

Abstract
Internalization of Staphylococcus aureus in bovine endothelial cells (BEC) is increased by tumor necrosis factor alpha stimulation and NF-κB activation. Because the phosphoinositide-3-kinase (PI3K)–Akt signaling pathway also modulates NF-κB activity, we considered whether the internalization of S. aureus by BEC is associated with the activity of PI3K and Akt. We found a time- and multiplicity of infection-dependent phosphorylation of Akt on Ser473 in BEC infected with S. aureus. This phosphorylation was inhibited by LY294002 (LY), indicating the participation of PI3K. Inhibition of either PI3K with LY or wortmannin, or Akt with SH-5, strongly reduced the internalization of S. aureus. Transfection of BEC with a dominant-negative form of the Akt gene significantly decreased S. aureus internalization, whereas transfection with the constitutively active mutant increased the number of internalized bacterium. Inhibition of PDK1 activity with OSU-03012 did not affect the level of S. aureus internalization, demonstrating that phosphorylation of Akt on Thr308 is not important for this process. Compared to the untreated control, the adherence of S. aureus to the surface of BEC was unaltered when cells were transfected or incubated with the pharmacological inhibitors. Furthermore, Akt activation by internalized S. aureus triggered a time-dependent phosphorylation of glycogen synthase kinase-3α (GSK-3α) on Ser21 and GSK-3β on Ser9 that was partially inhibited with SH-5. Finally, treatment of BEC with LY prior to S. aureus infection inhibited the NF-κB p65 subunit phosphorylation on Ser536, indicating the involvement of PI3K. These results suggest that PI3K-Akt activity is important for the internalization of S. aureus and phosphorylation of GSK-3α, GSK-3β, and NF-κB.
INTRODUCTION
Staphylococcus aureus is a Gram-positive bacterium widely distributed among humans and animals. In humans, S. aureus causes a variety of illnesses ranging from minor skin and soft tissue infections to life-threatening diseases such as endovascular infections, pneumonia, septic arthritis, endocarditis, osteomyelitis, and sepsis (21). This bacterium can also infect animals that serve as reservoirs with zoonotic implications (34). In bovine cattle, S. aureus is the main pathogenic bacterium causing mastitis, a disease characterized by mammary gland inflammation (45, 46) that causes important economic losses to dairy producers and represents a risk to the consumers because of contaminated milk.
S. aureus has been traditionally considered an extracellular bacterium, but different reports have demonstrated its ability to invade an array of nonprofessional phagocytic cells such as bovine epithelial cells (5), human and bovine endothelial cells (27, 42, 47), and human osteoblasts (9). This intracellular location potentially contributes to bacterial persistence in different diseases, evasion of the immune response, and protection from antibiotics activity. Efforts have been made to elucidate the molecular mechanisms that S. aureus uses to be internalized by its host cells. Different reports have pointed out that binding of the S. aureus fibronectin-binding protein to the integrin dimer α5β1 plays an important role in the internalization process (40, 49, 57). The data obtained with kidney epithelial cells and fibroblasts showed that S. aureus internalization requires polymerization of the actin cytoskeleton and the activation of the enzymes focal adhesion kinase (FAK) and Src (1, 2, 24). Moreover, internalized S. aureus has been shown to cause the recruitment of Rab5 in a cyclic and alternating manner, as well as the participation of tensin (54). Although the signaling pathway downstream of FAK activation has not been studied in detail, autophosphorylation of FAK on Y397 induced by S. aureus is the binding site for phosphoinositide-3-kinase (PI3K) and Src enzymes through their Src-homology 2 (SH2) domain (16).
The PI3K-Akt is a signaling pathway that is important in phagocytosis, regulation of the inflammatory response, and other activities, including vesicle trafficking and cytoskeletal reorganization (22, 63). PI3K is a heterodimeric protein with lipid kinase activity constituted by a catalytic subunit of 110 kDa (p110) and a regulatory subunit of 85 kDa (p85). When a ligand (i.e., growth factors or a bacterial molecular structure) binds to the cognate plasma membrane receptor, the SH2 domain of p85 recognizes the phosphorylated tyrosines on the cytosolic domain of the receptor. This causes an allosteric activation of p110 and the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) that is recognized by the enzymes Akt and the constitutively active 3′-phosphoinositide-dependent kinase 1 (PDK1) through their plekstrin homology domains (25). The interaction of Akt with PIP3 causes a change in the Akt conformation and phosphorylation of the residues Thr308 and Ser473 by PDK1 (4) and rictor-mTOR complex (53), respectively. Phosphorylation of these two residues causes the activation of Akt which in turn phosphorylates, among other substrates, the enzyme glycogen synthase kinase-3 (GSK-3). This enzyme is present in two constitutively active isoforms GSK-3α and GSK-3β that are structurally related but functionally nonredundant (17). Inactivation of GSK-3 is observed when the residues Ser21 in GSK-3α or Ser9 in GSK-3β, located in their regulatory N-terminal domains, are phosphorylated by Akt and other kinases (6, 7). Inhibition of GSK-3 by phosphorylation is important for the modulation of the inflammation and phagocytosis processes (13, 39).
Although several studies using different bacteria or bacterial virulence factors have documented the activation of the PI3K-Akt signaling pathway (3, 7, 20, 30, 33, 38, 50, 60), NF-κB (18, 20, 30, 38), and more recently GSK-3 (11, 18, 43, 44), none of them has reported the participation of the PI3K-Akt signaling pathway in the internalization of S. aureus. We have recently demonstrated that internalization of S. aureus by bovine endothelial cells (BEC) was increased by the soluble proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) through a process associated with the NF-κB activity state (47). However, the signaling pathway activated during the internalization of S. aureus was not elucidated. We show here that the number of S. aureus internalized by BEC decreased when cells were pretreated with specific inhibitors of PI3K and Akt, implying that activation of both enzymes is required for S. aureus internalization without affecting its adherence to the cell surface. In addition, confirmation of the results observed with the pharmacological inhibitors was obtained in BEC expressing a dominant-negative form of the Akt gene. Interestingly, activation of the PI3K-Akt signaling pathway by S. aureus produced a time-dependent phosphorylation of GSK-3α/GSK-3β and NF-κB p65 subunit that was blocked by specific inhibitors of Akt and PI3K, respectively. The data presented in this study indicate that internalization of S. aureus by BEC is a process associated with the activation of the PI3K-Akt signaling pathway, as well as with the PI3K-Akt-dependent phosphorylation of GSK-3α, GSK-3β, and NF-κB.
MATERIALS AND METHODS
Media and chemicals.
Ham F-12 (HF-12), Dulbecco modified Eagle medium, trypsin-EDTA, wortmannin (W), LY294002 (LY), Bradford reagent, bovine serum albumin (BSA), and lysostaphin were purchased from Sigma-Aldrich, Inc. (St. Louis, MO). Fetal calf serum (FCS) was acquired from Equitech-Bio, Inc. (Kerrville, TX). Penicillin G, streptomycin, and reduced serum Opti-MEM I medium were purchased from Gibco-BRL (Gaithersburg, MD). SH-5 was acquired from Enzo Life Sciences (Plymouth, PA), and OSU-03012 (OSU) was purchased from Cedarlane Labs (Burlington, NC). FuGENE transfection reagent and the 50× EDTA-free protease inhibitor cocktail were purchased from Roche Applied Science (Manheim, Germany).
Antibodies and plasmids.
