

Abstract
Porencephaly is a neurological disorder characterized by fluid-filled cysts or cavities in the brain that often cause hemiplegia. It has been suggested that porencephalic cavities result from focal cerebral degeneration involving hemorrhages. De novo or inherited heterozygous mutations in COL4A1, which encodes the type IV α1 collagen chain that is essential for structural integrity for vascular basement membranes, have been reported in individuals with porencephaly. Most mutations occurred at conserved Gly residues in the Gly-Xaa-Yaa repeats of the triple-helical domain, leading to alterations of the α1α1α2 heterotrimers. Here we report on two individuals with porencephaly caused by a heterozygous missense mutation in COL4A2, which encodes the type IV α2 collagen chain. Mutations c.3455G>A and c.3110G>A, one in each of the individuals, cause Gly residues in the Gly-Xaa-Yaa repeat to be substituted as p.Gly1152Asp and p.Gly1037Glu, respectively, probably resulting in alterations of the α1α1α2 heterotrimers. The c.3455G>A mutation was found in the proband's mother, who showed very mild monoparesis of the left upper extremity, and the maternal elder uncle, who had congenital hemiplegia. The maternal grandfather harboring the mutation is asymptomatic. The c.3110G>A mutation occurred de novo. Our study confirmed that abnormalities of the α1α1α2 heterotrimers of type IV collagen cause porencephaly and stresses the importance of screening for COL4A2 as well as for COL4A1.
Main Text
Porencephaly (MIM 175780) is a neurological disorder characterized by fluid-filled cysts or cavities in the brain.1 It has been suggested that porencephalic cysts are caused by a disturbance of vascular supply leading to cerebral degeneration.2, 3 Porencephaly clinically causes hemiplegia (most often), tetraplegia, epilepsy, and intellectual disability.4, 5 Monozygous twinning, maternal cardiac arrest or abdominal trauma, a deficient protein C anticoagulant pathway, or cytomegalovirus infections are risk factors for sporadic porencephaly.2, 6 Recently, mutations in the gene encoding type IV collagen α1 chain (COL4A1 [MIM 120130]) have been shown to cause familial porencephaly.7 Since then, de novo and inherited COL4A1 mutations have been reported,8, 9, 10 confirming that COL4A1 abnormalities are involved in both sporadic and familial porencephaly. Type IV collagens are basement membrane proteins that are expressed in all tissues including the vasculature. COL4A1 (α1 chain) and COL4A2 (α2 chain) are the most abundant type IV collagens, and form heterotrimers with 2:1 stoichiometry (α1α1α2).11 A mouse model of the heterozygous COL4A1 mutation (Col4a1+/Δex40) showed cerebral hemorrhage and porencephaly and displayed abnormalities of vascular basement membranes, such as uneven edges, inconsistent density, and highly variable thickness.7 In addition, a dominant negative effect of the Col4a1+/Δex40 mutation was demonstrated on collagen IV α1α1α2 heterotrimer assembly and its secretion.7 In humans, most mutations are substitutions of the conserved Gly residue in the Gly-Xaa-Yaa repeat of the triple-helical domain, and they have a dominant negative effect on heterotrimer formation.11, 12
COL4A2 (MIM 120090), which encodes the type IV α2 collagen chain, is a possible candidate for porencephaly because its mutations may affect the α1α1α2 heterotrimer. Supporting this idea, osteogenesis imperfecta type I-IV (MIM 166200, 166210, 259420, and 166220), which is characterized by abnormal bone fragility and low bone mass, is caused by mutations in both COL1A1 (MIM 120150) and COL1A2 (MIM 120160) that may interfere with formation of the collagen I α1α1α2 heterotrimer.13 Moreover, mice lines harboring Col4a2 point mutations (Col4a2ENU415, c.227G>T [p.Val31Phe]; Col4a2ENU4003 and Col4a2ENU4020, c.2073G>A [p.Gly646Asp]) showed abnormalities of the lens, cornea, and vascular stability.14 In the brains of the mutants, pseudocysts in the upper cortical plate, hemorrhages surrounding small blood vessels, and focal hemorrhagic necroses were observed, indicating that Col4a2 mutations cause abnormalities of the cerebral vasculature similar to those caused by Col4a1 mutations.7, 14 In this study, we screened for COL4A2 mutations in 35 Japanese individuals with porencephaly. Substitutions of a Gly residue in the Gly-Xaa-Yaa repeat were identified in two individuals (individuals 1 and 2). Clinical information and peripheral blood samples were obtained from their family members after obtaining written informed consent. Experimental protocols were approved by the Institutional Review Board of Yokohama City University School of Medicine.
