

Abstract
Leptospirosis is a widespread zoonosis characterized by multiple organ failure and variable host susceptibility toward pathogenic Leptospira strains. In this study, we put the role of inflammatory mediators in parallel with bacterial burdens and organ lesions by comparing a susceptible animal model, the hamster, and a resistant one, the Oncins France 1 (OF1) mouse, both infected with virulent Leptospira interrogans serovar Icterohaemorrhagiae strain Verdun. Histological observations evidenced edema, congestion, hemorrhage, and inflammatory infiltration in the organs of hamsters, in contrast to limited changes in mice. Using reverse transcription-quantitative PCR techniques, we showed that the relative Leptospira burden progressively increased in hamster tissues, while a rapid clearance was observed in mouse tissues. The early regulation of the proinflammatory mediators interleukin-1β (IL-1β), IL-6, tumor necrosis factor alpha, and cyclo-oxygenase-2 and the chemokines gamma interferon-inducible protein 10 kDa/CXCL10 and macrophage inflammatory protein-1α/CCL3 in mouse tissues contrasted with their delayed and massive overexpression in hamster tissues. Conversely, the induction of the anti-inflammatory cytokine IL-10 was faster in the resistant than in the susceptible animal model. The role of these cytokines in the pathophysiology of leptospirosis and the implications of their differential regulation in the development of this disease are discussed.
INTRODUCTION
Leptospirosis is a widespread zoonotic disease caused by pathogenic spirochetes of the genus Leptospira. It is transmitted to humans by direct or indirect exposure to contaminated urine from mammalian reservoir hosts such as rodents but also farm, wild, and domestic mammals (1). The clinical presentations of the disease are extremely variable, including asymptomatic or nonsevere forms of leptospirosis with fever, headache, and myalgia that can spontaneously resolve. However, the disease can also degenerate to more severe cases requiring hospitalization in an intensive care unit with severe sepsis and multiple organ failure, including hepatic dysfunctions with jaundice, acute renal failure, and hemorrhage, a presentation referred to as Weil's syndrome (18). Severe pulmonary forms of leptospirosis (SPFL) with acute respiratory distress syndrome (ARDS) and pulmonary hemorrhage are also reported and have high mortality rates, reaching 50% (5, 57, 65).
Several plasmatic inflammatory markers are increased during sepsis and organ failure, leading to or demonstrating a dysregulation of the inflammatory response to infectious pathogens. There is, notably, a massive release of inflammatory cytokines, such as tumor necrosis factor (TNF-α), interleukin-1β (IL-1β), IL-6, and IL-10, that induces organ dysfunction, contributing to fatal issues in septic shock (36, 37, 40). Moreover, C-C-type chemokines such as macrophage inflammatory protein-1α (MIP-1α)/CCL3 and MIP-1β/CCL4 or C-X-C-type chemokines such as IL-8/CXCL8 and gamma interferon (IFN-γ)-inducible protein 10 kDa (IP-10)/CXCL10 are also induced during sepsis (15, 42). Similar to sepsis, human leptospirosis was shown to be associated with high plasmatic levels of inflammatory cytokines such as IL-6, IL-10, TNF-α, and IL-12p40, a subunit of the inflammatory cytokine IL-12 (8, 58). Circulating levels of the chemokines IL-8/CXCL8, monokine induced by IFN-γ (Mig)/CXCL9, and IP-10/CXCL10 were also found to be increased in patients with leptospirosis (11, 58).
The diversity of the clinical forms of human leptospirosis may be associated with the variability of the host immune response, especially with different kinetics and/or intensities of cytokine expression. This was first evidenced by the measurement of elevated plasma concentrations of the proinflammatory cytokine TNF-α among leptospiremic patients with poor outcomes compared to the concentrations in the plasma of survivors or less severely affected patients (49, 50). More recently, IL-6, IL-8/CXCL8, and the acute-phase protein long pentraxin-3 (PTX-3) were all shown to be elevated in plasma from patients with severe leptospirosis compared to the plasma levels in survivors and were associated with fatal issues (59).