Rabbit monoclonal antibodies against phospho-Akt (Ser473 and Thr308), phospho-GSK-3α (Ser21), and GSK-3β, as well as the polyclonal antibodies against phospho-GSK-3β (Ser9), NF-κB p65, and phospho-p65 (Ser536) were purchased from Cell Signaling Technology (Boston, MA). Antibodies against hemagglutinin (HA) tag (rat) and calnexin (rabbit) were acquired from Roche and Sigma, respectively. pCMV5 and plasmids containing the constitutively active (pCMV5-Akt-CA) and dominant-negative (pCMV5-Akt-DN) forms of the Akt gene were purchased from Addgene (Cambridge, MA).
Bacterial strain, cell line, and culture conditions.
The strain of S. aureus, isolated from a clinical case of bovine mastitis, was obtained from the American Type Culture Collection (ATCC 27543). Bacteria were cultured overnight in 3 ml of Luria-Bertani (LB) medium at 37°C with continuous agitation. The inoculum for infection assays was prepared by adding 1 ml of this preculture to 49 ml of LB medium and grown at 37°C until the culture reached the initial-middle log phase (optical density at 600 nm of 0.3).
The endothelial cell line used was obtained from bovine umbilical veins and immortalized by transfection with an expression vector containing the E6E7 oncogenes of human papillomavirus 16 (BVE-E6E7) (14). This immortalized bovine endothelial cell line, referred to as BEC here, was grown and maintained in HF-12 supplemented with 10% FCS unless otherwise noted.
Bacterial internalization and attachment assays.
Quantitative analysis of intracellular S. aureus was done essentially as described by Oviedo-Boyso et al. (47) with minor modifications. In brief, 0.5 × 106 cells per well were seeded in 24-well plates (Corning-Costar, Inc., Corning, NY) in a medium containing 2 ml of HF-12 supplemented with 10% FCS, 100 U of penicillin G/ml, and 100 μg of streptomycin/ml and then cultured at 37°C in 5% CO2 and 95% air up to a confluence of 90 to 100%. For internalization assays, BEC were washed three times with phosphate-buffered saline (PBS) to remove FCS and antibiotics and then infected with 107 CFU of S. aureus/ml, which gives a multiplicity of infection (MOI) of 20. Then, infected cells were immediately centrifuged at 130 × g for 5 min, followed by incubation at 37°C for 40 min. After infection, extracellular noninternalized S. aureus was killed by incubation of BEC with 5 μg of lysostaphin/ml for 20 min, and the cells were washed three times with PBS, lifted with 250 μl of 0.25% trypsin–0.5 mM EDTA, and recovered by centrifugation at 3,500 rpm for 12 min in an Eppendorf centrifuge. The supernatant was discarded, and BEC were lysed by hypotonic shock in 250 μl of sterile deionized water containing 0.1% Triton X-100. Intracellular bacteria were cultured in LB agar at 37°C for 19 to 24 h, and the number of S. aureus CFU/ml was calculated by the counting plate technique. For the adherence assays, the procedure was identical except that the incubation of BEC with lysostaphin was omitted. Because in this case the number of CFU represented internalized and adherent S. aureus, we calculated the number of adherent bacteria by subtracting the number of intracellular ones to the total CFU counted for each condition tested. To test the effect of inhibitors on the internalization and adherence of S. aureus, BEC were preincubated for 30 min with LY294002 (LY), W, and SH-5 and for 15 min with OSU and then infected with bacteria in the presence of the inhibitors. The intracellular and adherent bacteria were recovered, cultured, and calculated according to the procedure described. The viability of BEC, evaluated by the trypan blue technique, was >95% in the presence of 50 μM LY, 100 nM W, 10 μM SH-5, or 2 μM OSU.
Transient transfection of BEC.
Cells were grown in 24-well plates to 60 to 70% confluence, and the culture medium was changed to HF-12 plus 10% FCS. Then, in order to have a similar protein expression 5 ng of pCMV5-Akt-CA or 200 ng of pCMV5-Akt-DN in 1.2 μl of FuGENE transfection reagent (ratio, 4:1 [FuGENE-plasmid]) were added to BEC in reduced serum Opti-MEM I medium according to the manufacturer's instructions. As a control, BEC were transfected with 300 ng of pCMV5. To maintain a constant amount of DNA in transfection assays, pCMV5 was added to pCMV5-Akt-CA or pCMV5-Akt-DN transfection mixtures to have a final amount of 300 ng of total DNA. After 24 h of incubation at 37°C in 5% CO2, we performed the internalization and the Western blot assays to quantitate the number of intracellular S. aureus and the level of expression of the two Akt mutants, respectively.
Protein extraction and Western blot analysis.
To test for the relative abundance of phosphorylated and nonphosphorylated proteins, BEC were grown in six-well culture plates to ca. 90% confluence before serum starvation for at least 4 h. In control and treated cells, the total protein (cytosolic plus nuclear) was obtained by washing the cells twice with cold PBS and lysing them with 80 μl of a cold lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Igepal CA-930, 10 mM sodium pyrophosphate, and 50 mM NaF, supplemented with 1 mM sodium orthovanadate and 1× protease inhibitor cocktail added immediately before lysing the cells. The lysates were centrifuged at 13,000 × g for 20 to 30 min at 4°C, and the supernatant was transferred to ice-cold Eppendorf tubes. The protein concentration was measured by the Bradford method (10) using BSA as standard. Then, 40 to 60 μg of protein was separated by electrophoresis in 10% sodium dodecyl sulfate-polyacrylamide gels and electroblotted to a 0.45-μm-pore-size nitrocellulose membrane (Bio-Rad) in a wet chamber at 250 to 300 mA for 1 h. The membranes were then probed with the indicated antibody, and the abundance of the phosphorylated forms of Akt, GSK-3α, or GSK-3β, and the NF-κB p65 subunit was detected with the Immobilon western chemiluminescent HRP substrate kit from Millipore (Billerica, MA). Membranes were exposed to an X-ray film (Kodak) with two intensifying screens (DuPont) at room temperature.
Statistical analysis.
For internalization and adherence, the data were normalized by calculating the ratio of S. aureus CFU/ml to the number of BEC/ml for each condition tested. In each experiment, the ratio obtained for each condition was referenced to the control condition that was arbitrarily assigned a value of 100%. For each condition, the error standard of the mean (n = 3) was calculated. The statistical significance was evaluated with the t test paired analysis by using the SigmaStat program (version 3.0; SPSS, Inc., Chicago, IL). Densitometric analysis of the bands was performed with the Image Processing and Analysis in Java Program ImageJ (http://rsbweb. nih.gov/ij).
RESULTS
Internalization of S. aureus by BEC involves the PI3K-dependent phosphorylation of Akt.
To investigate the host-cell signaling events involved in the internalization of S. aureus by BEC, we explored the role of the PI3K-Akt signaling pathway because of its well-known function in diverse cellular processes, including inflammation and cytoskeleton rearrangement. Since we have previously reported that S. aureus internalization by BEC is strongly inhibited by cytochalasin D (47), we determined whether S. aureus internalization in these cells causes the activation of PI3K and the subsequent phosphorylation of Akt. The data shown in Fig. 1 A indicated that S. aureus was able to induce a time-dependent Akt phosphorylation on Ser473 with a maximum at 40 min postinfection, followed by a gradual decrease with longer infection times of 60 and 120 min. A gradual increase in Akt phosphorylation was also observed when BEC were infected with different MOI values reaching the maximum activation at an MOI of 20 (Fig. 1B). These results allowed us to establish the conditions (MOI of 20 and infection time of 40 min) to evaluate the involvement of the PI3K-Akt signaling pathway in the internalization of S. aureus by BEC.
