

Abstract
Recent evidence suggests that smoking affects the composition of the disease-associated subgingival biofilm, yet little is known about its effects during the formation of this biofilm. The present investigation was undertaken to examine the contributions of smoking to the composition and proinflammatory characteristics of the biofilm during de novo plaque formation. Marginal and subgingival plaque and gingival crevicular fluid samples were collected from 15 current smokers and from 15 individuals who had never smoked (nonsmokers) following 1, 2, 4, and 7 days of undisturbed plaque formation. 16S rRNA gene cloning and sequencing were used for bacterial identification, and multiplex bead-based flow cytometry was used to quantify the levels of 27 immune mediators. Smokers demonstrated a highly diverse, relatively unstable initial colonization of both marginal and subgingival biofilms, with lower niche saturation than that seen in nonsmokers. Periodontal pathogens belonging to the genera Fusobacterium, Cardiobacterium, Synergistes, and Selenomonas, as well as respiratory pathogens belonging to the genera Haemophilus and Pseudomonas, colonized the early biofilms of smokers and continued to persist over the observation period, suggesting that smoking favors early acquisition and colonization of pathogens in oral biofilms. Smokers also demonstrated an early proinflammatory response to this colonization, which persisted over 7 days. Further, a positive correlation between proinflammatory cytokine levels and commensal bacteria was observed in smokers but not in nonsmokers. Taken together, the data suggest that smoking influences both the composition of the nascent biofilm and the host response to this colonization.
INTRODUCTION
Nearly 42% of periodontitis in the United States is attributable to tobacco smoking (50), and numerous studies have reported a critical role for smoking in increasing the risk for developing extensive and severe forms of this disease (3, 22, 26). With there being over 1 billion adult smokers worldwide (55), smoking-related periodontitis presents a significant global public health issue. Although there is a large body of evidence on the clinical effects of smoking on the periodontium, little is known about the biological mechanisms by which it increases the risk for disease. Bacteria in dental plaque are the primary etiological agents of periodontal diseases, and elucidating the effects of smoking on this ecosystem is critical to understanding the role of smoking in the etiopathogenesis of periodontal diseases.
Studies using molecular approaches for bacterial identification and characterization have demonstrated that the subgingival microbial profile associated with periodontitis in smokers is diverse and distinct from that in nonsmokers (44, 53, 57). Recent evidence also indicates that smoking cessation alters patterns of microbial recolonization following periodontal therapy, with a decrease in the levels of putative periodontal pathogens (12, 18). Further, evidence from the nasopharyngeal ecosystem indicates that smoking alters the microbial signatures of these communities, with a decrease in the commensal population and a concomitant increase in pathogens (7, 9). Together, these investigations suggest that the disease-associated subgingival biofilm in smokers is enriched for pathogenic bacteria. However, the effects of smoking on a health-compatible ecosystem have not been previously examined.
Bacteria colonize a tooth surface within a few minutes after its eruption into the oral cavity and form complex communities on the tooth surface and in the subgingival sulcus (36, 45). Early colonization of tooth-associated ecosystems is a specific and selective process, and the development of a mature community is influenced by these early interbacterial as well as host-bacterium interactions (28, 33, 42). In the gastrointestinal tract, it has been shown that early colonizers occupy this spatial niche in large numbers, thereby playing an important role in resisting colonization by pathogenic species. This phenomenon of niche saturation, whereby a few selected species occupy an ecological habitat and provide resistance to colonization of this niche by pathogenic organisms, along with metabolic synergism, plays a critical role in maintaining a healthy, stable community (6). Examining the effect of smoking on these nascent communities thus is an important initial step in understanding the etiopathogenic role of smoking in periodontal diseases.
Therefore, the purpose of the present investigation was to compare bacterial acquisition and colonization in current smokers and those who have never smoked during marginal and subgingival plaque formation over 7 days.
MATERIALS AND METHODS
Subject selection.
Approval for this study was obtained from the Office of Responsible Research Practices at The Ohio State University. Twenty-three periodontally healthy current smokers and 20 individuals who had never smoked (referred to here as nonsmokers) were recruited following clinical oral examination, and informed consent was obtained. Clinical health was defined as attachment loss of ≤1 mm and probe depths of ≤3 mm at all sites. Exclusion criteria included diabetes, HIV infection, use of immunosuppressant medications, bisphosphonates, or steroids, antibiotic therapy or oral prophylactic procedures within the last 3 months, and fewer than 20 teeth in the dentition.
Study procedures.
Both current smokers and nonsmokers received professional dental cleaning (oral prophylaxis) and oral hygiene instructions. All subjects returned 7 to 10 days after this initial visit, when gingival health was confirmed and oral prophylaxis was administered once again. Both current smokers and nonsmokers were instructed to wear stents to cover a total of 6 teeth in two contralateral quadrants while brushing. All subjects returned 24 h later, and clinical data and marginal plaque, subgingival plaque, and gingival crevicular fluid (GCF) samples were collected; following this, oral prophylaxis was administered to remove the bacterial biofilm (zero plaque). Subjects returned after 48 h, when 2-day-old samples were collected, followed by oral prophylaxis. A similar protocol was followed to collect 4-day-old and 7-day-old samples.
Data collection.
Smoking status and tobacco exposure were assessed by questionnaire and salivary cotinine measurements (2). All subjects were examined by two calibrated periodontists. Probe depths and attachment levels were recorded throughout the mouth on 6 sites per tooth using a PCP-UNC 15 probe. The Turesky modification of the Quigley and Hein plaque index (51) and the Löe and Silness gingival index (34) were used to record plaque levels and gingival health status, respectively.
Sample collection.
Marginal plaque samples were collected and pooled from the mesial surfaces (buccal and lingual) of all test teeth by scraping the teeth at the mesial gingival margin with a 204S scaler and wiping onto paper points. Subgingival plaque samples were collected from the same sites by inserting one sterile endodontic paper point (Caulk-Dentsply) into the each mesial site for 10 s, followed by scraping with a curette. Samples were placed in 1.5-ml microcentrifuge tubes and frozen until further analysis. GCF samples were collected prior to subgingival sampling by inserting Periopaper (Oraflow Inc., NY) into the mesial sulcus of test teeth for 30 s. GCF volume was measured with a Periotron 8000 micromoisture meter (Oraflow Inc., NY) and samples immediately frozen at −20°C.