Individual 1 is 7 years old and a product of nonconsanguineous healthy parents (Figure 1A, arrow). There was no abdominal traumatism associated with the pregnancy and delivery in the mother. The individual was born at 36 weeks' gestation with a planned Caesarean section because, at 31 weeks' gestation, an antenatal ultrasound scan revealed an enlarged right lateral ventricle. Apgar scores were 9 at 1 min and 10 at 5 min. He weighed 2,900 g (+1.09 standard deviation [SD]) and had a head circumference of 32.5 cm (+0.05 SD). His early development was delayed with poor left hand use and abnormal leg movement. Brain magnetic resonance imaging (MRI) at 6 months showed an enlarged right lateral ventricle. Abrupt vomiting and nausea followed by motionless arrest developed at the 10 months. An electroencephalogram (EEG) showed focal spikes in the right frontal region, and carbamazepine treatment was initiated at the 12 months. Rehabilitation was started at 10 months. The individual started rolling at 12 months, crawling at 18 months, and walking alone at 3 years. He had spastic triplegia (diplegia and left hemiplegia) showing hemiplegic and diplegic gait with fluent speech and normal word comprehension. At the 5 years of age, he underwent orthopedic surgery for foot deformity due to spastic paresis. An EEG showed spikes in the right occipital to posterior temporal region and midcentral region. A brain MRI at age 6 showed an enlarged right lateral ventricle, reduced volume of the right frontal white matter, and atrophic right cerebral peduncle and body of corpus callosum (Figures 2A–2C). His intelligent quotient [IQ] score, evaluated at 6 years with Wechsler Intelligence Scale for Children-Third Edition (WISC-III), was 74 (his performance IQ was 69 and his verbal IQ was 82). The individual is now 7 years old and attending a local school. He can walk with ankle foot orthosis and hand assist. The epilepsy is well controlled with carbamazepine and clobazam. He does not show hematuria, muscular cramps, or ophthalmic abnormalities. His mother was born at term without asphyxia after an uneventful pregnancy. She had convulsions at the age of 18 months, and anticonvulsant was started under a diagnosis of focal epilepsy. Seizures were well controlled and treatment was discontinued at the age of 13. She first realized clumsiness of the left hand when she started learning piano and recorder at the age of 9. When she was a junior high school student, she felt severe headaches, and abnormal findings were pointed out in the brain MRI study (detailed information was unavailable). However, she did not undergo any more examinations because the headaches disappeared and did not recur. Neurological examination at 31 years revealed very mild monoparesis of the left upper extremity. She had neither spasticity nor exaggerated tendon reflexes. The grip power of her right and left hands was 25 and 15 kg, respectively. Mirror movement was observed on the right hand. The brain MRI revealed a mildly enlarged right lateral ventricle and high signal intensity around the enlarged ventricular wall on a Fluid Attenuated Inversion Recovery (FLAIR) image, which is consistent with mild porencephaly or periventricular venous infarction (Figures 2D–2F). MR angiography showed no aneurysms. Of note, his maternal elder uncle also showed congenital left hemiplegia with an assisted walk, and his maternal granduncle had also been afflicted by congenital hemiplegia, suggesting a genetic predisposition in the family (Figure 1A).
Figure 1.
