

Abstract
By using whole-exome sequencing, we identified a homozygous guanine-to-adenine transition at the invariant −1 position of the acceptor site of intron 1 (c.97−1G>A) in solute carrier organic anion transporter family member 2A1 (SLCO2A1), which encodes a prostaglandin transporter protein, as the causative mutation in a single individual with primary hypertrophic osteoarthropathy (PHO) from a consanguineous family. In two other affected individuals with PHO from two unrelated nonconsanguineous families, we identified two different compound heterozygous mutations by using Sanger sequencing. These findings confirm that SLCO2A1 mutations inactivate prostaglandin E2 (PGE2) transport, and they indicate that mutations in SLCO2A1 are the pathogenic cause of PHO. Moreover, this study might also help to explain the cause of secondary hypertrophic osteoarthropathy.
Main Text
Primary hypertrophic osteoarthropathy (PHO [MIM 167100]), also named pachydermoperiostosis, is a rare genetic disease that affects both skin and bones. PHO is characterized by clubbed nails, periostosis, acroosteolysis, painful joint enlargement, and skin manifestations that include thickened facial skin, a thickened scalp, and coarse facial features.1 Uppal et al.2 revealed that homozygous mutations in HPGD (MIM 601688), which encodes 15-hydroxyprostaglandin dehydrogenase (15-PGDH), cause PHO and that increased levels of prostaglandin E2 (PGE2) are involved in the pathogenesis of PHO. Subsequently, a number of cases of PHO and isolated, congenital clubbed nails were found to display mutations in HPGD.3, 4, 5, 6, 7, 8 However, no HPGD mutation was detected in either the proband (14-year-old boy) or the 19-year-old brother of the proband from a consanguineous Moroccan family affected by PHO.9 We also failed to identify an HPGD mutation in the proband from a consanguineous Chinese family affected by PHO. These findings support genetic heterogeneity of PHO and suggest that a potential causative genetic mutation is located in these affected individuals. Researchers have recently applied exome sequencing to rare heritable diseases in order to successfully identify causative genetic mutations.10, 11, 12, 13, 14 Here, we report on a homozygous guanine-to-adenine transition at the invariant −1 position of the acceptor site of intron 1 (c.97−1G>A) in solute carrier organic anion transporter family member 2A1 (SLCO2A1) (MIM 601460, NM_005630.2), which encodes a prostaglandin transporter protein, as the causative mutation in a single PHO-affected individual from a consanguineous family. In two other PHO-affected individuals from two unrelated nonconsanguineous families, we identify two different compound heterozygous mutations. These findings confirm that SLCO2A1 mutations inactivate PGE2 transport, and they indicate that mutations in SLCO2A1 are the pathogenic cause of PHO.
The pedigrees of the three Han Chinese families with PHO are shown in Figure 1. In family 1, a 24-year-old man (proband, family1-P1, IV.4 in family 1 in Figure 1) is the only son of consanguineous parents who are first cousins. In families 2 and 3, the probands, a 27-year-old man (family2-P2; III.5 in family 2 in Figure 1) and a 21-year-old man (family3-P3; III.1 in family 3 in Figure 1), respectively, are the only sons of nonconsanguineous parents. The disease was diagnosed in the affected persons with the use of clinical and radiological criteria. Disease onset in the three individuals occurred at 12–15 years of age and manifested as digital clubbing, swelling of the knees, periostosis, and a progressive thickening and furrowing of facial skin (Figure 2 and Figure S1, available online). The individual from family 2 had a stomach hemorrhage due to a gastric ulcer at 21 years of age. Physical examination revealed that the individuals had no other secondary hypertrophic osteoarthropathy, such as heart or lung abnormities, Graves disease, or inflammatory bowel disease. No other affected individuals were found in the three families. This study was approved by the ethics committee of the Shanghai Sixth People's Hospital, (affiliated with Shanghai Jiao Tong University). Written informed consent was obtained from all of the subjects who contributed DNA and clinical information to the study. Families 1, 2, and 3 were from Jiangsu, Anhui, and Shandong, respectively. The numbers of participating individuals from families 1, 2, and 3 were 16, 20, and 3, respectively (Figure 1). Venous blood samples were obtained from 39 individuals (three affected and 36 unaffected) from three unrelated PHO families. We extracted genomic DNA from whole blood by using standard methods. All subjects are of Chinese Han ethnicity.
Figure 1.