DNA isolation.
Subgingival bacteria were separated from the paper points by adding 200 μl of phosphate-buffered saline (PBS) to the tubes and vortexing. Supragingival bacteria were separated from the points by bead beating for 60 s. The points were then removed, and DNA was isolated from both sample types with a Qiagen DNA MiniAmp kit (Qiagen, Valencia, CA) using the tissue protocol according to the manufacturer's instructions (31).
Amplification of 16S rRNA genes.
Bacterial 16S rRNA genes were amplified from the community DNA with universal bacterial primers A17 (5′-GTT TGA TCC TGG CTC AG-3′) and 317 (5′-AAG GAG GTG ATC CAG GC-3′) (Biosynthesis, Lewisville, TX). PCR was performed as previously described (31). The PCR products were purified with the QIAquick PCR purification kit (Qiagen, Valencia, CA).
Cloning and sequencing.
The 16S rRNA gene amplicons generated by PCR were cloned into Escherichia coli using a commercially available kit (TOPO TA cloning kit; Invitrogen, San Diego, CA). Inserts of the correct molecular size (≅1,500bp) were confirmed by PCR amplification. The products were purified with a Millipore kit (Millipore, Billerica, MA) and sequenced with an ABI Prism cycle sequencing kit (BigDye Terminator cycle sequencing kit) using an ABI 3730 instrument (31).
Sequence analysis.
Partial sequences of 1,200 to 1,400 bp were obtained from each amplicon. Sequences were depleted of chimeras using the Black Box Chimera Check program (B2C2) (Research and Testing Laboratory, Lubbock, TX). The remaining sequences were analyzed using the QIIME pipeline (8), in which sequences were clustered into species-level operational taxonomic units (s-OTUs) at 97% sequence similarity and assigned a taxonomic identity by alignment to a locally hosted version of the Greengenes database (13) using the Blastn algorithm. Phylogenetic trees were generated and visualized using FastTree (41). UniFrac and community diversity metrics were computed as previously described (35).
Cytokine assay.
Periopaper strips were thawed on ice, and GCF was eluted by adding 200 μl of elution buffer containing 50 mM Tris-HCl with 5 mM CaCl2, 0.2 mM NaCl (pH 7.6), 1 mg/liter antipain, 1 mg/liter aprotinin, 1 mg/liter leupeptin, 125 mg N-ethylaleimide, and 50 mg Zwittergent 3-12 and vortexing vigorously at 15-min intervals for an hour (21). Cytokine analysis was done using a commercially available multiplex bead-based assay designed to quantitate multiple cytokines (38). A panel of 27 cytokines was selected, including Th1 and Th2 cytokines (interleukin-2 [IL-2], IL-12, gamma interferon [IFN-γ], and IL-1ra), proinflammatory cytokines (IL-1β, IL-6, IL-12, and granulocyte-macrophage colony-stimulating factor [GM-CSF]), chemokines (IL-8, IFN-γ-induced protein 10 [IP-10], monocyte chemotactic protein 1 [MCP-1], macrophage inflammatory proteins [MIP-1α and MIP-1β], RANTES [regulated on activation, normal T expressed and secreted], and eotaxin), regulators of T cells and natural killer cells (IL-7 and IL-15), and growth factors (vascular endothelial growth factor [VEGF] and plasma-derived growth factor [PDGF]). Briefly, 27 distinct sets of fluorescently dyed beads (Bio-Rad Laboratories, Inc., Hercules, CA) were conjugated with monoclonal antibodies specific for each cytokine and incubated with 50 μl of GCF, and then 25 μl of biotinylated detection antibody and 50 μl of streptavidin-phycoerythrin reporter were sequentially added. The level of each cytokine was analyzed by measuring the fluorescence of each bead type as well as the fluorescent signal from the reporter on a Bio-Plex 200 flow cytometric detection system.
Statistical analysis.
A minimum of 100 sequences were identified from each sample. Comparisons of overall bacterial community composition were made using Fast UniFrac, a metric based on phylogenetic distances. Weighted UniFrac distances were used for principal-component analysis. A variance-stabilizing transformation was used to create a normal distribution of the data (39, 44). The proportion (p) of each s-OTU in the community of each subject was expressed as X = sin−1(√p), and repeated-measures analysis of variance (ANOVA) was used to compare the means of this transformed variable X between the two groups and over each time point. A “cytokine score” was computed to reduce intersubject variability in immune mediator levels (5). To achieve this, the absolute concentration of each analyte (nanograms per microliter of GCF) was normalized across all visits for each subject, such that the maximum value was 1. Thus, irrespective of intersubject variations in immune mediator levels, the values ranged from zero to one for each subject. A repeated-measures ANOVA was used to compare normalized scores between current smokers and nonsmokers. For both bacterial and cytokine levels, the interaction between group (smoker/nonsmoker) and the age of the plaque (day 1 to 7) was examined for significant differences between groups. Regression analysis with interaction term was used to examine the effect of smoking on host-bacterium relationships. Reported P values correspond to 2-tailed tests. All statistical analyses were carried out with JMP (SAS Institute Inc., Cary, NC).
RESULTS
Of the 23 subjects recruited, 8 withdrew from the study at various stages due to antibiotic use or inability to wear the stent for a prolonged period. Thus, 15 current smokers and 15 individuals who had never smoked completed the study, and samples from these subjects were included in the analysis. A power analysis revealed that the use of 15 subjects in each group will give 80 to 90% power with α = 0.05 to detect a difference of 1% between the 2 groups at any given time point (given a standard deviation [SD] of 1.4% based on our previous data [31]). The demographic characteristics of the subjects are shown in Table 1. There were no differences between the groups except for tobacco exposure, as measured by salivary cotinine levels (P < 0.05, 2-sample t test).