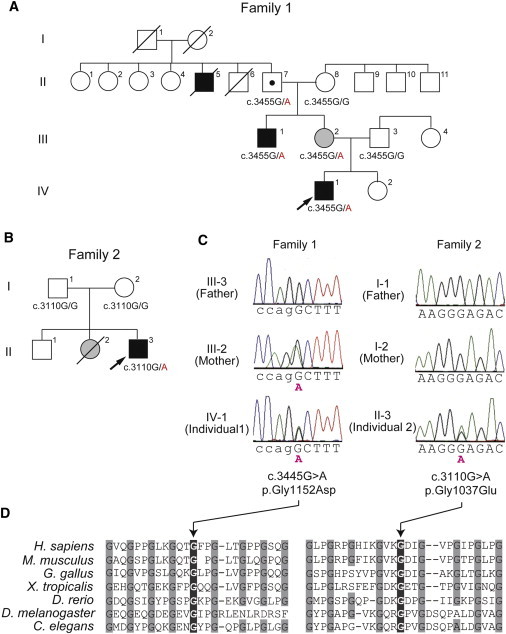
Pedigrees and COL4A2 Mutations in Individuals 1 and 2
Pedigrees of family 1(A) and family 2 (B). The arrows indicate the probands (Individual 1 in family 1 and individual 2 in family 2). The segregation of the COL4A2 mutations is shown. In family 1, the proband's mother (III-2) and maternal uncle (III-1) had mild monoparesis of the left upper extremity and congenital left hemiplegia and an assisted walk, respectively. The maternal grandfather (II-7) was healthy. The elder granduncle (II-5) was also afflicted by congenital hemiplegia and died in his 60s. (B) In family 2, the proband had a heterozygous mutation, but his parents did not have this mutation, indicating that the mutation occurred de novo. His elder sister (II-2) had intraventricular hemorrhage two days after birth but her DNA was unavailable.
(C) Electropherogram of family 1 (left) and family 2 (right). The intron and exon bases are in lower and upper cases, respectively. The c.3455G>A (p.Gly1152Asp) mutation in individual 1 was inherited from his mother. The c.3110G>A (p.Gly1037Glu) mutation in individual 2 occurred de novo.
(D) Multiple amino acid sequence alignments of COL4A2 proteins showing the evolutionarily conserved amino acids. The protein sequences obtained from the National Center for Biotechnology Information protein database are, NP_001837.2 (Homo sapiens), NP_034062.3 (Mus musculus), NP_001155862.1 (Gallus gallus), XP_002933063.1 (Xenopus tropicalis), XP_687811.5 (Danio rerio), AAB64082.1 (Drosophila melanogaster), and CAA80537.1 (Caenorhabditis elegans). The multiple sequence alignment was performed via the CLUSTALW website (see Web Resources). The positions of the conserved Gly residues in the Gly-X-Y repeats where the mutations occurred are highlighted with gray.
Figure 2.
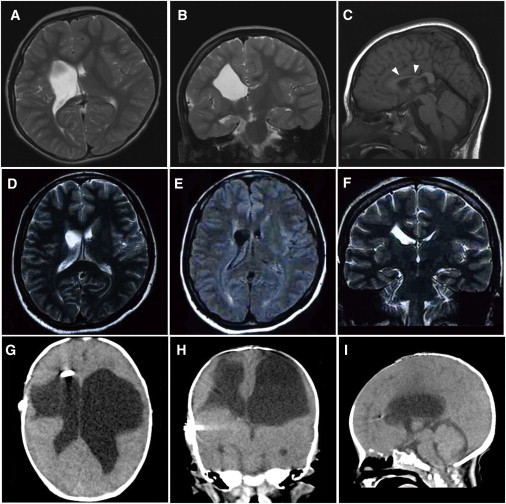
Brain Imaging in Individuals 1 and 2
(A–C) Brain MRIs of individual 1 at 6 years old; (A) T2-weighted axial image. (B) Coronal image. The images in (A) and (B) show an enlarged right lateral ventricle and a reduced volume of the right frontal white matter. (C) T1-weighted midline sagittal image showing atrophy of the body of the corpus callosum (arrowheads). The lesion responsible for the left leg paresis is not evident in these images.
(D–F) Brain MRIs of individual 1's mother at age 31. (D) T2-weighted axial and (F) coronal images show a mildly enlarged right lateral ventricle. (E) FLAIR axial image shows high signal intensity around the enlarged ventricular wall, which is consistent with mild porencephaly or periventricular venous infarction.