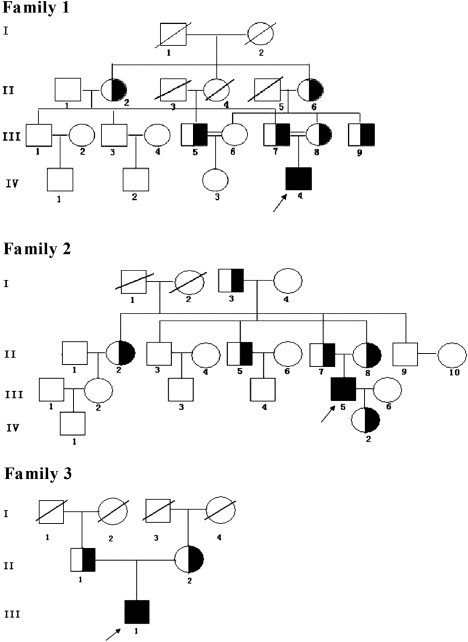
The Pedigrees of the Chinese Families Affected by PHO
Black symbols represent the affected individuals, open symbols represent the unaffected individuals, and the half-blackened symbols represent asymptomatic mutation carriers. Circles and squares indicate females and males, respectively. The arrows identify the probands in the families. Double lines indicate consanguineous marriages. All living individuals were the individuals available for genotyping in families 1, 2, and 3.
Figure 2.
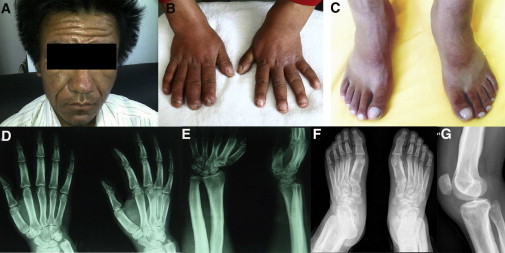
Clinical Images of the Affected Individual, Family1-P1
The images show the thickening and furrowing of facial skin (A) and the clubbing of fingernails and toenails (B and C). Hand radiographs show a loss of the normal tabulation of metacarpals and phalanges and cortical thickening of the metacarpals and the proximal and middle phalanges (D and E). A radiograph of the feet shows cortical thickening and acroosteolysis (F). A radiograph of a knee shows periosteal hyperostosis of the knee region and shows patellae sclerosis and sclerosis of both the distal femur and tibiofibula (G). All images are published with permission from the affected individual.
We first sequenced the exome of a single PHO-affected individual (family1-P1) from a consanguineous family. Exon-enriched DNA from the proband of family 1 was sequenced with the Illumina Genome Analyzer II platform according to the manufacturer's (Illumina's) instructions. The raw image files were processed with Illumina Base Calling Software 1.7 with default parameters, and the sequences of each individual were reported as 90 bp paired-end reads. Sequence reads were mapped to a reference genome (UCSC Genome Browser hg18 assembly) with SOAP2 (BGI-Shenzhen).15, 16, 17 The SOAPsnp results were filtered as follows: The base quality was equal to or more than 20, and the sequencing depth was between 4 and 200, whereas the estimated copy number was less than two, and the distance between two SNPs was more than 5 bp.12, 18 Approximately 1.26 gigabases (Gb) of high-quality data were aligned to the targeted regions for family1-P1 with a per-base mismatch rate of 0.37%, resulting in an average read depth of 33.53 for the individual exome (Table S1). We identified 31,738 SNPs, of which 7,038 were splice-site and nonsynonymous variants (SS-NSVs) in the target region (Table S2). By filtering the data with public SNP databases, including dbSNP129, the UCSC Genome Browser hg18 assembly, the 1000 Genomes Browser, and the YanHuang Genome Database (provided by the Beijing Genomics Institute), we identified 472 nonsynonymous (NS) and splice-site (SS) substitutions as candidate SNPs (Table S3). Because of the recessive nature of this consanguineous family affected by PHO, our subsequent validation analyses focused on homozygous nonsynonymous variants in coding sequences. In family1-P1, we identified 28 genes that contained homozygous missense, nonsense, or splicing mutations (Table S4). However, a thorough literature review suggested that among these genes, only SLCO2A1 appears to be a functional candidate. A previous study clearly demonstrated that SLCO2A1 is associated with the transport, clearance, and degradation of PGE2.19 Sanger sequencing confirmed a homozygous guanine-to-adenine transition at the invariant −1 position of the acceptor site of intron 1 (c.97−1G>A) in SLCO2A1 (Figure 3A) of the proband, and each of his healthy parents was found to have a heterozygous mutation at this site. We also confirmed by using Sanger sequencing that some relatives of the proband, including II-2, II-6, III-5, III-7, III-8, and III-9 in family 1, were heterozygous carriers (Figure 1). The identified mutation was not detected in chromosomes from 250 ethnically matched control individuals.