Table 1.
Demographic characteristics and tobacco exposure of sample population
| Characteristic | Value for group |
|
|---|---|---|
| Nonsmokers | Current smokers | |
| Mean age, yr | 21 | 20 |
| Gender | 8 males, 7 females | 12 males, 3 females |
| Pack yr tobacco use (mean ± SD)a | 0 | 1.46 ± 0.68 |
| Salivary cotinine, ng/ml (mean ± SD) | 0 | 27.3 ± 4.2 |
Number of packs per day multiplied by number of years of smoking.
The clinical features of the two groups are shown in Fig. 1. The plaque index increased significantly between day 1 and day 7 (P < 0.05 by repeated-measures ANOVA); however, there were no differences in gingival or plaque index between the two groups for any of the 4 observation periods (P > 0.05).
Fig. 1.
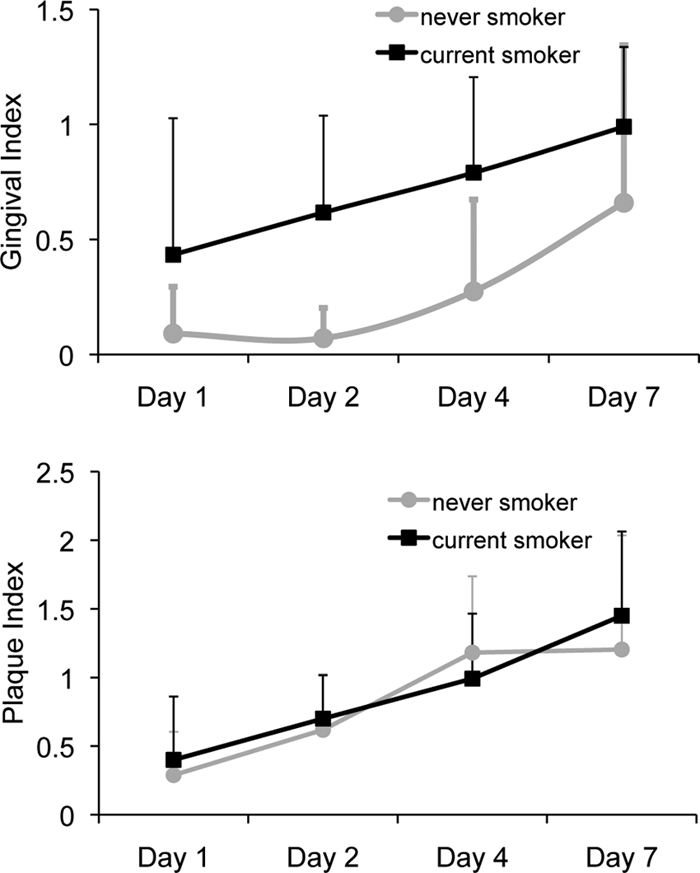
Gingival and plaque indices of 15 current smokers and 15 nonsmokers over 7 days of plaque accumulation. Both groups demonstrated an increase in plaque and gingival indices over the observation period.
A total of 12,078 high-quality, chimera-depleted sequences were used in all analyses. Overall, these sequences represented the phyla Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, Spirochaetes, and Synergistes, with Firmicutes accounting for 83.4% of all sequences. These sequences represented 158 and 271 s-OTUs in the marginal biofilms and 216 and 241 s-OTUs in the subgingival biofilms of nonsmokers and current smokers, respectively. Smokers demonstrated greater bacterial diversity than nonsmokers during the early days of bacterial colonization in both marginal and subgingival plaques (P < 0.05 by repeated-measures ANOVA) (Fig. 2). Moreover, smokers also demonstrated a decrease in bacterial diversity in both the marginal and the subgingival habitats over 7 days of plaque development (P < 0.05 by repeated-measures ANOVA). In nonsmokers, 71% of the community remained stable over the 7-day period, while in smokers, only 46% of the community was stable over the same period (data not shown). This was reflected by the statistically significant decrease in diversity in smokers, but not nonsmokers, from day 1 to day 7 in both habitats.
Fig. 2.

Shannon diversity index over 7 days in current smokers and nonsmokers. Results for subgingival biofilm are shown in the upper panel and those for marginal biofilm in the lower panel. Smokers demonstrated a decrease in diversity over 7 days (#, P < 0.05 by repeated-measures ANOVA) and showed a greater diversity than nonsmokers (*, P < 0.05; **, P < 0.01).
Principal-component analysis of unweighted UniFrac distances revealed significant clustering of bacterial sequences by tobacco exposure in both the subgingival and marginal communities (P < 0.05 by distance-based ANOVA with permutation) (Fig. 3A and B).
Fig. 3.

Clustering of microbial communities using principal-component analysis (PCoA) of unweighted UniFrac distances. (A) Subgingival communities; (B) marginal communities. Both communities demonstrated a significant clustering based on tobacco exposure.
Smokers demonstrated significantly divergent patterns of bacterial acquisition and colonization over 7 days compared to nonsmokers. This difference was apparent in both the marginal and subgingival microbiomes. When averaged over the 7-day observation period, the genera Streptococcus, Veillonella, Neisseria, Abiotrophia, and Selenomonas formed 70% of the subgingival community in nonsmokers, while in smokers, Streptococcus, Veillonella, Selenomonas, Campylobacter, Pseudomonas, Dialister, Abiotrophia, Neisseria, and Prevotella accounted for this number (Fig. 4). Moreover, during the first 7 days, smokers acquired several species/phylotypes belonging to the genera Lactobacillus, Fusobacterium, Centipeda, Pseudomonas, Leptotrichia, Synergistes, Propionibacterium, and Cardiobacterium, while these species were absent in nonsmokers. In marginal plaque (Fig. 5), smokers demonstrated consistently greater 7-day average levels of Streptococcus, Haemophilus, Kingella, Selenomonas, Lachnospira, and Pseudomonas and lower levels of Neisseria, Actinomyces, Rothia, and Lautropia than nonsmokers (P < 0.05 by repeated-measures ANOVA on transformed variable). Only smokers acquired the genera Lactobacillus, Treponema, and Pseudomonas during this observation period.