Animal models of leptospirosis also demonstrated the contribution of cytokines to the host response to leptospires. Indeed, we have previously shown an increase in expression of several inflammatory mediators, such as cyclo-oxygenase-2 (COX-2), IFN-γ, IL-1α, IL-4, IL-6, IL-10, IL12p40, and TNF-α, in the blood of hamsters infected with a 50% lethal dose (LD50) of virulent Leptospira interrogans serovar Icterohaemorrhagiae strain Verdun (55). The comparison between surviving and nonsurviving animals evidenced significant differences in COX-2, IL-1α, IL-10, and TNF-α mRNA levels at 3 days postinfection (p.i.), suggesting that the gene expression levels of these effectors might be predictors of a poor outcome in leptospirosis. Moreover, different expression profiles of the chemokines monocyte chemoattractant protein-1 (MCP-1)/CCL2, MIP-1α/CCL3, and IL-8/CXCL8 were noticed in tissue and serum samples between resistant BALB/c and susceptible C3H/HeJ mice inoculated with a high dose of pathogenic L. interrogans serovar Copenhageni (10). These results indicated that resistance to leptospirosis is partially correlated with the increase in MIP-1α/CCL3 levels observed in BALB/c mice, while an increasing and sustained expression of MCP-1/CCL2 and IL-8/CXCL8 in the lungs of C3H/HeJ mice correlated with the severity and progression of the disease.
The histopathological analysis of the lungs of hamsters infected with L. interrogans serovar Icterohaemorrhagiae showed hemorrhage, alveolar congestion, and infiltrated cellular areas associated with both TNF-α and IL-10 overexpression (26). In the kidneys of hamsters infected with L. interrogans serovar Pyrogenes, IP-10/CXCL10, IL-10, transforming growth factor β (TGF-β), and TNF-α were also overexpressed, suggesting a role of T cell recruitment in the immune response and the pathology of renal leptospirosis (21). Moreover, histological observations of kidneys from leptospiremic guinea pigs showed interstitial inflammatory infiltrates, confirming involvement of cellular immunity (33). Warthin-Starry (WS) silver staining of tissues realized in the same study also revealed large numbers of leptospires in kidneys, liver, and spleen but not in lungs from lethally infected animals. Thus, the burden of leptospires does not necessarily seem to be associated with lesions, especially in the lungs, as was concluded in a previous study in infected hamsters (54). However, evidence of a critical threshold of Leptospira density related to lethality was observed in susceptible models of leptospirosis in both mice (2) and guinea pigs (20). Similarly, a threshold bacteremia of 104 leptospires/ml was shown to prognosticate a poor outcome in humans (45, 52).
All these different studies focusing on human cases or using different in vivo animal models suggest that the pathophysiology of leptospirosis, especially the development of severe lesions in target organs of susceptible individuals, can be correlated with a sustained overexpression of cytokines and chemokines and Leptospira burden compared to asymptomatic or less susceptible animals. In this study, we aimed to better understand the variability of the host immune response to pathogenic Leptospira by comparing two animal models: the susceptible golden Syrian hamster and the resistant Oncins France 1 (OF1) mouse. For this purpose, we characterized the cytokine mRNA expression profiles, in parallel with a detailed histological analysis and the quantification of the relative Leptospira load in blood and target organ samples.
MATERIALS AND METHODS
Animals and Leptospira strain.
OF1 mice (Mus musculus) and golden Syrian hamsters (Mesocricetus auratus) whose genitors were initially purchased from Charles River Laboratories (Charles River Wiga GmbH, Sulzfeld, Germany) were bred at the Institut Pasteur in New Caledonia. Virulent Leptospira interrogans serovar Icterohaemorrhagiae strain Verdun was provided by the Reference Collection of the Institut Pasteur in Paris, France. Leptospires were cultivated in liquid Ellinghausen, McCullough, Johnson, and Harris (EMJH) medium at 30°C under aerobic conditions. The bacterial cell concentration was determined using a Petroff-Hausser counting chamber (Hausser Scientific). Virulence of leptospires was maintained by cyclic passages in 5- to 6-week-old golden Syrian hamsters after intraperitoneal (i.p.) injection. Protocols for animal manipulation and experiments were prepared and conducted according to the guidelines of the Animal Care and Use Committees of the Institut Pasteur and followed European Recommendation 2007/526/EC, which provides “guidelines for the accommodation and care of animals used for experimental and other scientific purposes.” The protocol was validated before the start of the experiments by the appropriate committees of the Institute Pasteur in New Caledonia.
Experimental infection of animals.