(G–I) CT images of individual 2 at 2 months of age. (G) Axial image. (H) Coronal image. (I) Sagittal image. The images in (G), (H), and (I) show an enlarged bilateral lateral ventricle and an extremely reduced volume of bilateral frontal white matter. The V-P shunt tube is also visible in the right lateral ventricle. The pontocerebellar structures seem to be normal.
Individual 2 is 1 year and 4 months old and a product of nonconsanguineous healthy parents (Figure 1B, arrow). There was no abdominal traumatism associated with the pregnancy and delivery in the mother. He was born at 35 weeks' gestation. His birth weight was 1,694 g (−2.36 SD) and his head circumference was 29 cm (−1.77 SD). Mild asphyxia was observed, and he had Apgar scores of 3 at 1 min and 7 at 5 min. An ultrasound scan at 6 hr after birth revealed a parenchymal hemorrhage of the right cerebral hemisphere with an enlarged left lateral ventricle. Because a blood test revealed significant increases in prothrombin time (29.3 s) and activated partial thromboplastin time (104.3 s), but not in D-dimer (0.7 μg/ml) at 1 day after birth, he was treated with a daily infusion of fresh frozen plasma for 12 days. At 37 days after birth, he underwent a ventricular-peritoneal shunt (V-P shunt) operation for progressive enlargement of the lateral ventricle. Computed tomography (CT) at 2 months of age showed an enlarged bilateral lateral ventricle and an extremely reduced volume of bilateral frontal white matter (Figures 2G–2I). Blood coagulation was normalized at 7 months. At the 7 months, the individual did not show any head control or rolling, and presented with abnormal posturing and spastic quadriplegia dominant on the left side of his body. With rehabilitation, he had full-range visual pursuit, a social smile, and incomplete head control. Although his spasticity improved, exaggerated deep tendon reflexes with synergic voluntary movement of the distal part of the extremities were recognized. An EEG at 1 year of age showed no epileptic discharges. His present developmental quotient is below 20. He did not show hematuria, muscular cramps, intracranial aneurysms, or cataracts. His elder sister was found to have an intraventricular hemorrhage two days after birth and underwent a V-P shunt. Her development was almost normal, and internal strabismus was noted. Unfortunately, she died in an accident at the age of four, and so her DNA was unavailable (Figure 1B).
Genomic DNA was isolated from peripheral blood leukocytes according to standard methods. DNA for mutation screening was amplified by illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare, Buckinghamshire, UK). The DNA of family members of individual 1 was isolated from saliva samples with Oragene (DNA Genotek Inc., Ontario, Canada). Exons 2 to 48 covering the entire COL4A2 coding region (GenBank accession number NM_001846.2) were examined by high-resolution melting curve (HRM) analysis or directly sequenced (for exon 46). The samples showing an aberrant melting curve pattern in the HRM analysis were sequenced. PCR primers and conditions are shown in Table S1, available online. All the mutations were verified with genomic DNA as a template. Two heterozygous mutations, c.3455G>A (p.Gly1152Asp) in individual 1 and c.3110G>A (p.Gly1037Glu) in individual 2, were identified. Both mutations occur at evolutionary conserved Gly residues in the Gly-X-Y repeats (Figure 1D), suggesting that the two mutations may alter the collagen IV α1α1α2 heterotrimers. These mutations were absent in 200 normal Japanese controls, and our evaluation with web-based prediction tools strongly suggested that these substitutions are pathogenic (Table S2). Screening for COL4A1 mutations was negative for both individuals (data not shown). The c.3455G>A mutation was found in the proband's mother and the maternal uncle, who showed very mild monoparesis of the left upper extremity and congenital left hemiplegia, respectively, and in maternal grandfather who is asymptomatic (Figures 1A and 1B). Therefore the c.3455G>A mutation can be considered as a pathogenic mutation with incomplete penetrance. The c.3110G>A mutation in individual 2 was not found in his parents, indicating that this mutation occurred de novo (Figure 1C).