Fig. 4.

Distribution by genus of clones in the subgingival biofilms of current smokers and nonsmokers over 7 days. Genera accounting for >0.5% of total clones are shown. Significant differences in the levels of several genera were observed between the two groups (by repeated-measures ANOVA on transformed variable, *, P < 0.05; **, P < 0.01).
Fig. 5.

Distribution by genus of clones in the marginal biofilms of current smokers and nonsmokers over 7 days. Genera accounting for >0.5% of total clones are shown. Significant differences in the levels of several genera were observed between the two groups (by repeated-measures ANOVA on transformed variable, *, P < 0.05; **, P < 0.01).
Smokers demonstrated significantly higher levels of G-CSF, IL-1β, IL-2, IL-8, IL-9, IL-12, IFN-γ, RANTES, tumor necrosis factor alpha (TNF-α), and VEGF than nonsmokers during one or more days of plaque development (P < 0.05 by repeated-measures ANOVA on normalized score) (Table 2). VEGF and G-CSF were consistently higher in smokers over the whole observation period. Nonsmokers demonstrated significantly higher levels of IL-15, IL-10, and MCP-1 than current smokers (P < 0.05).
Table 2.
Levels of 27 immune mediators in current smokers and nonsmokers over 7 days
| Immune mediator | Mean ng/μl ± SD (median ng/μl)a on day: |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 |
2 |
4 |
7 |
|||||
| Nonsmokers (n = 15) | Current smokers (n = 15) | Nonsmokers (n = 15) | Current smokers (n = 15) | Nonsmokers (n = 15) | Current smokers (n = 15) | Nonsmokers (n = 15) | Current smokers (n = 15) | |
| IL-1ra | 26,023 ± 8,869 (24,749) | 26,628 ± 15,772 (23,900) | 29,681 ± 10,782 (27,502) | 25,966 ± 11,427 (27,273) | 26,581 ± 8,008 (23,976) | 20,429 ± 15,893 (25,098) | 24,831 ± 8,074 (24,593) | 26,454 ± 11,817 (21,068) |
| IL-4 | 23 ± 16 (17) | 29 ± 19 (26) | 15 ± 13 (13) | 28 ± 14 (30) | 16 ± 13 (12) | 24 ± 15 (24) | 29 ± 19 (27) | 35 ± 15 (31) |
| IL-10 | 73 ± 93 (52) | 78 ± 105 (13) | 66 ± 62 (38) | 71 ± 89 (70) | 76 ± 79 (59) | 33 ± 59 (0) | 72 ± 66 (79) | 9 ± 21 (0)** |
| IL-13 | 93 ± 78 (69) | 114 ± 89 (77) | 110 ± 138 (42) | 90 ± 115 (68) | 68 ± 70 (40) | 109 ± 131 (90) | 92 ± 89 (75) | 74 ± 64 (41) |
| G-CSF | 33 ± 28 (21) | 133 ± 167 (88)*** | 29 ± 37 (12) | 113 ± 103 (126)** | 41 ± 60 (23) | 114 ± 82 (106)** | 61 ± 70 (46) | 130 ± 74 (86)* |
| IL-1b | 7,915 ± 3,843 (7,726) | 15,271 ± 9,302 (17,208)** | 8,437 ± 6,054 (5,920) | 14,023 ± 8,017 (15,334)** | 9,072 ± 4,303 (7,120) | 15,484 ± 8,353 (17,752)* | 13,471 ± 5,833 (12,106) | 20,109 ± 10,356 (16,045) |
| IL-2 | 35 ± 14 (37) | 59 ± 42 (50)* | 31 ± 15 (33) | 56 ± 33 (48)* | 32 ± 15 (30) | 61 ± 49 (61)* | 40 ± 23 (39) | 64 ± 36 (49) |
| IL-5 | 2 ± 2 (2) | 7 ± 6 (6) | 2 ± 4 (0) | 7 ± 7 (6) | 1 ± 2 (0) | 13 ± 30 (8) | 2 ± 2 (0) | 8 ± 5 (4) |
| IL-6 | 76 ± 69 (58) | 131 ± 189 (42) | 70 ± 53 (65) | 49 ± 152 (99) | 45 ± 27 (47) | 76 ± 86 (41) | 42 ± 30 (35) | 68 ± 59 (40) |
| IL-7 | 16 ± 10 (15) | 26 ± 24 (25) | 14 ± 10 (13) | 21 ± 18 (17) | 12 ± 8 (10) | 31 ± 43 (25) | 15 ± 10 (16) | 24 ± 16 (17) |
| IL-8 | 14,430 ± 7,584 (12,321) | 26,032 ± 17,105 (25,143)** | 13,992 ± 7,798 (14,029) | 23,060 ± 15,101 (20,555)* | 10,147 ± 6,030 (8,090) | 16,216 ± 16,686 (18,200) | 9,431 ± 4,803 (7,581) | 18,509 ± 8,840 (15,445) |
| IL-9 | 19 ± 22 (16) | 36 ± 38 (34)* | 9 ± 12 (0) | 12 ± 30 (4) | 9 ± 17 (0) | 21 ± 27 (0) | 11 ± 18 (0) | 14 ± 21 (1) |
| IL-12 | 33 ± 28 (36) | 134 ± 128 (74)** | 30 ± 45 (9) | 80 ± 145 (94) | 15 ± 24 (0) | 82 ± 53 (57)* | 21 ± 20 (19) | 71 ± 85 (32) |
| IL-15 | 101 ± 180 (30) | 0 ± 0 (0)** | 102 ± 121 (40) | 0 ± 0 (0)** | 85 ± 106 (57) | 73 ± 284 (0) | 58 ± 72 (0) | 0 ± 0 (0) |
| IL-17 | 29 ± 51 (12) | 21 ± 27 (3) | 12 ± 21 (0) | 14 ± 19 (0) | 19 ± 39 (0) | 15 ± 29 (12) | 30 ± 41 (10) | 15 ± 30 (0) |
| GM-CSF | 23 ± 20 (20) | 16 ± 14 (12) | 18 ± 14 (18) | 14 ± 11 (17) | 16 ± 15 (8) | 11 ± 9 (10) | 29 ± 21 (18) | 14 ± 10 (13) |
| IFN-γ | 21 ± 9 (17) | 41 ± 27 (27)** | 18 ± 9 (18) | 36 ± 25 (29)* | 18 ± 10 (18) | 30 ± 22 (32) | 26 ± 16 (29) | 40 ± 22 (28) |
| MCP-1 | 63 ± 80 (32) | 14 ± 45 (0)** | 13 ± 20 (0) | 0 ± 1 (0) | 32 ± 75 (0) | 12 ± 45 (0) | 5 ± 11 (0) | 5 ± 18 (0) |
| MIP-1a | 88 ± 58 (85) | 90 ± 93 (83) | 84 ± 66 (64) | 72 ± 50 (72) | 93 ± 66 (76) | 77 ± 52 (61) | 94 ± 51 (93) | 88 ± 61 (58) |
| MIP-1b | 3,045 ± 2,572 (2,925) | 2,517 ± 4,410 (1,031) | 1,549 ± 2,434 (1,030) | 621 ± 3,304 (698) | 474 ± 235 (420) | 442 ± 501 (339) | 536 ± 374 (407) | 334 ± 389 (125) |