Fig. 1.
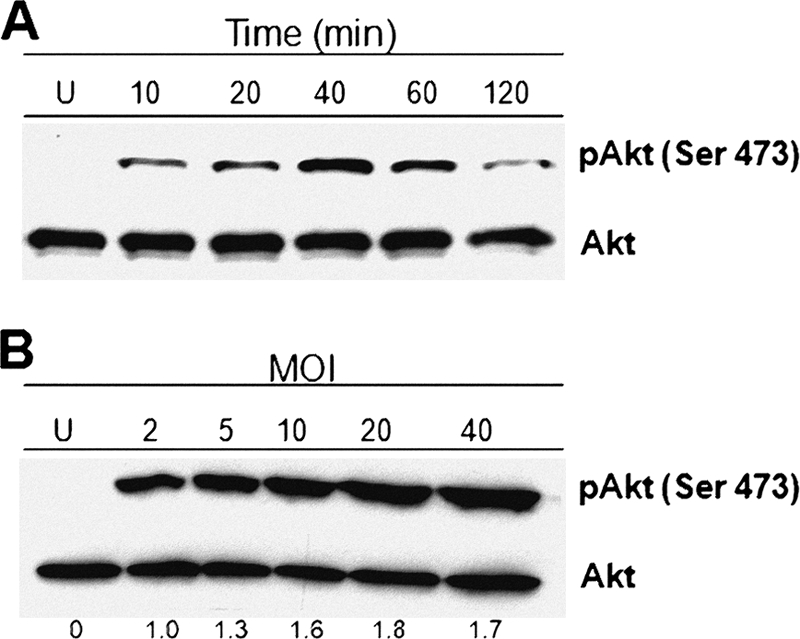
S. aureus activates the phosphorylation of Akt on Ser473 in BEC. (A) Cells were left uninfected (U) or infected with S. aureus at an MOI of 20 for 10 to 120 min. (B) Cells were left uninfected (U) or infected with S. aureus at MOIs of 2 to 40 for 40 min. After infection, the phosphorylation of Akt was analyzed by Western blotting. Detection of Akt isoforms 1 to 3 in each sample was performed to ensure equal protein loading. Blots are representative of three (A) and two (B) independent experiments. The numbers at the bottom of panel B indicate the relative band intensities obtained by densitometric analysis of each assay compared to the uninfected control.
To determine whether S. aureus mediates Akt phosphorylation via PI3K activity, we incubated BEC with increasing concentrations of LY, a specific inhibitor of PI3K. We observed that LY completely abolished the phosphorylation of Akt induced by infection of BEC with S. aureus (Fig. 2 A). We next sought to determine whether the inhibition of PI3K with LY or W affected the internalization and adherence of S. aureus to BEC. Inhibition of PI3K with increasing concentrations of both chemicals caused a concentration-dependent reduction of S. aureus internalization with maximal decreases of 90 and 95% observed in the presence of 50 μM and 100 nM LY and W, respectively (Fig. 2B and D). Although internalization of S. aureus by BEC was significantly reduced in the presence of LY and W, the adherence was not affected even at the highest concentrations used for both inhibitors (Fig. 2C and E). These data suggest that S. aureus is able to induce the PI3K-dependent phosphorylation of Akt on Ser473 and that PI3K activity is involved in the internalization of this bacterium.
Fig. 2.

Inhibition of PI3K activity abolishes phosphorylation of Akt and reduces S. aureus internalization by BEC. (A) Cells were left untreated and uninfected (U), were untreated (−), or were treated with 10, 25, or 50 μM LY294002 (LY) for 30 min and then infected with S. aureus at an MOI of 20 for 40 min. A control in which cells were only treated with 50 μM LY for 30 min was also included. After infection, the phosphorylation of Akt was analyzed by Western blotting. Detection of the Akt isoforms 1 to 3 in each sample was performed to ensure equal protein loading. The blot is representative of three independent experiments. (B and D) The number of internalized S. aureus was determined in untreated cells (U) or in cells treated with the indicated concentrations of LY and wortmannin (W). Extracellular S. aureus was killed by lysostaphin treatment. (C and E) The number of adherent S. aureus was analyzed in untreated cells (U) or in cells treated with the indicated concentrations of LY and W. In this case, the cells were washed three times with PBS to eliminate nonadherent bacteria. Untreated controls contained 0.1% dimethyl sulfoxide (DMSO). Bacteria were recovered from hypotonically lysed cells, cultured on LB agar for 19 to 24 h at 37°C, and counted. The data represent means ± the standard error of the mean (SEM; n = 3). *, P < 0.05; **, P < 0.001 (compared to the untreated control value).
The Akt activity is important for S. aureus internalization by BEC.
To explore if the activity of Akt is involved in S. aureus internalization, we incubated BEC with SH-5, a specific inhibitor of Akt, and evaluated both the phosphorylation level of Ser473 and the number of intracellular bacteria. Our results indicated that 5 μM SH-5 inhibited the phosphorylation of Akt by 35%, whereas 10 μM caused a reduction of 86% with respect to the untreated control (−) (Fig. 3 A). No effect on Akt phosphorylation was observed when BEC were incubated with 10 μM SH-5 alone (Fig. 3A). S. aureus internalization was 95% inhibited compared to the untreated control when BEC were incubated with SH-5 (Fig. 3B). In contrast, the adherence of S. aureus to BEC was unaffected in the presence of this inhibitor (Fig. 3C), as was the case when LY or W was added to inhibit the activity of PI3K.
Fig. 3.