Figure 3.
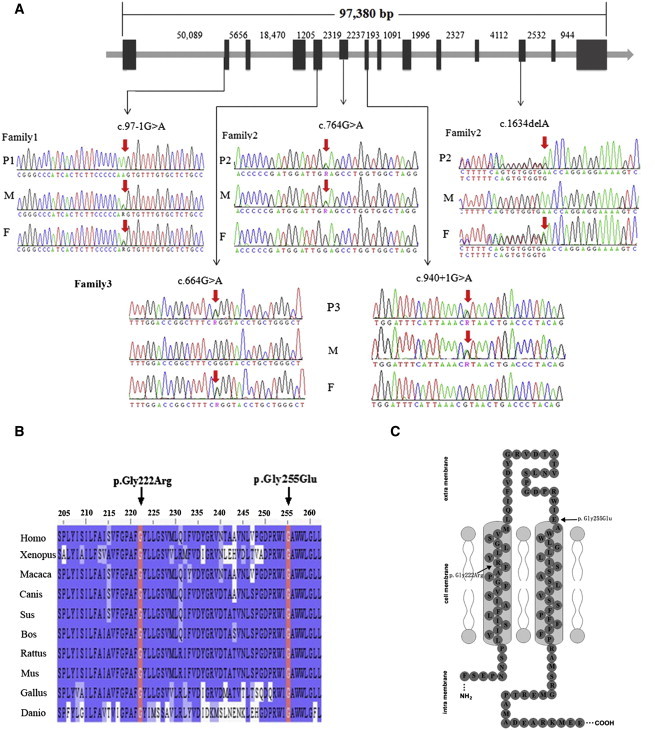
Locating and Sequencing the SLCO2A1 Mutations
(A) The location of the mutations identified in the SLCO2A1 gene. Family1-P1 (P1) had a homozygous mutation, and the father (F) and mother (M) were both heterozygous carriers. Family2-P2 (P2) and family3-P3 (P3) had compound heterozygous mutations, and their fathers and mothers were heterozygous carriers.
(B) Both the p.Gly222Arg and p.Gly255Glu heterozygous missense mutations occur at a highly conserved position in SLCO2A1, as shown by a comparison of the corresponding sequences of ten vertebrates. The bases that are identical to those in Homo sapiens are highlighted in blue. Abbreviations are as follows: Homo, Homo sapiens; Xenopus, Xenopus laevis; Macaca, Macaca mulatta; Canis, Canis lupus familiaris; Sus, Sus scrofa; Bos, Bos taurus; Rattus, Rattus norvegicus; Mus, Mus musculus; Gallus, Gallus gallus; and Danio, Danio rerio.
(C) Two missense mutations (p.Gly222Arg and p.Gly255Glu) in the transmembrane model of SLCO2A1. The mutations are indicated by arrows.
By using direct Sanger sequencing, we identified two unrelated PHO-affected individuals (family2-P2 and family3-P3) from two nonconsanguineous families as having compound heterozygous mutations in SLCO2A1: Family2-P2 had a missense mutation (c.764G>A [p.Gly255Glu]) in exon 6 combined with a frameshift mutation (c.1634delA) in exon 12, and family3-P3 had a missense mutation (c.664G>A [p.Gly222Arg]) in exon 5 combined with a guanine-to-adenine transition at the invariant +1 position of the acceptor site of intron 7 (c.940+1G>A) (Figure 3A). The single-base deletion in SLCO2A1 results in a frameshift and a premature termination codon (p.Asn545Thrfs∗15), and the c.940+1G>A mutation is predicted to cause an abnormal splicing event. The healthy parents of family2-P2 each carried either c.764G>A or c.1634delA as a heterozygous mutation, and we also confirmed that some relatives of family2-P2, including I-3, II-2, II-5, II-7, II-8, and IV-2, were heterozygous carriers (Figure 1). In addition, the healthy parents of family3-P3 were found to carry heterozygous mutations (c.664G>A or c.940+1G>A). Accordingly, all three genetic variants identified in the three PHO-affected individuals from three independent families were not spontaneous mutations, but rather were inherited. We detected none of the observed mutations in 250 unrelated, ethnically matched control individuals. These results indicate that in addition to HPGD mutations, the identified mutations in SLCO2A1 are the cause of PHO.