Five- to 6-week-old healthy mice and hamsters were i.p. injected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun at the LD100 (2 × 108 bacteria), previously determined in hamster (55), in 500 to 600 μl EMJH medium. Animals were stocked in individual cages at room temperature with sawdust bedding, fed their routine food, and watered with a bottle of sterile water during the experimentations. Kinetic analyses of gene expression and target organ histology were conducted by the study of injected individual hamsters and mice at each of the following time points: 3, 6, 12, 24, 48, and 72 h, plus additional points at 96 h for hamsters and at days 5 and 15 for mice. The effect of sterile EMJH medium on gene expression was similarly assessed by i.p. injection of 500 μl EMJH medium. Animals without injection were used as negative controls. At the time points mentioned above, 3 animals were anesthetized with chloroform (Prolabo, VWR International S.A.S, France) and whole blood (∼1.5 ml) was rapidly collected by cardiac puncture and conserved in PAXgene blood RNA tubes (PreAnalytiX; Qiagen, Australia) at room temperature for 2 h for stabilization of total RNA before storage at −20°C until RNA extraction. Immediately after blood collection, animals were euthanatized by atlanto-occipital dislocation and dissected. Macroscopic observations of the organ biopsy specimens were carried out concomitantly, and the specimens (pieces of ∼1 cm3) were placed in 1,500 μl RNAlater reagent (Sigma-Aldrich) for stabilization of total RNA at room temperature for 2 h before conservation at −20°C until RNA extraction. The remaining parts of the organs were fixed in 10% formalin and conserved at room temperature for 24 to 48 h until treatment for histological observations.
Histopathology and silver staining of Leptospira.
For each organ, formalin-fixed paraffin-embedded tissue blocks were subjected to hematoxylin-erythrosin-safran (HES) staining for tissue sections of 2 to 3.5 μm. Leptospires were visualized after silver impregnation following the Warthin-Starry (WS) protocol modified with pyrocatechol as described by Wuscher and Huerre (60). Lesions in target organs were graded (see Table 2) according to previously reported criteria (26, 33, 35, 53, 64).
Table 2.
Histological lesion grading in organs of animals inoculated with virulent L. interrogans serovar Icterohaemorrhagiae
| Organ and pathology | Lesion score at indicated times postinfection (days)a |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hamsters |
Mice |
|||||||||||
| 0.5 | 1 | 2 | 3 | 4 | 4.5 | 0.5 | 1 | 2 | 3 | 5 | 15 | |
| Liver | ||||||||||||
| Edema and congestion | − | 3 | 5 | 6 | 6 | 9 | − | − | − | − | − | − |
| Portal inflammation | − | 1 | 3 | 3 | 3 | 6 | − | 1 | − | 2 | 3 | 2 |
| Hepatocyte necrosis | − | 2 | 1 | 4 | 3 | 7 | − | − | − | 2 | 3 | 2 |
| Regeneration | − | 1 | − | 4 | 3 | 9 | − | − | 1 | 3 | 3 | − |
| Kupffer cell hyperplasia | − | − | 1 | 4 | 1 | 8 | − | − | − | − | − | − |
| Kidneys | ||||||||||||
| Interstitial nephritis | − | − | − | − | − | − | − | − | − | 2 | 1 | − |
| Congestion | − | − | − | 3 | 5 | 9 | − | − | − | 1 | − | − |
| Hemorrhage | − | − | − | − | − | 9 | − | − | − | − | − | − |
| Lungs | ||||||||||||
| Edema | − | 3 | 4 | 6 | 6 | 6 | − | 1 | 1 | 1 | 4 | − |
| Alveolar hemorrhage | − | 2 | 3 | 5 | 9 | 8 | − | 1 | − | − | 3 | − |
| Inflammation of alveolar septa | − | 2 | 3 | 3 | 5 | 6 | − | − | 1 | 1 | 3 | − |
Hamsters and mice were intraperitoneally infected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun. Scores were obtained from addition of individual scores (n = 3) after observation of HES-stained sections of each organ. −, no lesion; 1 or 2 (white), slight lesions; 3 or 4 (gray shading), moderate lesions; ≥5 (boldface), severe lesions.
Total RNA extraction.