Here we report two individuals with porencephaly who harbor COL4A2 mutations. In individual 2, the mutation occurred de novo. It is noteworthy that individual 2's elder sister also suffered from an intraventricular hemorrhage. A coincidental phenocopy in the sister is possible and would be consistent with de novo occurrence of the mutation. Alternatively, the sister might have the same mutation, which could be inherited from either one of the parents with a germline-mosaic mutation, though it was impossible to examine the sister because her sample is unavailable. Thus, with the present data, we concluded that the c.3110G>A mutation occurred de novo. On the other hand, the mutation in individual 1 was inherited from his mildly affected mother. In addition, congenital hemiplegia is observed in familial members of individual 1; the segregation of the c.3455G>A mutation is consistent with a dominant trait with incomplete penetrance. Such incomplete penetrance also has been reported in familial porencephalies with COL4A1 mutations,8, 9 suggesting that abnormalities of collagen IV α1α1α2 heterotrimers may conspire with other risk factors. The porencephalic cyst was unilateral in individual 1 and bilateral in individual 2, who required shunting, indicating variable severities caused by the different COL4A2 mutations. Most porencephalic cysts caused by COL4A1 mutations are unilateral;9 however, Meuwissen et al. recently reported de novo COL4A1 mutations in sporadic extensive bilateral porencephaly resembling hydranencephaly, indicating similar variable severities caused by COL4A1 mutations.10 Thus the involvement of COL4A1 and COL4A2 abnormalities should be considered in porencephaly and related pre- and perinatal cerebral hemorrhages, regardless of their severities.
It has been reported that COL4A1 mutations cause a variety of phenotypes, including porencephaly, infantile hemiplegia, and cerebral small vessel diseases involving both ischemic stroke and intracerebral hemorrhage with radiological features of lacunar infarction, and leukoaraiosis in adult individuals.9, 15, 16, 17, 18 The phenotypes in the central nervous system are often accompanied by ocular features (cataracts, retinal vessel tortuosity and hemorrhage, and defects of the anterior segment of the eye), nephropathy, and muscle cramps.9, 16, 17 Considering the common pathological mechanism between COL4A1 and COL4A2 mutations (abnormalities of collagen IV α1α1α2 heterotrimers), COL4A2 mutations also may be involved in small vessel diseases that can be manifested in adulthood. Supporting this idea, mice lines harboring Col4A2 point mutations showed cataracts, abnormalities of the lens and the cornea, and cerebral abnormalities.14 Thus it is important to identify mutations in both COL4A1 and COL4A2 in individuals with porencephaly as well as in asymptomatic carriers, for whom the prevention of stroke and genetic counseling are quite important. Identification of pathogenic mutations in individuals with porencephaly is of great interest for obstetricians and pediatricians, and for neurologists working for adult individuals.
In summary, we have identified mutations in COL4A2 as a genetic cause of both sporadic and familial porencephaly. Our data further support the importance of genetic testing in porencephaly and related pre- and perinatal cerebral hemorrhages for which the genetic predisposition is gradually being uncovered.
Acknowledgments
We would like to thank all the individuals and their families for their participation in this study. This work was supported by research grants from the Ministry of Health, Labour and Welfare (K.H., N. Miyake, H.O., M.K., N. Matsumoto, and H.S.), the Japan Science and Technology Agency (N. Matsumoto), the Strategic Research Program for Brain Sciences (N. Matsumoto), and a Grant-in-Aid for Scientific Research on Innovative Areas-(Foundation of Synapse and Neurocircuit Pathology)-from the Ministry of Education, Culture, Sports, Science and Technology of Japan (N. Matsumoto), a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (H.O., N. Matsumoto), a Grant-in-Aid for Young Scientist from Japan Society for the Promotion of Science (H.D., N. Miyake, H.S.) and a grant from the Takeda Science Foundation (N. Miyake and N. Matsumoto). This work has been done at the Advanced Medical Research Center, Yokohama City University, Japan.