| RANTES | 33 ± 16 (31) | 93 ± 203 (47)** | 24 ± 16 (26) | 173 ± 522 (57)*** | 26 ± 14 (30) | 32 ± 16 (38) | 43 ± 22 (45) | 70 ± 44 (38) |
| TNF-α | 21 ± 47 (0) | 55 ± 48 (58)* | 10 ± 17 (0) | 39 ± 49 (36)* | 14 ± 30 (0) | 63 ± 54 (63)** | 38 ± 53 (0) | 52 ± 52 (52) |
| VEGF | 344 ± 265 (261) | 1,655 ± 1,273 (1,870)*** | 318 ± 310 (155) | 1,298 ± 1,206 (1,299)** | 307 ± 362 (208) | 1,203 ± 892 (767)** | 367 ± 286 (307) | 1,309 ± 995 (1,012)** |
| PDGF-bb | 72 ± 68 (37) | 152 ± 151 (102) | 92 ± 144 (33) | 132 ± 141 (176) | 41 ± 40 (36) | 115 ± 83 (76) | 63 ± 56 (66) | 101 ± 89 (48) |
| IP-10 | 7,450 ± 14,862 (2,596) | 10,764 ± 8,695 (3,161) | 1,488 ± 2,626 (407) | 1,044 ± 12,030 (722) | 193 ± 250 (119) | 939 ± 975 (465) | 192 ± 324 (83) | 1,106 ± 1,813 (372) |
| Eotaxin | 32 ± 15 (31) | 56 ± 65 (12) | 29 ± 14 (25) | 49 ± 53 (23) | 26 ± 14 (24) | 36 ± 41 (29) | 34 ± 16 (37) | 29 ± 38 (13) |
| FGFBb | 55 ± 60 (47) | 101 ± 146 (18) | 52 ± 38 (43) | 56 ± 81 (1) | 52 ± 45 (36) | 70 ± 103 (6) | 61 ± 40 (57) | 68 ± 116 (2) |
Significant differences between the two groups (by 2-sample t test on normalized score) are highlighted in gray. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FGFB, fibroblast growth factor basic.
Significantly opposing patterns of interactions were observed between subgingival levels of Streptococcus and certain immune mediators among current smokers and nonsmokers (Fig. 6). In both groups, these interactions were significant (P < 0.05). In smokers, a positive correlation was observed between the levels of Streptococcus and IL-2, G-CSF, VEGF, and RANTES, whereas nonsmokers showed a negative correlation. On the other hand, a positive interaction was observed between the levels of Selenomonas and IL-2 and RANTES in nonsmokers but not in current smokers.
Fig. 6.

Best linear fits for coassociation between subgingival genera and inflammatory cytokines in current smokers and nonsmokers. P values highlighted in gray correspond to the null hypothesis that the two slopes are the same. P values corresponding to the null hypothesis that each slope is zero are also shown (*, P < 0.05; **, P < 0.01).
When these correlations were examined irrespective of smoking status, there was a negative correlation between levels of streptococci and the levels of these immune mediators, while a positive correlation was observed between IL-2, IL-9, RANTES, and Selenomonas.
DISCUSSION
In order to understand the temporal effects of smoking on oral bacterial colonization, the marginal and subgingival biofilms of 15 current smokers and 15 individuals who had never smoked were examined over 7 days of plaque development. Previous studies have demonstrated that the oral microbiome has several distinct microbial niches, e.g., the tooth surface, the subgingival sulcus, and oral mucosal surfaces (48, 56). The subgingival sulcus provides both a shedding mucosal surface (the sulcular epithelium) and a nonshedding surface (the tooth). Therefore, a tooth-associated habitat and a mucosa-associated habitat were selected to obtain a representational characterization of the oral microbiome. Combining an open-ended molecular approach with a multiplex immunological assay and an adequately powered, longitudinal clinical study design revealed a critical role for smoking in altering colonization in a health-compatible oral microbiome. To the best of our knowledge, this is the first study to demonstrate such an influence.
A “zero-plaque” model was used in the present study to examine the compositional and inflammatory changes that occur in the biofilm during undisturbed plaque formation. Traditionally, longitudinal evaluations of plaque formation have been limited by disruption of the biofilm when plaque samples are collected at specific intervals. Previous investigations have sought to overcome this by sampling different teeth at different time points (43). However, sampling different teeth at each time point may not be representational of the changes that occur in the mouth, since it is known that tooth morphology and position play a role in determining the composition of the biofilm (47). Therefore, in the present investigation, the sampling sites were professionally cleaned to remove the biofilm following sampling at each time point. By resetting the “sampling clock” after each visit, undisturbed plaque samples were collected at 1, 2, 4, and 7 days. Samples collected immediately following oral prophylaxis did not demonstrate the presence of bacterial DNA (data not shown), indicating that de novo colonization occurred following each visit.