SLCO2A1 encodes an organic anion-transporting polypeptide (OATP2A1) (NP_005621.2) (named prostaglandin transporter [PGT]) that is located on chromosome 3q21. SLCO2A1 is organized into 14 exons that encode a 643 amino acid, 12-transmembrane-domain organic anion cell-surface transporter.19, 20, 21 SLCO2A1 is widely expressed in various peripheral tissues and in the brain of several mammalian species, including humans.19, 20, 21 To date, an exact understanding of the role of OATP2A1 in prostaglandin (PG) metabolism remains unclear. Many researchers have postulated three possible roles for OATP2A1.19, 20, 21, 22 First, OATP2A1 might mediate the efflux of newly synthesized PGs from cells. Second, OATP2A1 might mediate epithelial PG transport. Third, OATP2A1 could mediate PG clearance and degradation. Indeed, many studies have determined that the magnitude of the effects of PGs depends not only on their production but also on their metabolism. Kanai et al.20 identified OATP2A1 and found that OATP2A1 removes PGs from the extracellular compartment. PGT delivers PGs to cytoplasmic HPGD, resulting in their oxidation and inactivation.20, 23, 24 However, until now, no disease has been reported as being associated with a SLCO2A1 mutation. In this study, among the three individuals with PHO, we detected a homozygous splice-site mutation (c.97−1G>A), a compound heterozygous missense (c.664G>A [p.Gly222Arg]) plus splice-site (c.940+1G>A) mutation, and a compound heterozygous missense (c.764G>A [p.Gly255Glu]) plus frameshift (c.1634delA) mutation in SLCO2A1. Both the p.Gly222Arg and p.Gly255Glu heterozygous missense mutations are at highly conserved positions (Figure 3B) and are likely to be functionally damaging (Figure 3C). To illustrate the effects of the two missense mutations in exons, we modeled the structure of SLCO2A1 on the crystal structure of a glycerol-3-phosphate transporter (GlpT, PDB ID 1pw4). A molecular model of SLCO2A1 was constructed with the SWISS-MODEL server and Swiss-PdbViewer,25, 26 which retrieved the template structure, 1pw4, from PDB.27 The resulting models were evaluated on the basis of the energy values that were calculated with the MODELER28 and SWISS model-structure assessment tools. The mutant structure was also built with Swiss-PdbViewer.25, 26 PGE2 was docked in the SWISS dock server, which is based on the EADock DSS pipeline.29 Structural graphics and visualization were both based on PyMol and Swiss-PdbViewer. Although the identity was only 16%, all of the 12 helix regions were covered. According to the predicted structure, the mutant Gly222Arg is localized at helix 5 (H5). The p.Gly255Glu mutation is located in the connecting loop of H5 and the periplasmic end of helix 6 (H6). PGE2 docks to the modeled structure and appears to occupy a pocket similar to that of GlpT (Figure 4A). Two significant features of the p.Gly222Arg mutation could make the membrane transporter dysfunctional. First, the p.Gly222Arg mutation could disturb the positive electrostatic potential of the substrate-binding surface by adding an additional positively charged residue.27, 30 Second, the larger side chain could break the rocker switch of H127 by terminating the helical structure of H1 and changing the N-Cα dihedral angles (φ) of Tyr48 from −68.8° to −92.8° and the Cα-C angles (ψ) from −24.6° to 84.0° (Figures 4B and 4C). Although the p.Gly255Glu mutation is not predicted to affect the function of SLCO2A1, the mutation might alter the structure of the sixth transmembrane domain as a result of the substitution of a macromolecular hydrophilic amino acid (Glu) for a micromolecular hydrophobic amino acid (Gly). Furthermore, in the protein with the latter mutation, we found another heterozygous single-base-deletion (c.1634delA), which would result in a frameshift and a premature termination codon (p.Asn545Thrfs∗15). Therefore, we predicted that this single-base-deletion would lead to a loss of the eleventh and twelfth transmembrane domains. Similarly, although family3-P3 carried a predicted functional heterozygous p.G222R mutation, the mutation occurred in combination with a c.940+1G>A splice-site mutation. The latter mutation would result in the skipping of exon 7, leading to a frameshift and a premature termination codon (p.Arg288Glyfs∗6). To assess the effect of the c.97−1G>A mutation in SLCO2A1 on splicing, we amplified the cDNA fragments between exons 1 and 3 with RT-PCR of family1-P1 and the control individual. Total RNA was extracted from the conchal cartilage of family1-P1 and from the articular cartilage of the control individual's knee, respectively. The expected 340 bp fragment was only seen in the control individual, whereas an extra short 202 bp fragment was found in the affected individual. Furthermore, we confirmed that exon 2 was skipped, which lead to the loss of 46 acid residues in SLCO2A1 (p.Val33_Glu78del) (Figure S2). All the assay results were consistent with the conclusion that autosomal-recessive PHO can be caused by the loss of SLCO2A1 function as a result of either a homozygous mutation or compound heterozygous mutations.
