS-II, below first percentile).
The ASD sister (F2-004) was diagnosed at 3 years, 9 months. As part of a research protocol, she was assessed at the age of 5 years, 8 months with the ADI-R and the ADOS Module 1, and she was found to have autism. Her nonverbal-communication ability was assessed with Leiter-R and was found to be in the low-average range (12th percentile) for her age group, whereas her VABS-II assessment indicated extremely low functioning (below first percentile). Assessment of her receptive and expressive language was attempted with the OWLS measure, but it could not be completed. Receptive-vocabulary testing (PPVT-4) could not be completed either.
The unaffected brother (F2-005) was assessed at the age of 4 years, 10 months with a social communication questionnaire (SCQ), which indicated that he did not have ASD (total score: 3).
These three children were born to nonconsanguineous parents of French Canadian descent; neither parent has an ASD diagnosis. The mother has asthma and was found not to have a BAP, i.e., she was not alexithymic on the Toronto Alexithymia Scale (TAS-20; total score: 29). The father is color blind and was also found to be non-BAP (TAS-20; total score: 41). Clinical information does not exist on male deletion carrier F2-006.
In a third family, a male proband (F3-003, Figure 1) with a ∼336 kb deletion (chr14: 78,503,451–78,839,469 [hg18]) overlapping exons 10–12 of NRXN3 alpha and exon 1 of NRXN3 beta was also identified (Figure 2). This individual originated from a collection of 9,400 patients tested at Guy's Hospital, where samples were collected by pediatricians and other health specialists, as well as by genetics centers both in and outside the region (SE. Thames).48 In all, 1,368 cases were reported to have ASD.
F3-003 was diagnosed with autism in early childhood, and clinical testing with an ADI-R at age 13 confirmed this finding. He displayed aggression, self-harm, obsessive behaviors, persecutory and delusional ideas, suicidal ideation, and sleep problems. The father, who also carried the NRXN3 deletion, was assessed by clinical review and was found to have BAP as well as learning disabilities, depression, and alcohol-related issues (Table 1).
A fourth family was identified, in which the male proband (F4-003, Figure 1) had an NRXN3 deletion of ∼247 kb (chr14: 78,766,127–79,013,263 [hg18]); qPCR confirmed that the deletion overlapped exon 13 of NRXN3 alpha and exons 1–2 of NRXN3 beta (Figure 2, Figure S1, and Table S7). FISH analysis of the proband and parents determined that the deletion arose de novo (Figure S2). F4-003 originated from a collection of 13,082 patients tested at the Mayo Clinic; 1,796 of these patients were referred to the Mayo Clinic as a result of their having been diagnosed with autism or pervasive developmental disorder (PDD). FMR-1 testing showed a normal repeat number.
In family 4, the male proband (F4-003, Table 1) was born at 38 weeks and weighed 8 lbs, 4 oz. The pregnancy was complicated by insulin-controlled gestational diabetes and hypertension. He was delivered via a cesarean section necessitated by maternal hemorrhaging. The neonatal course was uneventful. During the period from 19 to 24 months of age, the parents first noted that their son was not easily consoled, developed temper tantrums, started to bang his head, developed difficulty making eye contact, and started to develop some stiffness and rigidity. Between the ages of 2 and 3, he could not keep to a schedule, developed extreme reactions to taste and touch, and had trouble falling asleep. At 2 years, 10 months of age, he received a clinical diagnosis of ASD, speech language delays, and sleep-onset disorder. At 4 years, 6 months of age, the Caregiver-Teacher Report Form (C-TRF) for Ages 1.5–5 was completed; his scores indicated that he had internalizing and externalizing problems. His score on the DSM-Oriented Scales for Boys was in the clinical range (above the 97th percentile), indicating pervasive developmental problems. At 6 years, 10 months of age, he had near-normal speech, no longer banged his head, and had no oppositional defiance. Most concerning were his high levels of anxiety and his temper tantrums in unfamiliar situations. An individualized educational plan was put in place for math, reading, and writing, and he had a one-on-one educational assistant. Although he was in good general health, he received occupational therapy for his difficulty with thumb flexion and his hypotonia of the hands and chest.
To examine whether potential sequence-level mutations, as well as CNVs, in NRXN3 could be associated with ASD, we sequenced all coding exons and intron-exon splice sites in NRXN3 in another 350 unrelated ASD cases. Several inherited missense mutations, but no truncating mutations, were identified in four additional ASD individuals (Table S8). All of the mutations were inherited (three were maternal, one was paternal) and occurred within conserved amino acids, but the effect of these changes on the function of the resulting protein was not elucidated. Three of the four missense mutations were also present in the unaffected siblings.