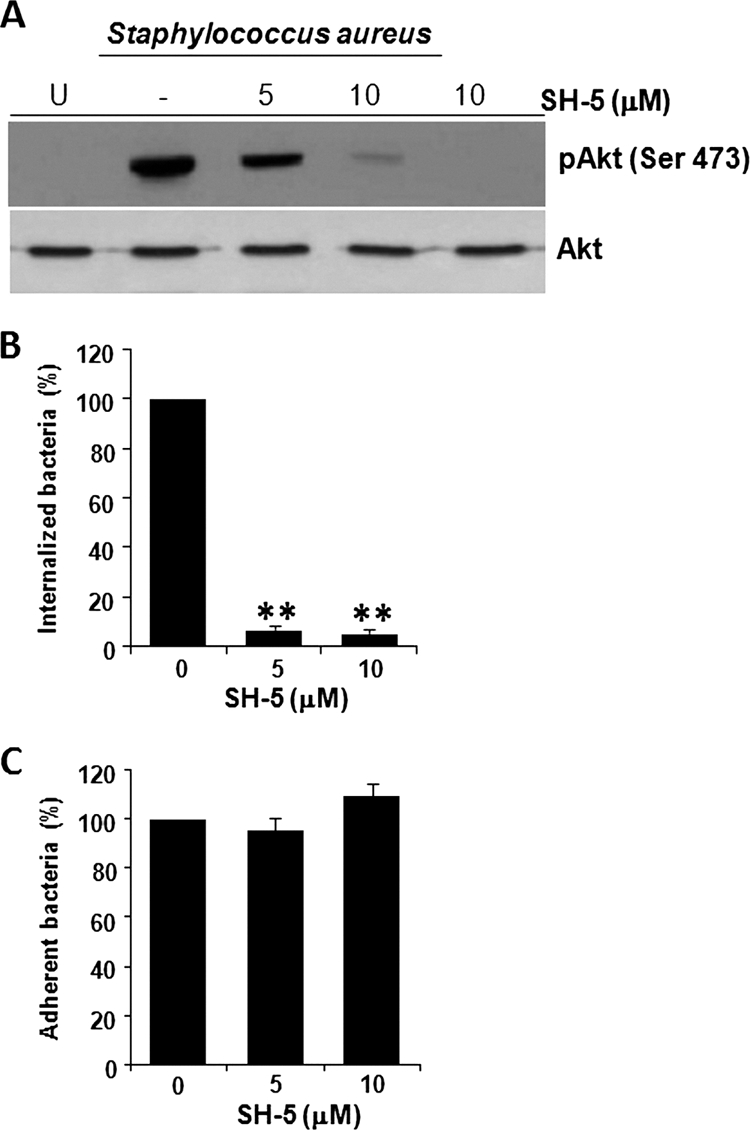
Inhibition of Akt phosphorylation on Ser473 reduces the S. aureus internalization by BEC. (A) Cells were left untreated and uninfected (U), were left untreated (−), or were treated with 5 or 10 μM SH-5 for 30 min and then infected with S. aureus at an MOI of 20 for 40 min. A control in which cells were only treated with 10 μM SH-5 for 30 min was also included. After infection, the phosphorylation of Akt was analyzed by Western blotting. Detection of the Akt isoforms 1 to 3 in each sample was performed to ensure equal protein loading. The blot is representative of three independent experiments. (B) The number of internalized S. aureus was analyzed in untreated cells (column 0) or in cells treated with the indicated concentrations of SH-5. Extracellular S. aureus was killed by lysostaphin treatment. (C) The number of adherent S. aureus was analyzed in nontreated cells (column 0) or in cells treated with the indicated concentrations of SH-5. In this case the cells were washed three times with PBS in order to eliminate nonadherent bacteria. Untreated controls contained 0.1% DMSO. Bacteria were recovered from hypotonically lysed cells, cultured on LB agar for 19 to 24 h at 37°C, and counted. The data represent means ± the SEM (n = 3). **, P < 0.001 (compared to the untreated control value).
It is known that Akt becomes fully activated when the residues Thr308 and Ser473 are phosphorylated by PDK1 and rictor-mTOR complex, respectively (4, 53). To determine whether phosphorylation of Thr308 is important for S. aureus internalization, we incubated BEC with 0.5 or 2 μM OSU, an inhibitor of PDK1 that specifically inhibits the phosphorylation of Akt on Thr308 (66), prior to infection with S. aureus. As expected, OSU did not inhibit the phosphorylation of Ser473 induced by S. aureus even at the highest concentration used, whereas it completely abolished the phosphorylation of Thr308 (Fig. 4 A and B). Interestingly, neither internalization nor adherence of S. aureus was affected in BEC pretreated with OSU (Fig. 4C and D).
Fig. 4.

Inhibition of Akt phosphorylation on Thr308 does not reduce the S. aureus internalization or adherence by BEC. (A and B) Detection of Akt phosphorylated on Ser473 or Thr308 in cells left untreated and uninfected (U), in untreated cells (−), or in cells treated with 0.5 or 2 μM OSU-03012 (OSU) for 15 min and then infected with S. aureus at an MOI of 20 for 40 min. A control in which cells were only treated with 2 μM OSU for 15 min was also included. After infection, the phosphorylation of Akt was analyzed by Western blotting. Detection of the Akt isoforms 1 to 3 in each sample was performed to ensure equal protein loading. The blots are representative of three independent experiments. (C) The number of internalized S. aureus was evaluated in untreated cells (column −) or in cells treated with the indicated concentrations of OSU. Extracellular S. aureus was killed by lysostaphin treatment. (D) The number of adherent S. aureus was evaluated in untreated cells (column −) or in cells treated with the indicated concentrations of OSU. In this case, the cells were washed three times with PBS in order to eliminate nonadherent bacteria. Bacteria were recovered from hypotonically lysed cells, cultured on LB agar for 19 to 24 h at 37°C, and counted. The data represent means ± the SEM (n = 3). The numbers at the bottom of panel A indicate the relative band intensities obtained by densitometric analysis of each assay compared to the uninfected control.
To confirm that Akt plays a central role in the S. aureus internalization, we expressed the constitutively active (CA) and dominant-negative (DN) forms of the Akt gene in BEC. Cells were transfected with pCMV5 plasmids containing the Akt-CA or Akt-DN mutants tagged with HA and after 24 h of incubation, the presence of the corresponding proteins was detected by Western blotting with antibodies that recognize the HA tag. We observed similar expression levels for the CA and DN mutants, while no signal was detected when BEC were left untransfected (U) or transfected with the vector alone (pCMV) (Fig. 5 A). Internalization of S. aureus was significantly reduced (∼46%) or increased (∼65%) in BEC expressing the Akt-DN or Akt-CA, respectively (Fig. 5B). The adherence of S. aureus to the BEC surface remained unaltered in transfected cells compared to the values of the untreated and pCMV controls (Fig. 5C). Altogether, these results demonstrated that Akt activity is associated with S. aureus internalization and that phosphorylation of Akt on Ser473, but not on Thr308, appears to be essential for this process.
Fig. 5.