Figure 4.
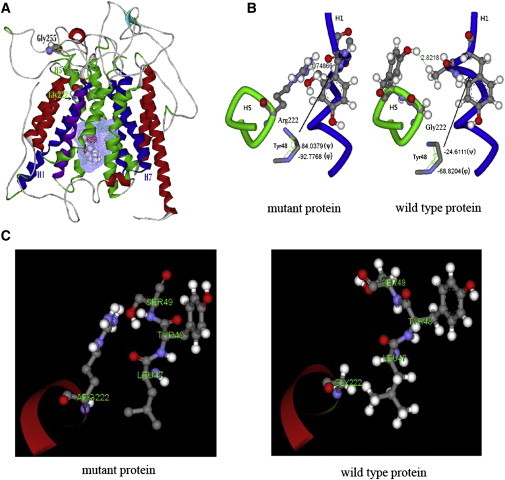
Protein Modeling of the SLCO2A1 p.Gly222Arg Mutation
(A) The wild-type structure of SLCO2A1. H1, H5, and H6 represent three of the twelve helices in the protein structure. Two amino acids of interest, Gly222 and Gly255, are labeled and shown in the Corey, Pauling, Koltun (CPK) model. The mesh grid surface and the CPK molecule represent docked PGE2 in the middle of the structure.
(B and C) The effect of the p.Gly222Arg mutant on the interaction between H1 and H5. Gly222 of H5 and the neighboring portion of H1 are shown in green and blue, respectively. The shortest distances between these two parts of H1 and H5 are 1.74 Å and 2.82 Å at the labeled positions in the p.Gly222Arg mutant and wild-type proteins, respectively. The symbols Φ and ψ represent the dihedral angles of Tyr48 in H1.
Uppal et al.2 revealed that PHO pathogenesis occurs through increased levels of PGE2. In this study, plasma and urine samples were collected from three affected individuals and 36 unaffected individuals from three unrelated families. Early-morning plasma and urine samples were collected after the individuals had fasted, and the samples were immediately frozen. The concentrations of PGE2 and 13,14 dihydro,15 keto metabolites (PGE-M) were measured in three assays at different sample dilutions with competitive enzyme-linked immunosorbent assays (Cayman Chemical), and data were analyzed with the manufacturer's analysis tools. We observed that urinary PGE2 levels were significantly elevated in the three affected individuals, who were found to have PGE2 levels that were sixteen times higher than those of the unaffected controls. However, no significant difference in urinary PGE2 levels was found between the heterozygous individuals from families 1 and 2 and the unaffected controls (Table 1). Unexpectedly, we found that the concentration of urinary PGE-M was significantly higher in the three affected individuals than in the heterozygous carriers and unaffected controls. However, compared with the levels in the unaffected control individuals, the urinary PGE-M concentrations in the three affected individuals were only six times higher. We also found that plasma PGE-M levels were higher in the affected individuals than in the heterozygous carriers and unaffected controls. Our results differ from those associated with homozygous HPGD mutations, which cause a significant increase in urinary PGE2 levels and a decrease in urinary PGE-M levels in affected individuals.2 The difference might be attributable to the fact that the affected individuals with SLCO2A1 mutations had normal 15-PGDH function, but this speculation will require further investigation. Our findings demonstrate that in affected individuals with SLCO2A1 mutations, a portion of PGE2 is cleared and degraded, which indicates the inactivation of PGE2 transport by SLCO2A1. To determine the role of PGT in ductus arteriosus closure, Chang et al.31 used global PGT-knockout mice (PGT−/−). They observed that urinary PGE2 levels were significantly higher in PGT−/− mice than in PGT+/+ mice and that plasma PGE2 metabolite concentrations decreased significantly in PGT−/− mice. These results are consistent with the fact that PGT-null mice fail to metabolize PGE2. However, they did not describe the pathogenesis of PHO in PGT−/− mice. Clarifying whether the PHO phenotype is indeed restricted to humans with a loss of SLCO2A1 will require detailed bone analyses in PGT−/− mice. Our findings are partially consistent with the above results, and the affected individuals with mutations showed significantly higher urinary PGE2 concentrations and relatively low plasma PGE-M levels. In fact, patent ductus arteriosus (PDA), as well as other congenital heart defects, is common in affected individuals with HPGD mutations. However, by using echocardiography, we found neither PDA nor other heart defects in the three affected individuals with SLCO2A1 mutations. We found that the three affected individuals with SLCO2A1 mutations seemed to be the most severely affected in the skin, bones, and knees; however, HPGD mutations were associated with mild PHO, characterized by either the absence of pachydermia or the presence of mild periostosis.3, 7, 9 Of course, to assess phenotypic differences between affected individuals with HPGD mutations and those with SLCO2A1 mutations, we need more cases with SLCO2A1 mutations.