CNVs within genes that encode several neuronal synaptic proteins have been implicated in ASD and other neurological disorders.8,49 The NRXN3 deletions identified here are very rare events in the general population. There are reported deletions overlapping exons of NRXN3 alpha in three female controls and exon 1 of NRXN3 beta in 1 of 15,122 examined controls (DNA was not available for experimental validation of these computational predictions). Taken together, the frequency of exonic deletions at the NRXN3 locus is significantly higher in ASD cases than in controls (4/4,322 cases versus 4/15,122 controls; Fisher's exact test, one-tailed p = 0.039). Furthermore, three of the four deletions in the ASD cases affect both the alpha and beta isoforms of NRXN3, whereas none of the deletions in the controls do. This difference is also statistically significant (3/4,322 ASD cases versus 0/15,122 controls; Fisher's exact test, one-tailed p = 0.0055). These findings parallel what is observed in NRXN1, for which deletions, while extremely rare, have been reported in a few control individuals.19,21,24,26 CNVs present in control individuals might be indicative of reduced penetrance of NXRN3 CNVs (perhaps modified by the nondeleted allele or other genes), the presence of CNVs within control individuals in regions where they do not destroy NRXN3 function, a lack of rigorous testing of the neuropsychiatric phenotype in the controls, or false-positive calls.50
Our data support the hypothesis that hemizygous deletions involving NRXN3 might be involved in the manifestation of an ASD or BAP phenotype. Perhaps most compelling, the deletion in family 4 arose de novo, lending strong support for a causative relationship between the loss of NRXN3 and the development of ASD.
Family 1 carried a deletion that overlapped exon 1 of NRXN3 alpha; we speculate that this deletion might predispose affected individuals to a mild ASD or Asperger syndrome phenotype in the absence of confounding genomic changes such as trisomy 21, as was observed in the Down-syndrome-affected sister. Because NRXN3 alpha is translated from a start site located in exon 2,51,52 it is possible that the full-length NRXN3 alpha could be produced despite the deletion. The deletion of exon 1 might decrease transcription of the gene or diminish the stability of messenger RNA. Interestingly, the same CNV deletion is present in the mother, who does not have a formal ASD diagnosis but who self-identified as possessing several ASD-like characteristics, including anxiety and social difficulties, as well as auditory and language-processing difficulties. Thus, there might be variable expressivity of the NRXN3 deletion. It is also possible that there are additional, currently unidentified, contributing genetic or environmental factors that could lead to a clinical ASD phenotype. Although the paternally inherited missense mutations found in the proband are predicted to be benign, they might work in conjunction with the maternally inherited NRXN3-alpha deletion to result in the observed ASD phenotype of the proband.
In contrast, families 2, 3, and 4 carried deletions that removed multiple exons of both the alpha and beta isoforms of NRXN3. The children carrying these deletions presented with a more severe form of ASD than that identified in family 1. This observation, coupled with the absence of deletions affecting both isoforms of NRXN3 in our controls, leads us to speculate that the loss of the alpha isoform in addition to the loss of the beta isoform might result in a more severe ASD phenotype. It is perhaps especially interesting to note that the apparently unaffected carrier father and paternal grandfather in family 2 carried the same deletion and missense mutations as the ASD-affected children, yet they had not been diagnosed with ASD or BAP. Conversely, the proband of family 3 presented with autism, whereas the carrier father was diagnosed with BAP. The complexities of penetrance and variable expressivity of NRXN3 in ASD might be partially explained by gene-dosage balance and functional redundancies of the neurexin gene family.53 Although all three neurexin genes have similar exonic nucleotide sequences, exon-intron structure, and patterns of alternative splicing for mRNAs, NRXN1 and NRXN3 have the greatest sequence and protein identity. Expression patterns vary between the neurexin genes and their alpha and beta isoforms. NRXN3 is expressed throughout the brain, but the other neurexins show differential locations and intensities of expression.54
Ultimately, fully understanding genotype and phenotype correlations will require knowledge of the diploid expression patterns of the neurexin genes, other interacting molecules, and perhaps nongenetic factors that might impede their homeostasis. Our discovery of rare NRXN3 deletions in ASD provides a set of reference cases for the evaluation of and comparison to other patients, both enabling the molecular diagnosis of autism and providing potential new targets for therapeutic intervention.