Over 12,000 near-full-length, high-quality sequences were analyzed, allowing high-resolution phylogenetic comparisons to be made. Sanger sequencing was used as the method of choice for this open-ended investigation, because even though deep-sequencing methodologies provide a greater depth of coverage, it has been demonstrated that short-read “tag” sequences do not provide the same stringency of phylogenetic classification as longer sequence reads (27).
UniFrac distances were used to quantify the similarities between each pair of samples. This methodology uses 16S rRNA gene sequences from each community to create a common phylogenetic tree, from which branch lengths for each community are computed. Thus, a lower UniFrac value indicates that the lineages that make up both communities are closely related, while higher values indicate a greater phylogenetic difference. Principal-coordinate analysis of UniFrac distances was used to examine the contributions of smoking to the compositions of the marginal and subgingival plaque communities. Our data revealed a high level of partitioning between current smokers and nonsmokers irrespective of the age of the biofilm, suggesting that tobacco exposure influences bacterial composition during all stages of plaque development. The data also indicated the need for further studies examining the long-term effects of smoking on the oral microbiome.
Bacterial colonization of the plaque biofilm occurs in a specific temporal sequence, leading to the formation of a health-compatible climax community (29). A central characteristic of a healthy, stable community is niche occupation or niche saturation, whereby a few selected species occupy an ecological habitat and provide resistance to colonization of this niche by pathogenic organisms (6). Recent evidence indicates that increased species diversity within an ecosystem drives further speciation (17). Smokers demonstrated a greater diversity than nonsmokers during initial colonization of both marginal and subgingival plaques. These microbial communities also demonstrated lower stability over the 7-day period than those of nonsmokers. The predominant genera found in nonsmokers (Streptococcus, Neisseria, and Veillonella) were previously reported to be abundant members of health-associated subgingival and supragingival communities (1, 32). Also, the levels of these genera did not change significantly during the 7-day observation period. Taking the findings together, it appears that periodontal health is associated with large numbers of a few species that stably saturate this niche. This phenomenon of niche saturation did not occur to the same extent in smokers, who were colonized by several types of genera within 24 h, many of which have been associated with disease (30, 46). The communities in smokers also demonstrated greater fluctuation than those in nonsmokers. These characteristics are similar to those of the community associated with periodontitis in smokers (24, 44, 52, 53). Our data suggest that an initial lack of niche saturation lowers colonization resistance and increases the susceptibility of this ecosystem to future colonization by pathogenic organisms. Further studies are needed to examine the long-term effects of smoking on subgingival and marginal biofilms.
Smokers were colonizers by pathogens within 24 hours of biofilm development, and these organisms persisted during the observation period. While the implications of acquiring periodontal pathogens in the biofilm are obvious, these individuals also acquired systemic pathogens such as Haemophilus and Pseudomonas. Members of both of these genera are prolific biofilm formers and have been associated with refractory and antibiotic-resistant disease (15, 19). It is known that smokers are at a higher risk for developing invasive antibiotic-resistant infections (4, 7, 9, 16, 23, 37), and the results of the present study suggest that smoking increases susceptibility to disease by promoting early acquisition and colonization of biofilm-forming pathogens.
Periodontal disease occurs due to an exaggerated host response to a bacterial trigger. Although it is known that smokers exhibit higher levels of certain proinflammatory cytokines in disease (3, 20), the baseline immune response of these high-risk individuals to normal bacterial colonization is not known. Examining a panel of immune mediators known to be important in the etiology of periodontal disease allowed for the quantification of the effects of smoking on the host response to early bacterial colonization. The levels of immune mediators exhibited a large interindividual variation, as reported by other investigators (54). A variance-stabilizing transformation was created to overcome these large interindividual differences. This has been previously validated on serum levels of immune mediators (5, 10). In the present study, onset of clinical disease occurred earlier in smokers than in nonsmokers, suggesting a prematurely amplified immune response to bacterial colonization. This clinical picture was corroborated by the immunological findings, with smokers demonstrating significantly higher levels of proinflammatory immune mediators (e.g., IL-1β, IL-2, IL-8, G-CSF, etc.) than nonsmokers from the first day. It is possible that this early elevation in immune mediator levels may persist during biofilm maturation, thereby increasing susceptibility to periodontitis in smokers; this warrants further investigation using long-term studies on plaque development and maturation.
Several opposing patterns of host-bacterium coassociations were observed in smokers and nonsmokers. The most robust of these associations were between streptococci and IL-2, RANTES, VEGF, and G-CSF and between the selenomonads and IL-2 and RANTES. Smokers not only demonstrated greater subgingival levels of streptococci than nonsmokers but also displayed a strong, positive correlation between a highly proinflammatory response and these bacterial levels. In contrast, nonsmokers showed an equally strong but negative correlation between bacterial levels and a proinflammatory response. Streptococci are early commensal colonizers of the subgingival and supragingival habitats (14, 29) and are abundant members of these communities in health (1, 31, 32, 58). Our data suggest that in nonsmokers, these commensals play an immunomodulatory role in the developing biofilm, primarily by attenuating the proinflammatory response. Our results are in agreement with emerging evidence demonstrating that streptococci exert a powerful anti-inflammatory effect on oral mucosal cells (11, 25). Our data also suggest that this immunomodulation is reversed in smokers. A strong correlation was also observed between the genus Selenomonas and the proinflammatory response in nonsmokers but not in smokers, providing further evidence of an altered host-bacterium interaction in these high-risk individuals. Selenomonas spp. are putative periodontal pathogens that have been associated with both gingivitis and periodontitis and are abundant members of these disease-associated communities (30, 40, 46, 49). Thus, the data support a critical role for smoking in modulating host-bacterium interactions in the oral cavity. However, it is not clear from this study whether smoking exerts this effect by decreasing the host response to bacteria, by altering the immunomodulatory behavior of the commensals, or by a combination of both. Further studies using in vitro or ex vivo methods are required to elucidate this mechanism.