Table 1.
Measurement of the Concentration of Urinary PGE2, Urinary PGE-M, and Plasma PGE-M in the Affected Individuals, the Heterozygous Carriers, and the Unaffected Controls
| Urinary PGE2 (ng/mmol creatinine) | Urinary PGE-M (ng/mmol creatinine) | Plasma PGE-M (pg/ml) | |
|---|---|---|---|
| Affected individuals (n = 3) | 439.3 ± 234.9 | 200.0 ± 123.1 | 46.9 ± 14.4 |
| Heterozygous carriers (n = 10) | 30.0 ±20.1 | 18.9 ± 6.5 | 18.2 ± 10.5 |
| Unaffected controls (n = 5) | 26.8 ± 7.5 | 30.9 ± 16.9 | 20.2 ± 7.7 |
| p value | 0.003 | 0.000 | 0.044 |
Three separate early-morning urine samples were measured in the individuals with homozygous or compound heterozygous SLCO2A1 mutations and the unaffected controls. The standard deviations represent the intra-assay variation of the three assays with different sample dilutions.
Although Uppal et al. found the pathogenic mechanism of PHO to be associated with increased concentrations of plasma PGE2,2 the cause of secondary hypertrophic osteoarthropathy (SHO) has not been completely elucidated. Coggins et al.32 said that explaining the pathogenesis of SHO without a primary insult to the lungs or pulmonary blood flow, as is found in Graves disease and inflammatory bowel disease, is very difficult to do on the basis of the HPGD mutation in PHO. Our findings suggest that the dysregulated levels of PGE2 in Graves disease and inflammatory bowel disease might be due to the inactivation of SLCO2A1 rather than to the loss of 15-PGDH function. Graves disease and inflammatory bowel disease are autoimmune disorders, and many previous studies have shown the dysregulation of PGE2 in these diseases by using in vitro and in vivo techniques.33, 34 In fact, this PGE2 transport is the rate-limiting step of PGE2 inactivation, at least in the lungs.35 We speculate that the production of cytokines in inflammatory cells in Graves disease and inflammatory bowel disease might influence the SLCO2A1-associated transmembrane transport of PGE2. Therefore, determining whether SHO associated with Graves disease or inflammatory bowel disease is caused by inactivated SLCO2A1 resulting from either mutations in SLCO2A1 or other causes will require further study.
In summary, our findings confirm that SLCO2A1 mutations inactivate PGE2 transport, and they indicate that mutations in SLCO2A1 are the pathogenic cause of PHO. Moreover, these results might also help to explain the cause of SHO. This study shows the advantage of using exome sequencing to identify genetic mutations associated with inherited disorders, especially in cases of small families and sporadic occurrence.
Acknowledgments
We are grateful to our patients and their families for participating in this study. We acknowledge technical assistance from Shanghai Genesky Bio-Teck Co., Ltd. This study was supported by the National Natural Science Foundation of China (81170803, 81070692, 81000360, 30771019, and 30800387), the Program of Shanghai Subject Chief Scientist (08XD1403000), the Science and Technology Commission of Shanghai Municipality (10D21950100 and 08411963100), and the Shanghai Rising-Star Program (11QA1404900). We wish to thank two anonymous reviewers for their helpful comments, which improved the manuscript and aided our further research.