Acknowledgments
We thank the WTCCC and the SAGE (Study of Addiction: Genetics and Environment) consortium and A. Fiebig, A. Franke, and S. Schreiber at PopGen (University of Kiel, Kiel, Germany) and A. Stewart, R. McPherson, and R. Roberts of the University of Ottawa Heart Institute (University of Ottawa, Ottawa, Canada), for providing control data. This work was supported by The Centre for Applied Genomics, Genome Canada and the Ontario Genomics Institute, the Canadian Institutes for Health Research, the Canadian Institute for Advanced Research, the McLaughlin Centre, the Canada Foundation for Innovation, the Ontario Ministry of Research and Innovation, Autism Speaks, NeuroDevNet, and the Hospital for Sick Children Foundation. A.C.L. holds an Ontario graduate scholarship from the Ontario Ministry of Education and Training and a NeuroDevNet doctoral fellowship. S.W.S. holds the GlaxoSmithKline-CIHR chair in Genome Sciences at the University of Toronto and the Hospital for Sick Children.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
Database of Single Nucleotide Polymorphisms (dbSNP), http://www.ncbi.nlm.nih.gov/SNP/
Online Mendelian Inheritance in Man, (OMIM), http://www.omim.org/
References
- 1.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L., Autism Genome Project Consortium Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy D., Ronemus M., Yamrom B., Lee Y.H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Cook E.H., Jr., Scherer S.W. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 9.Scherer S.W., Dawson G. Risk factors for autism: translating genomic discoveries into diagnostics. Hum. Genet. 2011;130:123–148. doi: 10.1007/s00439-011-1037-2. [DOI] [PubMed] [Google Scholar]
- 10.Jamain S., Quach H., Betancur C., Råstam M., Colineaux C., Gillberg I.C., Soderstrom H., Giros B., Leboyer M., Gillberg C., Bourgeron T., Paris Autism Research International Sibpair Study Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsäter H. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moessner R., Marshall C.R., Sutcliffe J.S., Skaug J., Pinto D., Vincent J., Zwaigenbaum L., Fernandez B., Roberts W., Szatmari P., Scherer S.W. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berkel S., Marshall C.R., Weiss B., Howe J., Roeth R., Moog U., Endris V., Roberts W., Szatmari P., Pinto D. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 14.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T., Autism Consortium Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 15.Noor A., Whibley A., Marshall C.R., Gianakopoulos P.J., Piton A., Carson A.R., Orlic-Milacic M., Lionel A.C., Sato D., Pinto D. Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci Transl Med. 2010;2:49ra68. doi: 10.1126/scitranslmed.3001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen Y., Dies K.A., Holm I.A., Bridgemohan C., Sobeih M.M., Caronna E.B., Miller K.J., Frazier J.A., Silverstein I., Picker J., Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–e735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guilmatre A., Dubourg C., Mosca A.L., Legallic S., Goldenberg A., Drouin-Garraud V., Layet V., Rosier A., Briault S., Bonnet-Brilhault F. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bremer A., Giacobini M., Eriksson M., Gustavsson P., Nordin V., Fernell E., Gillberg C., Nordgren A., Uppströmer A., Anderlid B.M. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2011;156:115–124. doi: 10.1002/ajmg.b.31142. [DOI] [PubMed] [Google Scholar]
- 19.Ching M.S., Shen Y., Tan W.H., Jeste S.S., Morrow E.M., Chen X., Mukaddes N.M., Yoo S.Y., Hanson E., Hundley R., Children's Hospital Boston Genotype Phenotype Study Group Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2010;153B:937–947. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H.G., Kishikawa S., Higgins A.W., Seong I.S., Donovan D.J., Shen Y., Lally E., Weiss L.A., Najm J., Kutsche K. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 23.Kirov G., Gumus D., Chen W., Norton N., Georgieva L., Sari M., O'Donovan M.C., Erdogan F., Owen M.J., Ropers H.H., Ullmann R. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet. 2008;17:458–465. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 24.Kirov G., Grozeva D., Norton N., Ivanov D., Mantripragada K.K., Holmans P., Craddock N., Owen M.J., O'Donovan M.C., International Schizophrenia Consortium. Wellcome Trust Case Control Consortium Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vrijenhoek T., Buizer-Voskamp J.E., van der Stelt I., Strengman E., Sabatti C., Geurts van Kessel A., Brunner H.G., Ophoff R.A., Veltman J.A., Genetic Risk and Outcome in Psychosis (GROUP) Consortium Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 2008;83:504–510. doi: 10.1016/j.ajhg.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rujescu D., Ingason A., Cichon S., Pietiläinen O.P., Barnes M.R., Toulopoulou T., Picchioni M., Vassos E., Ettinger U., Bramon E., GROUP Investigators Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman J.M., Baross A., Delaney A.D., Ally A., Arbour L., Armstrong L., Asano J., Bailey D.K., Barber S., Birch P. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am. J. Hum. Genet. 2006;79:500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D., Cheng L., Qian Y., Alliey-Rodriguez N., Kelsoe J.R., Greenwood T., Nievergelt C., Barrett T.B., McKinney R., Schork N. Singleton deletions throughout the genome increase risk of bipolar disorder. Mol. Psychiatry. 