In summary, smokers demonstrated a highly diverse, relatively unstable initial colonization of both marginal and subgingival biofilms, with lower niche saturation than that seen in nonsmokers. Several pathogens are acquired within the first 24 hours of biofilm formation in smokers and continue to persist over the observation period, suggesting that smoking alters bacterial acquisition and colonization in oral biofilms in favor of periodontopathogens. Smokers demonstrated a highly proinflammatory response to this early colonization, which persisted over 7 days. Opposing patterns of coassociations between predominant genera and proinflammatory cytokines were observed between current smokers and nonsmokers, providing evidence of altered host-bacterium interactions. Thus, the present investigation suggests a critical role for smoking in altering the oral microbiome in health, and further studies are required to examine the long-term effects of smoking on this host-associated ecosystem.
ACKNOWLEDGMENT
This study was supported by a research grant from Philips Oral Healthcare.
Footnotes
Published ahead of print on 22 August 2011.
REFERENCES
- 1. Aas J. A., Paster B. J., Stokes L. N., Olsen I., Dewhirst F. E. 2005. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 43:5721–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahijevych K. L., Wewers M. E. 1994. Patterns of cigarette consumption and cotinine levels among African American women smokers. Am. J. Respir. Crit. Care Med. 150:1229–1233 [DOI] [PubMed] [Google Scholar]
- 3. Apatzidou D. A., Riggio M. P., Kinane D. F. 2005. Impact of smoking on the clinical, microbiological and immunological parameters of adult patients with periodontitis. J. Clin. Periodontol. 32:973–983 [DOI] [PubMed] [Google Scholar]
- 4. Aronson M. D., Weiss S. T., Ben R. L., Komaroff A. L. 1982. Association between cigarette smoking and acute respiratory tract illness in young adults. JAMA 248:181–183 [PubMed] [Google Scholar]
- 5. Bauer J. W., et al. 2006. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 3:e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brockhurst M. A., Colegrave N., Hodgson D. J., Buckling A. 2007. Niche occupation limits adaptive radiation in experimental microcosms. PLoS One 2:e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brook I., Gober A. E. 2005. Recovery of potential pathogens and interfering bacteria in the nasopharynx of smokers and nonsmokers. Chest 127:2072–2075 [DOI] [PubMed] [Google Scholar]
- 8. Caporaso J. G., et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charlson E. S., et al. 2010. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One 5:e15216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Correia L. C., et al. 2010. Prognostic value of cytokines and chemokines in addition to the GRACE score in non-ST-elevation acute coronary syndromes. Clin. Chim Acta 411:540–545 [DOI] [PubMed] [Google Scholar]
- 11. Cosseau C., et al. 2008. The commensal Streptococcus salivarius K-12 downregulates the innate immune responses of human epithelial cells and promotes host-microbe homeostasis. Infect. Immun. 76:4163–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delima S. L., McBride R. K., Preshaw P. M., Heasman P. A., Kumar P. S. 2010. Response of subgingival bacteria to smoking cessation. J. Clin. Microbiol. 48:2344–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeSantis T. Z., et al. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diaz P. I., et al. 2006. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl. Environ. Microbiol. 72:2837–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drenkard E., Ausubel F. M. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743 [DOI] [PubMed] [Google Scholar]
- 16. El Ahmer O. R., et al. 1999. The effect of cigarette smoke on adherence of respiratory pathogens to buccal epithelial cells. FEMS Immunol. Med. Microbiol. 23:27–36 [DOI] [PubMed] [Google Scholar]
- 17. Emerson B. C., Kolm N. 2005. Species diversity can drive speciation. Nature 434:1015–1017 [DOI] [PubMed] [Google Scholar]
- 18. Fullmer S. C., Preshaw P. M., Heasman P. A., Kumar P. S. 2009. Smoking cessation alters subgingival microbial recolonization. J. Dent. Res. 88:524–528 [DOI] [PubMed] [Google Scholar]
- 19. Galli J., et al. 2007. Biofilm formation by Haemophilus influenzae isolated from adeno-tonsil tissue samples, and its role in recurrent adenotonsillitis. Acta Otorhinolaryngol. Ital. 27:134–138 [PMC free article] [PubMed] [Google Scholar]
- 20. Giannopoulou C., Kamma J. J., Mombelli A. 2003. Effect of inflammation, smoking and stress on gingival crevicular fluid cytokine level. J. Clin. Periodontol. 30:145–153 [DOI] [PubMed] [Google Scholar]
- 21. Golub L. M., et al. 2008. Subantimicrobial-dose doxycycline modulates gingival crevicular fluid biomarkers of periodontitis in postmenopausal osteopenic women. J. Periodontol. 79:1409–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gomes S. C., et al. 2006. Periodontal status in smokers and never-smokers: clinical findings and real-time polymerase chain reaction quantification of putative periodontal pathogens. J. Periodontol. 77:1483–1490 [DOI] [PubMed] [Google Scholar]
- 23. Gryczynska D., Kobos J., Zakrzewska A. 1999. Relationship between passive smoking, recurrent respiratory tract infections and otitis media in children. Int. J. Pediatr. Otorhinolaryngol. 49(Suppl. 1):S275–S278 [DOI] [PubMed] [Google Scholar]
- 24. Haffajee A. D., Socransky S. S. 2001. Relationship of cigarette smoking to the subgingival microbiota. J. Clin. Periodontol. 28:377–388 [DOI] [PubMed] [Google Scholar]