Published online: December 22, 2011
Footnotes
The supplemental Data include two figures and four tables and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Browser, http://browser.1000genomes.org/
NimbleGen Sequence Capture Human Exome 2.1M Array, http://www.nimblegen.com/products/seqcap/arrays/exome/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
RefSeq 36.3, ftp://ftp.ncbi.nih.gov/genomes/MapView/Homo_sapiens/sequence/BUILD.36.3
UCSC Genome Browser, http://genome.ucsc.edu/
YanHuang Project, http://yh.genomics.org.cn/
Supplemental Data
References
- 1.Castori M., Sinibaldi L., Mingarelli R., Lachman R.S., Rimoin D.L., Dallapiccola B. Pachydermoperiostosis: an update. Clin. Genet. 2005;68:477–486. doi: 10.1111/j.1399-0004.2005.00533.x. [DOI] [PubMed] [Google Scholar]
- 2.Uppal S., Diggle C.P., Carr I.M., Fishwick C.W., Ahmed M., Ibrahim G.H., Helliwell P.S., Latos-Bieleńska A., Phillips S.E., Markham A.F., et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat. Genet. 2008;40:789–793. doi: 10.1038/ng.153. [DOI] [PubMed] [Google Scholar]
- 3.Tariq M., Azeem Z., Ali G., Chishti M.S., Ahmad W. Mutation in the HPGD gene encoding NAD+ dependent 15-hydroxyprostaglandin dehydrogenase underlies isolated congenital nail clubbing (ICNC) J. Med. Genet. 2009;46:14–20. doi: 10.1136/jmg.2008.061234. [DOI] [PubMed] [Google Scholar]
- 4.Yüksel-Konuk B., Sırmacı A., Ayten G.E., Özdemir M., Aslan İ., Yılmaz-Turay Ü., Erdoğan Y., Tekin M. Homozygous mutations in the 15-hydroxyprostaglandin dehydrogenase gene in patients with primary hypertrophic osteoarthropathy. Rheumatol. Int. 2009;30:39–43. doi: 10.1007/s00296-009-0895-6. [DOI] [PubMed] [Google Scholar]
- 5.Seifert W., Beninde J., Hoffmann K., Lindner T.H., Bassir C., Aksu F., Hübner C., Verbeek N.E., Mundlos S., Horn D. HPGD mutations cause cranioosteoarthropathy but not autosomal dominant digital clubbing. Eur. J. Hum. Genet. 2009;17:1570–1576. doi: 10.1038/ejhg.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diggle C.P., Carr I.M., Zitt E., Wusik K., Hopkin R.J., Prada C.E., Calabrese O., Rittinger O., Punaro M.G., Markham A.F., Bonthron D.T. Common and recurrent HPGD mutations in Caucasian individuals with primary hypertrophic osteoarthropathy. Rheumatology (Oxford) 2010;49:1056–1062. doi: 10.1093/rheumatology/keq048. [DOI] [PubMed] [Google Scholar]
- 7.Sinibaldi L., Harifi G., Bottillo I., Iannicelli M., El Hassani S., Brancati F., Dallapiccola B. A novel homozygous splice site mutation in the HPGD gene causes mild primary hypertrophic osteoarthropathy. Clin. Exp. Rheumatol. 2010;28:153–157. [PubMed] [Google Scholar]
- 8.Bergmann C., Wobser M., Morbach H., Falkenbach A., Wittenhagen D., Lassay L., Ott H., Zerres K., Girschick H.J., Hamm H. Primary hypertrophic osteoarthropathy with digital clubbing and palmoplantar hyperhidrosis caused by 15-PGHD/HPGD loss-of-function mutations. Exp. Dermatol. 2011;20:531–533. doi: 10.1111/j.1600-0625.2011.01248.x. [DOI] [PubMed] [Google Scholar]
- 9.Harifi G., Brancati F., Dallapicola B., El Hassani S. Primary hypertrophic osteoarthropathy: A new family supporting genetic heterogeneity. Joint Bone Spine. 2011;78:218–219. doi: 10.1016/j.jbspin.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A., et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C., et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Y., Li Y., Zhang D., Zhang H., Li Y., Lu F., Liu X., He F., Gong B., Cai L., et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 2011;7:e1002084. doi: 10.1371/journal.pgen.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puente X.S., Quesada V., Osorio F.G., Cabanillas R., Cadiñanos J., Fraile J.M., Ordóñez G.R., Puente D.A., Gutiérrez-Fernández A., Fanjul-Fernández M., et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 2011;88:650–656. doi: 10.1016/j.ajhg.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Greef J.C., Wang J., Balog J., den Dunnen J.T., Frants R.R., Straasheijm K.R., Aytekin C., van der Burg M., Duprez L., Ferster A., et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011;88:796–804. doi: 10.1016/j.ajhg.