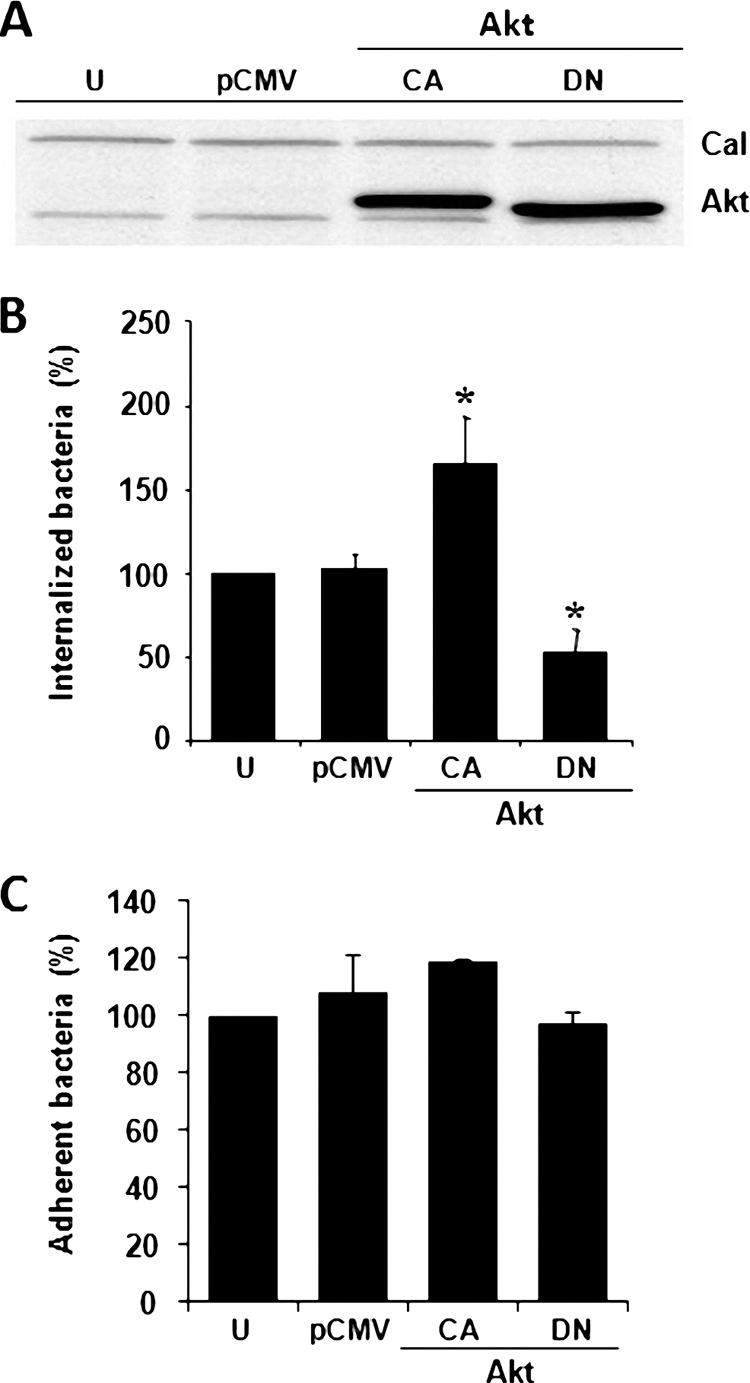
The number of internalized but not adherent S. aureus was altered in BEC transfected with mutants of Akt. (A) Cells were left untransfected (U), were transfected with pCMV5 vector (300 ng) only (pCMV), or were transfected with pCMV5 containing the constitutively active (CA) Akt (5 ng) or the dominant-negative (DN) Akt (200 ng) gene forms. The amount of total DNA transfected was kept constant at 300 ng by the addition of 295 ng of pCMV5 to the Akt-CA or 100 ng of pCMV5 to the Akt-DN. Expression of Akt-CA and Akt-DN (Akt) was analyzed 24 h after transfection by Western blotting with an antibody against the HA tag. Evidence of equal protein loading was obtained by detection of calnexin (Cal). (B) The number of internalized S. aureus was quantitated in untransfected cells (U) or in cells after 24 h of transfection as described in panel A. Extracellular S. aureus was killed by lysostaphin treatment. (C) The number of adherent S. aureus was quantitated in untransfected cells (U) or in cells after 24 h of transfection as described in panel A. In this case, the cells were washed three times with PBS in order to eliminate nonadherent bacteria. Bacteria were recovered from hypotonically lysed cells, cultured on LB agar for 19 to 24 h at 37°C, and counted. The data represent means ± the SEM (n = 3). *, P < 0.05 (compared to the untransfected control value).
Phosphorylation of GSK-3α and GSK-3β in BEC infected with S. aureus depends on the Akt activity.
Akt phosphorylates many substrates that modulate diverse cellular functions such as proliferation, differentiation, and apoptosis (37). One of those substrates is GSK-3 that, among its multiple functions, is involved in the regulation of the inflammatory process caused by whole bacteria or bacterial virulence factors (7). Because GSK-3 is a constitutively active enzyme present in two isoforms whose activities are inhibited by Akt phosphorylation (32), we sought to determine whether the activation of Akt by S. aureus leads to the phosphorylation of GSK-3α and GSK-3β. When BEC were infected with S. aureus at various times, we observed the phosphorylation of GSK-3α and GSK-3β (Fig. 6 A and C) with maxima at 40 to 60 min for GSK-3α and 40 min for GSK-3β, a finding that is in agreement with the time for maximal phosphorylation of Akt (see Fig. 1A). S. aureus-induced phosphorylation of GSK-3α and GSK-3β was strongly reduced by treatment of BEC with SH-5 prior to S. aureus infection (Fig. 6B and D). Compared to the uninfected and untreated (U) control, a slight decrease in the phosphorylation of both isoforms were observed when BEC were treated with SH-5 alone, suggesting an inhibition of basal Akt activity. These data indicate that phosphorylation of the isoforms GSK-3α and GSK-3β induced by S. aureus proceeds via Akt.
Fig. 6.

S. aureus activates the phosphorylation of GSK-3α and GSK-3β in BEC through the activation of Akt. (A and C) Cells were left uninfected (U) or were infected with S. aureus at an MOI of 20 for 10 to 120 min. (B and D) Cells were left untreated and uninfected (U), were untreated (−), or were treated with 10 μM SH-5 for 30 min and then infected with S. aureus at an MOI of 20 for 40 min. A control in which cells were only treated with 10 μM SH-5 for 30 min was also included. After infection, the phosphorylation of GSK-3α on Ser-21 or GSK-3β on Ser-9 was analyzed by Western blotting. Detection of total GSK-3β in each sample was performed to ensure equal protein loading. The blots are representative of two (A and C) or three (B and D) independent experiments. Numbers at the bottom of panels A and C indicate the relative band intensities obtained by densitometric analysis of each assay compared to the uninfected control.
Phosphorylation of the NF-κB p65 subunit in BEC infected with S. aureus depends on the PI3K activity.
Full transactivation potential of NF-κB, one of the main transcription factors involved in the inflammation process, is the result of its phosphorylation and subsequent translocation from the cytoplasm to the nucleus. Moreover, in certain cell types the activation of the PI3K–Akt–GSK-3 signaling pathway leads to the regulation of NF-κB activity (39). To explore whether the activation of PI3K and Akt induced by S. aureus causes the phosphorylation of NF-κB, BEC were infected with S. aureus for 10 to 120 min and phosphorylation of p65 on Ser536 was detected by Western blotting. The phosphorylation of Ser536 reached the maximum level at 40 min postinfection, followed by a marked decrease at 120 min (Fig. 7 A). Phosphorylation of p65 on Ser536 was dependent on the PI3K activity because incubation of BEC with LY for 30 min prior to infection with S. aureus caused a significant reduction of phosphorylation (Fig. 7B). However, no effect on the nuclear translocation of p65 was observed when BEC were infected with S. aureus (data not shown). These results suggest that S. aureus caused a PI3K-dependent NF-κB p65 subunit phosphorylation on Ser536 without affecting its nuclear translocation.
Fig. 7.
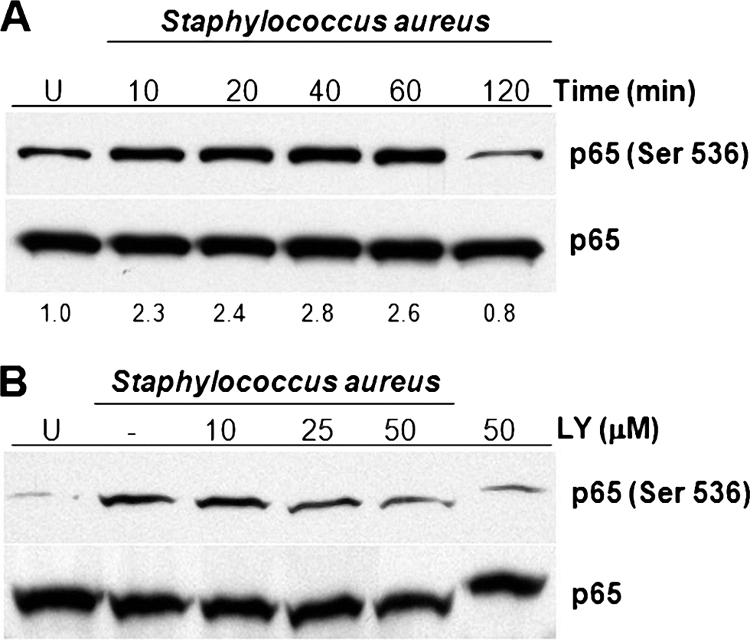
Inhibition of PI3K activity reduces the level of phosphorylation of NF-κB p65 on Ser536 in BEC infected with S. aureus. (A) Cells were left uninfected (U) or were infected with S. aureus at an MOI of 20 for 10 to 120 min. (B) Cells were left untreated and uninfected (U), were untreated (−), or were treated with 10, 25, or 50 μM LY294002 (LY) for 30 min, and then infected with S. aureus at an MOI of 20 for 40 min. A control in which cells were only treated with 50 μM LY for 30 min was also included. After infection, the phosphorylation of NF-κB p65 on Ser536 was analyzed by Western blotting. Detection of total NF-κB p65 in each sample was performed to ensure equal protein loading. The blots are representative of three independent experiments. Numbers at the bottom of A indicate the relative band intensity obtained by densitometric analysis of each assay compared to the uninfected control.