Tissue RNA isolation was performed with commercial kits and instruments from Roche Applied Science (New Zealand). Approximately 25 mg of tissue was weighed and placed into MagNA Lyser Green Beads tubes with 400 μl lysis buffer from a High Pure RNA tissue kit, before being submitted to 2 pulses of 50 s at 6,500 rpm in a MagNA Lyser instrument. Lysates were then collected after centrifugation for 2 min at 14,000 rpm, and total RNA was then immediately isolated using the spin column-based High Pure RNA tissue kit. Briefly, samples were treated with the provided DNase I for 15 min at room temperature, and after the washing steps, 100 μl of tissue RNA was eluted following the manufacturer's recommendations. Total RNA from blood was extracted using a PAXgene blood RNA system from PreAnalytiX (Qiagen, Australia). Samples were successively treated with the provided proteinase K and DNase I for 10 min at 55°C and for 15 min at room temperature, respectively. Following the washing steps, 80 μl of blood RNA was eluted according to the manufacturer's instructions. After purification, 40 and 50 μl of blood RNA and tissue RNA were treated for 30 min at 37°C with 1 and 1.2 μl of Turbo DNase (Turbo DNA-free kit; Ambion, Applied Biosystems), respectively, for elimination of residual genomic DNA, following the manufacturer's recommendations. Before storage at −80°C, purified RNA was quantified by measurement of the optical density at 260 nm (OD260) using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and the quality of nucleic acids was verified by measurement of the OD260/OD280 ratio.
RT.
DNA-free total RNA extracted from liver, kidneys, and blood (1 μg) and pulmonary tissue (500 ng) was reverse transcribed into cDNA using random hexamer primers from a Transcriptor First Strand cDNA synthesis kit (Roche Applied Science, New Zealand). The reaction mix was composed of 5 μl of buffer (5×), 2.5 μl of deoxynucleoside triphosphates (10 mM each), 2.5 μl of primer (6,000 pmol/ml), 0.625 μl of RNase inhibitor (40 U/ml), 0.625 μl of reverse transcriptase (20 U/ml), an adequate volume of RNA, and PCR-grade H2O adjusted to a final volume of 25 μl. Reverse transcription (RT) was conducted on a GeneAmp PCR system 9700 instrument (Applied Biosystems) and consisted of the activation step at 25°C for 10 min, followed by the RT step at 55°C for 30 min and the inactivation of enzyme at 85°C for 5 min. The cDNA synthesized was conserved at −20°C until quantitative PCR (qPCR) assays.
Quantitative PCR and amplification program.
Primers (Table 1) (30) were purchased from Eurogentec (Seraing, Belgium) after design using LightCycler primer probe design software (version 2.0; Roche Applied Science, New Zealand) or the free online Primer3 software (version 0.4.0) (43) using available mRNA sequences retrieved from GenBank (NCBI). Each qPCR was carried out in duplicate with 2 or 1 μl cDNA in a 20- or 10-μl final volume for cytokines or Leptospira 16S rRNA expression, respectively, and was conducted on a LightCycler 480 II instrument using LightCycler 480 software (version 1.5.0) and the LightCycler 480 SYBR green I master kit (Roche Applied Science, New Zealand). The amplification program consisted of an activation step at 95°C for 10 s, an annealing step at 60°C for 5 s, and an elongation step at 72°C for 12 s for 45 cycles for animal gene expression. For Leptospira 16S rRNA, it consisted of the same steps of 95°C for 8 s, 62°C for 5 s, and 72°C for 12 s for 50 cycles. A single acquisition of fluorescence for calculation of the threshold cycle (CT) was processed during the elongation step. The specificity of amplification was verified by size visualization of the PCR product (Table 1) after electrophoresis on a 1.8% agarose gel (Sigma-Aldrich, Germany) in 1× TBE (Tris-borate-EDTA) for 30 to 45 min at 120 V and by analysis of the melting curves of the PCR products (melting temperatures in Table 1).
Table 1.