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi X., Liang Y., Huerta-Sanchez E., Jin X., Cuo Z.X., Pool J.E., Xu X., Jiang H., Vinckenbosch N., Korneliussen T.S., et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R., Li Y., Kristiansen K., Wang J. SOAP: Short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 17.Li Y., Vinckenbosch N., Tian G., Huerta-Sanchez E., Jiang T., Jiang H., Albrechtsen A., Andersen G., Cao H., Korneliussen T., et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 18.Wang J.L., Yang X., Xia K., Hu Z.M., Weng L., Jin X., Jiang H., Zhang P., Shen L., Guo J.F., et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–3518. doi: 10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- 19.Lu R., Kanai N., Bao Y., Schuster V.L. Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT) J. Clin. Invest. 1996;98:1142–1149. doi: 10.1172/JCI118897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanai N., Lu R., Satriano J.A., Bao Y., Wolkoff A.W., Schuster V.L. Identification and characterization of a prostaglandin transporter. Science. 1995;268:866–869. doi: 10.1126/science.7754369. [DOI] [PubMed] [Google Scholar]
- 21.Schuster V.L. Prostaglandin transport. Prostaglandins Other Lipid Mediat. 2002;68-69:633–647. doi: 10.1016/s0090-6980(02)00061-8. [DOI] [PubMed] [Google Scholar]
- 22.Schuster V.L. Molecular mechanisms of prostaglandin transport. Annu. Rev. Physiol. 1998;60:221–242. doi: 10.1146/annurev.physiol.60.1.221. [DOI] [PubMed] [Google Scholar]
- 23.Nomura T., Lu R., Pucci M.L., Schuster V.L. The two-step model of prostaglandin signal termination: In vitro reconstitution with the prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol. Pharmacol. 2004;65:973–978. doi: 10.1124/mol.65.4.973. [DOI] [PubMed] [Google Scholar]
- 24.Chi Y., Khersonsky S.M., Chang Y.T., Schuster V.L. Identification of a new class of prostaglandin transporter inhibitors and characterization of their biological effects on prostaglandin E2 transport. J. Pharmacol. Exp. Ther. 2006;316:1346–1350. doi: 10.1124/jpet.105.091975. [DOI] [PubMed] [Google Scholar]
- 25.Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 26.Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y., Lemieux M.J., Song J., Auer M., Wang D.N. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301:616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 28.Fiser A., Sali A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 29.Grosdidier A., Zoete V., Michielin O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins. 2007;67:1010–1025. doi: 10.1002/prot.21367. [DOI] [PubMed] [Google Scholar]
- 30.Meier-Abt F., Mokrab Y., Mizuguchi K. Organic anion transporting polypeptides of the OATP/SLCO superfamily: Identification of new members in nonmammalian species, comparative modeling and a potential transport mode. J. Membr. Biol. 2005;208:213–227. doi: 10.1007/s00232-005-7004-x. [DOI] [PubMed] [Google Scholar]
- 31.Chang H.Y., Locker J., Lu R., Schuster V.L. Failure of postnatal ductus arteriosus closure in prostaglandin transporter-deficient mice. Circulation. 2010;121:529–536. doi: 10.1161/CIRCULATIONAHA.109.862946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coggins K.G., Coffman T.M., Koller B.H. The Hippocratic finger points the blame at PGE2. Nat. Genet. 2008;40:691–692. doi: 10.1038/ng0608-691. [DOI] [PubMed] [Google Scholar]
- 33.Zifroni A., Treves A.J., Sachar D.B., Rachmilewitz D. Prostanoid synthesis by cultured intestinal epithelial and mononuclear cells in inflammatory bowel disease. Gut. 1983;24:659–664. doi: 10.1136/gut.24.7.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konuk E.B., Konuk O., Misirlioglu M., Menevse A., Unal M. Expression of cyclooxygenase-2 in orbital fibroadipose connective tissues of Graves' ophthalmopathy patients. Eur. J. Endocrinol. 2006;155:681–685. doi: 10.1530/eje.1.02280. [DOI] [PubMed] [Google Scholar]
- 35.Ivanov A.I., Romanovsky A.A. Prostaglandin E2 as a mediator of fever: Synthesis and catabolism. Front. Biosci. 2004;9:1977–1993. doi: 10.2741/1383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.