DISCUSSION
The PI3K-Akt signaling pathway integrates a variety of extracellular signals for the control of diverse cellular responses such as cell growth and proliferation, glucose homeostasis, survival, and apoptosis. Immune responses in cancer are also regulated by the PI3K-Akt signaling (65). Regulation of actin cytoskeleton rearrangement by PI3K has also been shown to play an essential role in the phagocytosis of pathogenic microorganisms by macrophages (63), formation of pseudopods, and phagosome maturation (59, 64). Furthermore, a correlation of PI3K-Akt signaling pathway activity with the cellular internalization of Chlamydia pneumonia, Listeria monocytogenes, Pseudomonas aeruginosa, Helicobacter pylori, Legionella pneumophila, Salmonella spp., and Streptococcus pyogenes has been established (3, 19, 33, 43, 50, 56, 60). We propose the addition of S. aureus to this list because we have obtained evidence indicating that internalization of this bacterium by endothelial cells activates the PI3K-Akt signaling pathway, which in turn leads to the phosphorylation of GSK-3 and NF-κB.
Several reports have demonstrated that S. aureus is able to invade nonprofessional phagocytic cells such as epithelium and endothelium (5, 9, 27, 42, 47). The data in the present study indicated that internalization of S. aureus was associated with a time- and MOI-dependent Akt phosphorylation on Ser473 (Fig. 1A and B) that was mediated by the activity of PI3K because treatment of endothelial cells with LY abolished Akt phosphorylation (Fig. 2A). We also observed that treatment of BEC with LY and W caused a significant reduction of S. aureus internalization by BEC without altering its adherence to the cell surface (Fig. 2). These results demonstrate that the activity of PI3K-Akt pathway in BEC is only involved in the internalization but not the adherence of S. aureus to the cell surface. Similar data have been obtained with Cronobacter sakazakii, an opportunistic pathogenic bacterium that causes neonatal sepsis and meningitis. The infection of human brain microvascular endothelial cells (HBMEC) with this bacterium causes an increase in Akt phosphorylation and its invasion is blocked by treatment of HBMEC with PI3K inhibitors (36). The molecular mechanism of internalization used by S. aureus is also similar to that used by C. pneumonia, an intracellular pathogen. The invasion but not the binding to the surface of HEp2 epithelial cells by C. pneumoniae requires Akt phosphorylation, which reaches a maximum level at 40 min postinfection and is mediated by the activity of PI3K (19). Likewise, PI3K activity is essential for Campylobacter jejuni invasion of the human embryonic intestinal cell line INT407 (29) and for L. pneumophila invasion of macrophages (60).
Evidence of Akt participation in the S. aureus internalization by BEC was obtained by incubating the endothelial cells with SH-5, a specific inhibitor of the Akt activity. We found that SH-5 caused a significant reduction of Akt phosphorylation on Ser473 induced by S. aureus, and this reduction correlated with a strong decrease in the internalization of S. aureus, without altering its adherence to the cell surface (Fig. 3). To confirm the results obtained with SH-5, we genetically modified BEC by overexpressing the CA and DN forms of Akt. The expression of the Akt-DN caused a marked reduction of S. aureus internalization, while expression of Akt-CA significantly increased it without affecting the adherence (Fig. 5). Similarly, the internalization, but not the adherence, of P. aeruginosa was reduced when MDCK cells were pretreated with SH-5 or HeLa cells were transfected with small interfering RNAs specific to silence the Akt gene (33). Taken together, these results indicate that PI3K-Akt is one of the main signaling pathways involved in the internalization of several pathogenic bacteria in phagocytic and nonphagocytic cells.
Full activation of Akt is achieved when both Ser473 and Thr308 are phosphorylated (22). We have demonstrated that S. aureus is able to induce Akt phosphorylation on these residues (Fig. 4A and B) and that treatment of BEC with OSU effectively blocks the phosphorylation of Thr308 without affecting the phosphorylation of Ser473. More importantly, inhibition of Thr308 phosphorylation does not affect the internalization or adherence of S. aureus (Fig. 4C and D), indicating that internalization of this bacterium is associated with phosphorylation of Akt on Ser473 but not on Thr308. Although differential phosphorylation of these Akt residues has been observed in acute myeloid leukemia (26), this is the first report in which the phosphorylation of only one Akt residue is important for the internalization of a pathogenic bacterium. Interestingly, Jacinto et al. (31) have proposed that dual Akt phosphorylation is not necessarily required for all Akt functions. Further studies are necessary to precisely define the physiological importance of each Akt phosphorylation site and in particular the participation of phosphorylated Thr308 in different cellular processes.
The Akt activated by phosphorylation gives rise to downstream phosphorylation and inactivation of GSK-3, a constitutively active kinase involved in diverse cellular functions such as metabolism and regulation of the innate immune response (7, 12, 35). In the present study we found that S. aureus caused a time-dependent phosphorylation of GSK-3α and GSK-3β (Fig. 6A and C) with time peaks similar to the time for maximal Akt phosphorylation (Fig. 1). Moreover, treatment of BEC with SH-5 revealed that phosphorylation of both GSK-3 isoforms was dependent on Akt activity (Fig. 6B and D). Phosphorylation of paxillin by ERK/GSK-3 and rearrangement of actin cytoskeleton has already been observed (13, 41). It is likely that when S. aureus is internalized by BEC, the Akt-dependent phosphorylation of GSK-3 may lead to phosphorylation of paxillin and reorganization of the actin cytoskeleton, which favors the entry of bacteria. GSK-3 has also been associated with the regulation of pro- and anti-inflammatory cytokines production through TLR signaling (39). Interestingly, phosphorylation of GSK-3 by Akt turns off its catalytic activity, resulting in the activation of pathways that are normally repressed by GSK-3 (15). In this context, Cheng et al. (18) reported that inhibition of GSK-3β blocks NF-κB activation, TNF-α production, and iNOS/NO biosynthesis but increases IL-10 production when the microglia is stimulated with heat-inactivated S. aureus.