2009;14:376–380. doi: 10.1038/mp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley W.E., Raelson J.V., Dubois D.Y., Godin E., Fournier H., Privé C., Allard R., Pinchuk V., Lapalme M., Paulussen R.J., Belouchi A. Hotspots of large rare deletions in the human genome. PLoS ONE. 2010;5:e9401. doi: 10.1371/journal.pone.0009401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sundaram S.K., Huq A.M., Wilson B.J., Chugani H.T. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology. 2010;74:1583–1590. doi: 10.1212/WNL.0b013e3181e0f147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zweier C., de Jong E.K., Zweier M., Orrico A., Ousager L.B., Collins A.L., Bijlsma E.K., Oortveld M.A., Ekici A.B., Reis A. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 2009;85:655–666. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Etherton M.R., Blaiss C.A., Powell C.M., Südhof T.C. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. USA. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gauthier J., Siddiqui T.J., Huashan P., Yokomaku D., Hamdan F.F., Champagne N., Lapointe M., Spiegelman D., Noreau A., Lafrenière R.G. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 2011;130:563–573. doi: 10.1007/s00439-011-0975-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Südhof T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Missler M., Zhang W., Rohlmann A., Kattenstroth G., Hammer R.E., Gottmann K., Südhof T.C. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- 36.Lionel A.C., Crosbie J., Barbosa N., Goodale T., Thiruvahindrapuram B., Rickaby J., Gazzellone M., Carson A.R., Howe J.L., Wang Z. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3:95ra75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- 37.Pinto D., Darvishi K., Shi X., Rajan D., Rigler D., Fitzgerald T., Lionel A.C., Thiruvahindrapuram B., Macdonald J.R., Mills R. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011;29:512–520. doi: 10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuchiya K.D., Shaffer L.G., Aradhya S., Gastier-Foster J.M., Patel A., Rudd M.K., Biggerstaff J.S., Sanger W.G., Schwartz S., Tepperberg J.H. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet. Med. 2009;11:866–873. doi: 10.1097/GIM.0b013e3181c0c3b0. [DOI] [PubMed] [Google Scholar]
- 39.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krawczak M., Nikolaus S., von Eberstein H., Croucher P.J., El Mokhtari N.E., Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 42.Stewart A.F., Dandona S., Chen L., Assogba O., Belanger M., Ewart G., LaRose R., Doelle H., Williams K., Wells G.A. Kinesin family member 6 variant Trp719Arg does not associate with angiographically defined coronary artery disease in the Ottawa Heart Genomics Study. J. Am. Coll. Cardiol. 2009;53:1471–1472. doi: 10.1016/j.jacc.2008.12.051. [DOI] [PubMed] [Google Scholar]
- 43.Zogopoulos G., Ha K.C., Naqib F., Moore S., Kim H., Montpetit A., Robidoux F., Laflamme P., Cotterchio M., Greenwood C. Germ-line DNA copy number variation frequencies in a large North American population. Hum. Genet. 2007;122:345–353. doi: 10.1007/s00439-007-0404-5. [DOI] [PubMed] [Google Scholar]
- 44.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., Wellcome Trust Case Control Consortium Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bierut L.J., Agrawal A., Bucholz K.K., Doheny K.F., Laurie C., Pugh E., Fisher S., Fox L., Howells W., Bertelsen S., Gene, Environment Association Studies Consortium A genome-wide association study of alcohol dependence. Proc. Natl. Acad. Sci. USA. 2010;107:5082–5087. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feuk L., Kalervo A., Lipsanen-Nyman M., Skaug J., Nakabayashi K., Finucane B., Hartung D., Innes M., Kerem B., Nowaczyk M.J. Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am. J. Hum. Genet. 2006;79:965–972. doi: 10.1086/508902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahn J.W., Mann K., Walsh S., Shehab M., Hoang S., Docherty Z., Mohammed S., Mackie Ogilvie C. Validation and implementation of array comparative genomic hybridisation as a first line test in place of postnatal karyotyping for genome imbalance. Mol. Cytogenet. 2010;3:9. doi: 10.1186/1755-8166-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toro R., Konyukh M., Delorme R., Leblond C., Chaste P., Fauchereau F., Coleman M., Leboyer M., Gillberg C., Bourgeron T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 50.Lee C., Scherer S.W. The clinical context of copy number variation in the human genome. Expert Rev. Mol. Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 51.Rowen L., Young J., Birditt B., Kaur A., Madan A., Philipps D.L., Qin S., Minx P., Wilson R.K., Hood L., Graveley B.R. Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]
- 52.Tabuchi K., Südhof T.C. Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics. 2002;79:849–859. doi: 10.1006/geno.2002.6780. [DOI] [PubMed] [Google Scholar]
- 53.Ramocki M.B., Zoghbi H.Y. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ullrich B., Ushkaryov Y.A., Südhof T.C. Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron. 1995;14:497–507. doi: 10.1016/0896-6273(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J., Feuk L., Duggan G.E., Khaja R., Scherer S.W. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet. Genome Res. 2006;115:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- 56.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.