Published online: December 29, 2011
Footnotes
Supplemental Data include two tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Clustal W, http://www.genome.jp/tools/clustalw/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Supplemental Data
References
- 1.Berg R.A., Aleck K.A., Kaplan A.M. Familial porencephaly. Arch. Neurol. 1983;40:567–569. doi: 10.1001/archneur.1983.04050080067013. [DOI] [PubMed] [Google Scholar]
- 2.Govaert P. Prenatal stroke. Semin. Fetal Neonatal Med. 2009;14:250–266. doi: 10.1016/j.siny.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Hunter A. In: Human Malformations and related Anomalies. Re S., Jg H., editors. Oxford University Press; New York: 2006. Porencephaly; pp. 645–654. [Google Scholar]
- 4.Mancini G.M., de Coo I.F., Lequin M.H., Arts W.F. Hereditary porencephaly: clinical and MRI findings in two Dutch families. Eur. J. Paediatr. Neurol. 2004;8:45–54. doi: 10.1016/j.ejpn.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Vilain C., Van Regemorter N., Verloes A., David P., Van Bogaert P. Neuroimaging fails to identify asymptomatic carriers of familial porencephaly. Am. J. Med. Genet. 2002;112:198–202. doi: 10.1002/ajmg.10452. [DOI] [PubMed] [Google Scholar]
- 6.Moinuddin A., McKinstry R.C., Martin K.A., Neil J.J. Intracranial hemorrhage progressing to porencephaly as a result of congenitally acquired cytomegalovirus infection—an illustrative report. Prenat. Diagn. 2003;23:797–800. doi: 10.1002/pd.688. [DOI] [PubMed] [Google Scholar]
- 7.Gould D.B., Phalan F.C., Breedveld G.J., van Mil S.E., Smith R.S., Schimenti J.C., Aguglia U., van der Knaap M.S., Heutink P., John S.W. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 8.Breedveld G., de Coo I.F., Lequin M.H., Arts W.F., Heutink P., Gould D.B., John S.W., Oostra B., Mancini G.M. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J. Med. Genet. 2006;43:490–495. doi: 10.1136/jmg.2005.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanfranconi S., Markus H.S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke. 2010;41:e513–e518. doi: 10.1161/STROKEAHA.110.581918. [DOI] [PubMed] [Google Scholar]
- 10.Meuwissen M.E., de Vries L.S., Verbeek H.A., Lequin M.H., Govaert P.P., Schot R., Cowan F.M., Hennekam R., Rizzu P., Verheijen F.W., et al. Sporadic COL4A1 mutations with extensive prenatal porencephaly resembling hydranencephaly. Neurology. 2011;76:844–846. doi: 10.1212/WNL.0b013e31820e7751. [DOI] [PubMed] [Google Scholar]
- 11.Khoshnoodi J., Pedchenko V., Hudson B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008;71:357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel J., Prockop D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991;20:137–152. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 13.Gajko-Galicka A. Mutations in type I collagen genes resulting in osteogenesis imperfecta in humans. Acta Biochim. Pol. 2002;49:433–441. [PubMed] [Google Scholar]
- 14.Favor J., Gloeckner C.J., Janik D., Klempt M., Neuhäuser-Klaus A., Pretsch W., Schmahl W., Quintanilla-Fend L. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–736. doi: 10.1534/genetics.106.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vahedi K., Alamowitch S. Clinical spectrum of type IV collagen (COL4A1) mutations: a novel genetic multisystem disease. Curr. Opin. Neurol. 2011;24:63–68. doi: 10.1097/WCO.0b013e32834232c6. [DOI] [PubMed] [Google Scholar]
- 16.Sibon I., Coupry I., Menegon P., Bouchet J.P., Gorry P., Burgelin I., Calvas P., Orignac I., Dousset V., Lacombe D., et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann. Neurol. 2007;62:177–184. doi: 10.1002/ana.21191. [DOI] [PubMed] [Google Scholar]
- 17.Alamowitch S., Plaisier E., Favrole P., Prost C., Chen Z., Van Agtmael T., Marro B., Ronco P. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology. 2009;73:1873–1882. doi: 10.1212/WNL.0b013e3181c3fd12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gould D.B., Phalan F.C., van Mil S.E., Sundberg J.P., Vahedi K., Massin P., Bousser M.G., Heutink P., Miner J.H., Tournier-Lasserve E., John S.W. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.