NF-κB is one of the major transcriptional factors that regulate the inflammatory response (8), and its activity is modulated by microbial pathogens (51). The transactivation potential of this factor could be enhanced by phosphorylation of p65 (48, 52, 55). We found here that S. aureus caused the phosphorylation of p65 on Ser536 as early as 10 min postinfection and that this phosphorylation was partially inhibited by LY, indicating a dependency on PI3K (Fig. 7A and B). Several bacteria interfere with the activation of NF-κB as one of the strategies to evade the immune response and guarantee their intracellular survival (62). Phosphorylation of p65 on Ser536 has also been observed in gastric epithelial cells infected with H. pylori (61), and it has recently been shown that inhibition of NF-κB activity is associated with a reduction of P. aeruginosa internalization into human respiratory epithelial cell lines and HeLa cervical cancer cells (23). Taking into consideration that inhibition of the Akt activity also diminishes P. aeruginosa internalization (33), it is likely that a link between the PI3K-Akt signaling pathway and NF-κB activation may exist and that they coordinately function during bacteria internalization. A role of GSK-3β in NF-κB function was demonstrated during mammalian development by disrupting the GSK-3β gene in murine embryonic stem cells (28). However, Steinbrecher et al. (58) did not observe any TNF-α-induced change in nuclear localization of p65 associated with a GSK-3β activity loss in GSK-3-null mouse embryonic fibroblasts. In the present study we observed that, although S. aureus internalization activated the PI3K-dependent phosphorylation of p65, it did not apparently affect cytoplasmic and nuclear distribution of this NF-κB subunit (data not shown). Thus, it is likely that nuclear translocation of the NF-κB p65 subunit in BEC is not a process directly related to the phosphorylation inactivation of GSK-3 caused by S. aureus.
Based on the results presented here and data published elsewhere (47), we propose that the PI3K-Akt signaling pathway activated by S. aureus and the NF-κB nuclear translocation induced by TNF-α are important for S. aureus internalization by BEC. In the diagram shown in Fig. 8, positive symbols indicate activation of S. aureus internalization, whereas question marks indicate an as-yet-undefined role on S. aureus internalization of the PI3K- and Akt-dependent phosphorylation of the NF-κB p65 subunit and GSK-3 isoforms. The role of the NF-κB p65 subunit and GSK-3α/GSK-3β phosphorylation in the internalization of this bacterium was not determined here. Future experiments will be designed to clarify this point.
Fig. 8.

Schematic diagram summarizing the main results obtained in the present study (A) and in a previous report by Oviedo-Boyso et al. in 2008 (47) (B).
ACKNOWLEDGMENTS
We thank Wanyin Deng and Amit Bahvsar for critical reading and valuable suggestions to the manuscript. We also thank Inimex Pharmaceuticals, Inc., for sharing part of their research on S. aureus.
This study was supported by grants from Coordinación de la Investigación Científica, Universidad Michoacana de San Nicolás de Hidalgo, to V.M.B.-A. and by grants from the Canadian Institutes for Health Research (CIHR) to B.B.F. B.B.F. is a CIHR Distinguished Investigator, a Howard Hughes Medical Institute International Research Scholar, and UBC Peter Wall Distinguished Professor. V.M.B.-A. was a recipient of a sabbatical scholarship from Consejo Nacional de Ciencia y Tecnología (CONACYT), Mexico. R.C.-V. and A.H.-M. are receiving doctoral scholarships from CONACYT. H.B.Y. is supported by a CIHR postdoctoral fellowship.
Footnotes
Published ahead of print on 15 August 2011.
REFERENCES
- 1. Agerer F., Michel A., Ohlsen K., Hauck C. R. 2003. Integrin-mediated invasion of Staphylococcus aureus into human cells requires Src family protein-tyrosine kinases. J. Biol. Chem. 278:42524–42531 [DOI] [PubMed] [Google Scholar]
- 2. Agerer F., et al. 2005. Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalization. J. Cell Sci. 118:2189–2200 [DOI] [PubMed] [Google Scholar]
- 3. Allen L. H., Allgood J. A., Han X., Wittine L. M. 2005. Phosphoinositide 3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. J. Leukoc. Biol. 78:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allessi D. R., et al. 1997. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 7:776–789 [DOI] [PubMed] [Google Scholar]
- 5. Bayles K. W., et al. 1998. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect. Immun. 66:336–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beaulieu J. M., Gainetdinov R. R., Caron M. G. 2007. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28:166–172 [DOI] [PubMed] [Google Scholar]
- 7. Beurel E., Michalek S. M., Jope R. S. 2010. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 31:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonizzi G., Karin M. 2004. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25:280–288 [DOI] [PubMed] [Google Scholar]
- 9. Bost K. L., et al. 1999. Staphylococcus aureus infection of mouse or human osteoblasts induces high levels of interlekin-6 and interleukin-12 production. J. Infect. Dis. 180:1912–1920 [DOI] [PubMed] [Google Scholar]
- 10. Bradford M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 11. Burnham C. A., Shokoples S. E., Tyrrell G. J. 2007. Invasion of HeLa cells by group B streptococcus requires the phosphoinositide-3-kinase signaling pathway and modulates phosphorylation of host-cell Akt and glycogen synthase kinase-3. Microbiology 153:4240–4252 [DOI] [PubMed] [Google Scholar]
- 12. Buttrick G. J., Wakefield J. G. 2008. PI3-K and GSK-3. Cell Cycle 7:2621–2625 [DOI] [PubMed] [Google Scholar]
- 13. Cai X., Li M., Vrana J., Schaller M. D. 2006. Glycogen synthase kinase 3- and extracellular signal-regulated kinase-dependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Mol. Cell. Biol. 26:2857–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cajero-Juárez M., Åvila B., et al. 2002. Immortalization of bovine umbilical vein endothelial cells: a model for the study of vascular endothelium. Eur. J. Cell Biol. 81:1–8 [DOI] [PubMed] [Google Scholar]
- 15. Cantley L. C. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655–1657 [DOI] [PubMed] [Google Scholar]
- 16. Cary L. A., Guan J. L. 1999. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 4:D103–D113 [DOI] [PubMed] [Google Scholar]
- 17. Chan M. M. P., Cheung B. K. W., Li J. C. B., Chan L. L. Y., Lau A. S. Y. 2009. A role for glycogen synthase kinase-3 in antagonizing mycobacterial immune evasion by negatively regulating IL-10 induction. J. Leukoc. Biol. 86:283–291 [DOI] [PubMed] [Google Scholar]
- 18. Cheng Y. L., et al. 2009. Staphylococcus aureus induces microglial inflammation via a glycogen synthase kinase 3β-regulated pathway. Infect. Immun. 77:4002–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coombes B. K., Mahony J. B. 2002. Identification of MEK- and phosphoinositide 3-kinase-dependent signaling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell. Microbiol. 4:447–460 [DOI] [PubMed] [Google Scholar]
- 20. Cossart P., Roy C. R. 2010. Manipulation of host membrane machinery by bacterial pathogens. Curr. Opin. Cell Biol. 22:547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. David M. Z., Daum R. S. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deane J. A., Fruman D. A. 2004. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu. Rev. Immunol. 22:563–598 [DOI] [PubMed] [Google Scholar]
- 23. Emam A., Carter W. G., Lingwood C. 2010. Glycolipid-dependent, protease sensitive internalization of Pseudomonas aeruginosa into cultured human respiratory epithelial cells. Open Microbiol. J. 4:106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fowler T., Johansson S., Wary K. K., Höök M. 2003. Src kinase has a central role in in vitro cellular internalization of Staphylococcus aureus. Cell. Microbiol. 5:417–426 [DOI] [PubMed] [Google Scholar]