Details and sequence of primers used for qPCR assays
| Gene | GenBank accession no.a | Sequence (5′–3′)b | Sizec | Tmd | Efficiencye |
|---|---|---|---|---|---|
| Hamster β-actin | AF046210 | (F) TCTACAACGAGCTGCG | 357 | 88.33 | 1.802 ± 0.0438 |
| (R) CAATTTCCCTCTCGGC | |||||
| Murine β-actin | NM_007393 | (F) AAGAGAAGCTGTGCTATGTT | 251 | 86.25 | 1.660 ± 0.0960 |
| (R) GTTGGCATAGAGGTCTTTACG | |||||
| Hamster MIP-1α/CCL3 | AY819019.1 | (F) CTCCTGCTGCTTCTTCTA | 210 | 84.97 | 1.856 ± 0.0619 |
| (R) TGGGTTCCTCACTGACTC | |||||
| Murine MIP-1α/CCL3 | NM_011337.2 | (F) TCAGACACCAGAAGGATAC | 159 | 84.64 | 1.780 ± 0.0325 |
| (R) CTGAGAAGACTTGGTTGC | |||||
| Hamster IP-10/CXCL10 | AY007988.1 | (F) CTCTACTAAGAGCTGGTCC | 150 | 83.38 | 1.900 ± 0.0629 |
| (R) CTAACACACTTTAAGGTGGG | |||||
| Murine IP-10/CXCL10 | NM_021274.1 | (F) CTTAACCACCATCTTCCCAA | 152 | 76.62 | 1.581 ± 0.0339 |
| (R) GATGACACAAGTTCTTCCA | |||||
| Hamster COX-2 | AF345331 | (F) CAACTCCCTTGGGTGTGA | 173 | 81.71 | 1.905 ± 0.0407 |
| (R) TCCTCGTTTCTGATCTGTCT | |||||
| Murine COX-2 | NM_011198.3 | (F) GGGTTAAACTTCCAAAGGAG | 176 | 81.98 | 1.950 ± 0.0242 |
| (R) CTCACGAGGCCACTGATA | |||||
| Hamster GAPDH | DQ403055 | (F) CCGAGTATGTTGTGGAGTCTA | 170 | 85.67 | 1.938 ± 0.0493 |
| (R) GCTGACAATCTTGAGGGA | |||||
| Murine GAPDH | NM_008084 | (F) TCATCCCAGAGCTGAACG | 213 | 86.39 | 1.853 ± 0.0242 |
| (R) GGGAGTTGCTGTTGAAGTC | |||||
| Hamster IL-1β | AB028497.1 | (F) ATCTTCTGTGACTCCTGG | 156 | 85.29 | 1.852 ± 0.0442 |
| (R) GGTTTATGTTCTGTCCGT | |||||
| Murine IL-1β | NM_008361 | (F) GTGTGGATCCCAAGCAATAC | 161 | 83.35 | 1.660 ± 0.0936 |
| (R) GTTGTTTCCCAGGAAGACAG | |||||
| Hamster IL-6 | AB028635 | (F) AGACAAAGCCAGAGTCATT | 252 | 83.21 | 1.945 ± 0.0435 |
| (R) TCGGTATGCTAAGGCACAG | |||||
| Murine IL-6 | NM_031168 | (F) CAATTCCAGAAACCGCT | 268 | 84.10 | 1.832 ± 0.0194 |
| (R) GCAAGTGCATCATCGT | |||||
| Hamster IL-10 | AF046210 | (F) TGGACAACATACTACTCACTG | 308 | 85.50 | 1.871 ± 0.0573 |
| (R) GATGTCAAATTCATTCATGGC | |||||
| Murine IL-10 | NM_010548 | (F) ATTCCCTGGGTGAGAAG | 259 | 83.45 | 1.891 ± 0.0468 |
| (R) CTCTGTCTAGGTCCTGG | |||||
| Hamster TNF-α | AF046215 | (F) AACGGCATGTCTCTCAA | 278 | 88.05 | 1.849 ± 0.0388 |
| (R) AGTCGGTCACCTTTCT | |||||
| Murine TNF-α | NM_013693 | (F) CAACGGCATGGATCTCA | 325 | 87.80 | 1.832 ± 0.0499 |
| (R) GGACTCCGCAAAGTCT | |||||
| Leptospira 16S rRNAf | (F) GGCGGCGCGTCTTAAACATGf | 331 | 86.50 | 1.849 ± 0.0129 | |
| (R) TTCCCCCCATTGAGCAAGATTf |
Accession number of mRNA sequence in GenBank (NCBI) used for primer design.
(F) and (R), forward and reverse primer sequences, respectively.
PCR product size (in base pairs).
Tm, PCR product melting temperature (in °C).
Efficiency for PCR was determined by elaboration of standard curves as described in Materials and Methods.
As described by Mérien et al. (30).
Gene expression analysis.