- 25. Hasegawa Y., et al. 2007. Gingival epithelial cell transcriptional responses to commensal and opportunistic oral microbial species. Infect. Immun. 75:2540–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hujoel P. P., del Aguila M. A., DeRouen T. A., Bergstrom J. 2003. A hidden periodontitis epidemic during the 20th century? Community Dent. Oral Epidemiol. 31:1–6 [DOI] [PubMed] [Google Scholar]
- 27. Huse S. M., et al. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kolenbrander P. E., Andersen R. N., Moore L. V. 1989. Coaggregation of Fusobacterium nucleatum, Selenomonas flueggei, Selenomonas infelix, Selenomonas noxia, and Selenomonas sputigena with strains from 11 genera of oral bacteria. Infect. Immun. 57:3194–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kolenbrander P. E., Palmer R. J. J. 2004. Human oral bacterial biofilms, p. 85–117 In Ghannoum M., O'Toole G. (ed.), Microbial biofilms. ASM Press, Washington, DC [Google Scholar]
- 30. Kumar P. S., et al. 2003. New bacterial species associated with chronic periodontitis. J. Dent. Res. 82:338–344 [DOI] [PubMed] [Google Scholar]
- 31. Kumar P. S., Griffen A. L., Moeschberger M. L., Leys E. J. 2005. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J. Clin. Microbiol. 43:3944–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumar P. S., et al. 2006. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J. Clin. Microbiol. 44:3665–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Listgarten M. A. 1976. Structure of the microbial flora associated with periodontal health and disease in man. A light and electron microscopic study. J. Periodontol. 47:1–18 [DOI] [PubMed] [Google Scholar]
- 34. Loe H., Silness J. 1963. Periodontal disease in pregnancy. I. Prevalence and severity. Acta Odontol. Scand. 21:533–551 [DOI] [PubMed] [Google Scholar]
- 35. Lozupone C., Lladser M. E., Knights D., Stombaugh J., Knight R. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5:169–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marsh P. D., Bradshaw D. J. 1995. Dental plaque as a biofilm. J. Ind. Microbiol. 15:169–175 [DOI] [PubMed] [Google Scholar]
- 37. Nuorti J. P., et al. 2000. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N. Engl. J. Med. 342:681–689 [DOI] [PubMed] [Google Scholar]
- 38. Offenbacher S., et al. 2010. Changes in gingival crevicular fluid inflammatory mediator levels during the induction and resolution of experimental gingivitis in humans. J. Clin. Periodontol. 37:324–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osborne J. W. 2002. Notes on the use of data transformations. Practical Assessment Res. Eval. 8:(6). [Google Scholar]
- 40. Paster B. J., et al. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183:3770–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Price M. N., Dehal P. S., Arkin A. P. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26:1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramberg P., Sekino S., Uzel N. G., Socransky S., Lindhe J. 2003. Bacterial colonization during de novo plaque formation. J. Clin. Periodontol. 30:990–995 [DOI] [PubMed] [Google Scholar]
- 43. Salvi G. E., et al. 2005. Experimental gingivitis in cigarette smokers: a clinical and microbiological study. J. Clin. Periodontol. 32:441–447 [DOI] [PubMed] [Google Scholar]
- 44. Shchipkova A. Y., Nagaraja H. N., Kumar P. S. 2010. Subgingival microbial profiles of smokers with periodontitis. J. Dent. Res. 89:1247–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Socransky S. S., Haffajee A. D. 2005. Periodontal microbial ecology. Periodontology 2000 38:135–187 [DOI] [PubMed] [Google Scholar]
- 46. Socransky S. S., Haffajee A. D., Cugini M. A., Smith C., Kent R. L., Jr 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144 [DOI] [PubMed] [Google Scholar]
- 47. Sreenivasan P. K., et al. 2010. Regional differences within the dentition for plaque, gingivitis, and anaerobic bacteria. J. Clin. Dent. 21:13–19 [PubMed] [Google Scholar]
- 48. Takahashi N. 2005. Microbial ecosystem in the oral cavity: metabolic diversity in an ecological niche and its relationship with oral diseases. Int. Congr. Ser. 1284:103 [Google Scholar]
- 49. Tanner A., Maiden M. F., Macuch P. J., Murray L. L., Kent R. L., Jr 1998. Microbiota of health, gingivitis, and initial periodontitis. J. Clin. Periodontol. 25:85–98 [DOI] [PubMed] [Google Scholar]
- 50. Tomar S. L., Asma S. 2000. Smoking-attributable periodontitis in the United States: findings from NHANES III National Health and Nutrition Examination Survey. J. Periodontol. 71:743–751 [DOI] [PubMed] [Google Scholar]
- 51. Turesky S., Gilmore N. D., Glickman I. 1970. Reduced plaque formation by the chloromethyl analogue of victamine C. J. Periodontol. 41:41–43 [DOI] [PubMed] [Google Scholar]
- 52. Van der Velden U., et al. 2003. Effect of smoking and periodontal treatment on the subgingival microflora. J. Clin. Periodontol. 30:603–610 [DOI] [PubMed] [Google Scholar]
- 53. van Winkelhoff A. J., Bosch-Tijhof C. J., Winkel E. G., van der Reijden W. A. 2001. Smoking affects the subgingival microflora in periodontitis. J. Periodontol. 72:666–671 [DOI] [PubMed] [Google Scholar]
- 54. van Zyl-Smit R. N., Zwerling A., Dheda K., Pai M. 2009. Within-subject variability of interferon-γ assay results for tuberculosis and boosting effect of tuberculin skin testing: a systematic review. PLoS One 4:e8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. WHO 2009. Report on the global tobacco epidemic. WHO, Geneva, Switzerland [Google Scholar]
- 56. Winkelhoff A. J., Steenbergen T. J. M., Graaff J. 1985. Ecology of the oral cavity. Antonie Van Leeuwenhoek 51:599 [Google Scholar]
- 57. Zambon J. J., et al. 1996. Cigarette smoking increases the risk for subgingival infection with periodontal pathogens. J. Periodontol. 67:1050–1054 [DOI] [PubMed] [Google Scholar]
- 58. Zaura E., Keijser B. J., Huse S. M., Crielaard W. 2009. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9:259. [DOI] [PMC free article] [PubMed] [Google Scholar]