- 25. Fresno-Vara J. A., et al. 2004. PI3K/Akt signaling pathway and cancer. Cancer Treat. Rev. 30:193–204 [DOI] [PubMed] [Google Scholar]
- 26. Gallay N., et al. 2009. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23:1029–1038 [DOI] [PubMed] [Google Scholar]
- 27. Hamill R. J., Vann J. M., Proctor R. A. 1986. Phagocytosis of Staphylococcus aureus by cultured bovine aortic endothelial cells: model for postadherence events in endovascular infections. Infect. Immun. 54:833–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoeflich K. P., et al. 2000. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 406:86–90 [DOI] [PubMed] [Google Scholar]
- 29. Hu L., McDaniel J. P., Kopecko D. J. 2006. Signal transduction events involved in human epithelial cell invasion by Campylobacter jejuni 81-176. Microb. Pathog. 40:91–100 [DOI] [PubMed] [Google Scholar]
- 30. Ireton K., Payrastre B., Cossart P. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J. Biol. Chem. 274:17025–17032 [DOI] [PubMed] [Google Scholar]
- 31. Jacinto E., et al. 2006. SIN1/MIP1 maintains rictor-mTor complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127:125–137 [DOI] [PubMed] [Google Scholar]
- 32. Jope R. S., Johnson G. V. 2004. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29:95–102 [DOI] [PubMed] [Google Scholar]
- 33. Kierbel A., Gassama-Diagne A., Mostov K., Engel J. N. 2005. The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol. Biol. Cell 16:2577–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leonard F. C., Markey B. K. 2008. Methicillin-resistant Staphylococcus aureus in animals: a review. Vet. J. 175:27–36 [DOI] [PubMed] [Google Scholar]
- 35. Kim L., Kimmel A. R. 2006. GSK3 at the edge: regulation of developmental specification and cell polarization. Curr. Drug Targets 7:1411–1419 [DOI] [PubMed] [Google Scholar]
- 36. Li Q., et al. 2010. PI3K-dependent host cell actin rearrangements are required for Cronobacter sakazakii invasion of human brain microvascular endothelial cells. Med. Microbiol. Immunol. 199:333–340 [DOI] [PubMed] [Google Scholar]
- 37. Liu P., Cheng H., Roberts T. M., Zhao J. J. 2009. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Disc. 8:627–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mansell A., Khelef N., Cossart P., O'Neill L. A. 2001. Internalin B activates nuclear factor-κB via Ras, phosphoinositide 3-kinase, and Akt. J. Biol. Chem. 276:43597–43603 [DOI] [PubMed] [Google Scholar]
- 39. Martin M., Rehani K., Jope R. S., Michalek S. M. 2005. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6:777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Massey R. C., et al. 2001. Fibronecting-binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to fibronectin and invasion of endothelial cells. Cell. Microbiol. 3:839–851 [DOI] [PubMed] [Google Scholar]
- 41. May R. C., Machesky L. M. 2001. Phagocytosis and the actin cytoskeleton. J. Cell Sci. 114:1061–1077 [DOI] [PubMed] [Google Scholar]
- 42. Menzies B. E., Kourteva I. 1998. Internalization of Staphylococcus aureus by endothelial cells induces apoptosis. Infect. Immun. 66:5994–5998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Molero C., et al. 2009. Addressing the effects of Salmonella internalization in host cell signaling on a reverse-phase protein array. Proteomics 9:3652–3665 [DOI] [PubMed] [Google Scholar]
- 44. Nakayama M., et al. 2009. Helicobacter pylori VacA-induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J. Biol. Chem. 284:1612–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oliveira M., Bexiga R., Nunes S. F., Vilela C. L. 2011. Invasive potential of biofilm-forming staphylococci bovine subclinical mastitis isolates. J. Vet. Sci. 12:95–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oviedo-Boyso J., et al. 2007. Innate Immune response of bovine mammary gland to pathogenic bacteria responsible for mastitis. J. Infect. 54:399–409 [DOI] [PubMed] [Google Scholar]
- 47. Oviedo-Boyso J., et al. 2008. Internalization of Staphylococcus aureus by bovine endothelial cells is associated with the activity state of NF-κB and modulated by the proinflammatory cytokines TNF-α and IL-1β. Scand. J. Immunol. 67:169–176 [DOI] [PubMed] [Google Scholar]
- 48. Ozes O. N., et al. 1999. NF-κB activation by tumor necrosis factor requires the Akt serine-threonine kinase. Nature 401:82–85 [DOI] [PubMed] [Google Scholar]
- 49. Peacock S. J., Foster T. J., Cameron B. J., Berendt A. R. 1999. Bacterial fibronectin-binding proteins and endothelial cell surface fibronectin mediate adherence of Staphylococcus aureus to resting human endothelial cells. Microbiology 145:3477–3486 [DOI] [PubMed] [Google Scholar]
- 50. Pizarro-Cerdá J., Cossart P. 2004. Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat. Cell Biol. 6:1026–1033 [DOI] [PubMed] [Google Scholar]
- 51. Rahman M. M., McFadden G. 2006. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sale E. M., Sale G. J. 2008. Protein kinase B: signaling roles and therapeutic targeting. Cell. Mol. Life Sci. 65:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sarbassov D. D., Gertin D. A., Ali S. M., Sabatini D. M. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 54. Schröder A., et al. 2006. Staphylococcus aureus fibronectin binding protein A induces motile attachment sites and complex actin remodeling in living endothelial cells. Mol. Biol. Cell 17:5198–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schwabe R. F., Brenner D. A. 2002. Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G204–G211 [DOI] [PubMed] [Google Scholar]
- 56. Siemens N., Patenge N., Otto J., Fiedler T., Kreikemeyer B. 2011. Streptococcus pyogenes M49 plasminogen/plasmin-binding facilitates keratinocyte invasion via integrin-ILK pathways and protects from macrophage killing. J. Biol. Chem. 286:21612–21622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sinha B., et al. 1999. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin α5β1. Cell. Microbiol. 1:101–117 [DOI] [PubMed] [Google Scholar]
- 58. Steinbrecher K. A., Wilson W., Cogswell P. C., Baldwin A. S. 2005. Glycogen synthase kinase 3 functions to specify gene-specific, NF-κB-dependent transcription. Infect. Immun. 25:8444–8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Swanson J. A., Hoppe A. D. 2004. The coordination of signaling during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 76:1093–1103 [DOI] [PubMed] [Google Scholar]
- 60. Tachado S. D., Samrakandi M. M., Cirillo J. D. 2008. Non-opsonic phagocytosis of Legionella pneumophila by macrophages is mediated by phosphatidylinositol 3-kinase. PLoS One 3:e3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takeshima E., et al. 2009. NF-κB activation by Helicobacter pylori requires Akt-mediated phosphorylation of p65. BMC Microbiol. 9:36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62. Tato C. M., Hunter C. A. 2002. Host-pathogen interactions: subversion and utilization of the NF-κB pathway during infection. Infect. Immun. 70:3311–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Underhill D. M., Ozinsky A. 2002. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20:825–852 [DOI] [PubMed] [Google Scholar]
- 64. Vieira O. V., Botelho R. J., Grinstein S. 2002. Phagosome maturation: aging gracefully. Biochem. J. 366:689–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wickenden J. A., Watson C. J. 2010. Key signaling nodes in mammary gland development and cancer: signaling downstream of PI3 kinase in mammary epithelium: a play in three Akts. Breast Cancer Res. 12:202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhu J., et al. 2004. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 64:4309–4318 [DOI] [PubMed] [Google Scholar]