The level of expression of each target gene was normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin as housekeeping genes (HKG), using qbasePLUS software (version 1.5; Biogazelle, Belgium), considering the corresponding amplification efficiencies (AEs) (Table 1). The AE for each set of primers was determined by generating standard curves from serial dilutions of a known concentration of purified DNA specific for the target of each reference gene. This specific DNA was isolated from agarose gels with a MinElute gel extraction kit (Qiagen, Australia) after amplification of hamster or mouse total cDNA with related pairs of primers. The quantity (in copy number/μl of the extracted DNA) was calculated by classic methods using the Avogadro constant after measuring the concentrations at the OD260. For immune mediators, a relative normalized expression ratio was calculated as the ratio of the level of expression in infected animals to the level of expression in 5 to 9 noninjected animals used as calibrators, depending on the genes and tissues. For Leptospira quantification, the normalized expression ratio of 16S rRNA was divided by the same ratio for injected individuals at 3 h postinfection. The levels of gene expression were expressed as relative normalized expression ratios and were compared between individual samples and animal species. Differences between the two models were evaluated using a mouse/hamster (m/h) ratio, obtained by dividing the relative normalized expression of target genes in mice by the one for the corresponding genes in hamsters. An m/h ratio greater than 1 represents higher target gene expression in mice than hamsters, and conversely, an m/h ratio less than 1 represents higher target gene expression in hamsters.
RESULTS
Fatal outcome observed in hamsters as opposed to recovery in mice.
Experiments were conducted for 5 days on susceptible hamsters injected with the LD100 (2 × 108 virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun). Animals actually died (2/3) and one was euthanized because it was moribund (1/3) at day 4.5 p.i. Moribund or dead hamsters presented evidence of hemorrhage in the respiratory tract (blood at the nostril level). Gross observations of organs confirmed pulmonary hemorrhage in infected hamsters at day 2 p.i. that became worse at 4 days p.i. (Fig. 1). Adipose tissues were yellowish, evidencing an icterus (data not shown). In contrast, injection of the same inoculum in mice did not induce any visible pathological sign or macroscopic lesion in the animals by the end of the experiment at day 15 p.i. (data not shown). Consequently, the kinetics in hamsters ended at day 4 p.i., while it was extended to 15 days for mice.
Fig. 1.
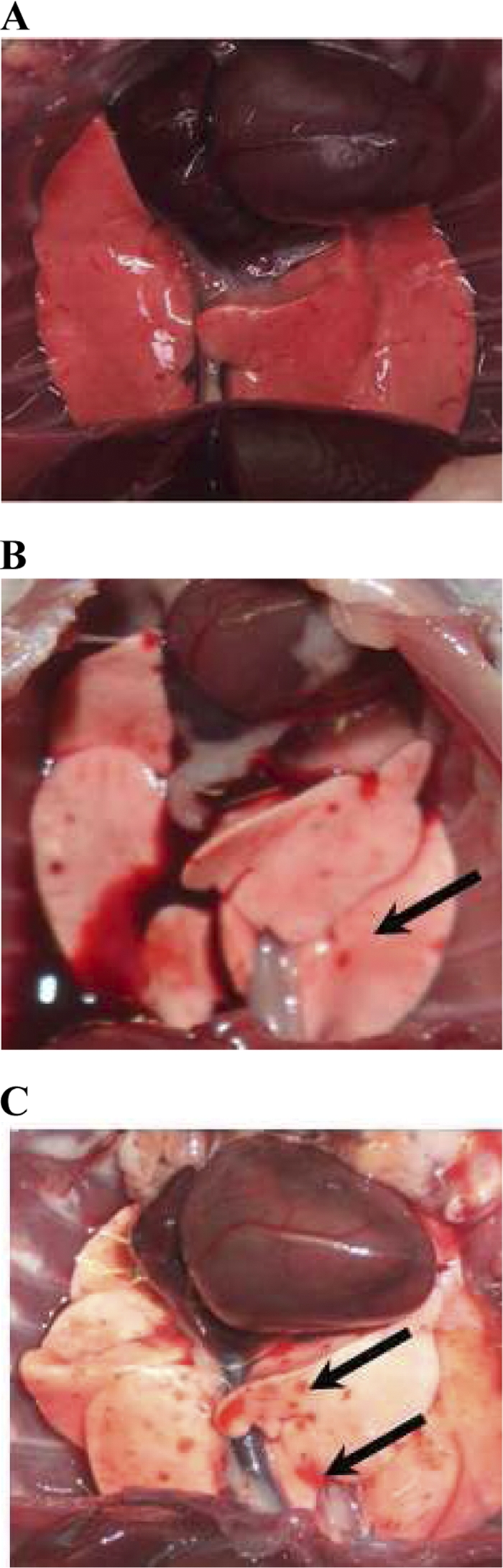
Lungs of a noninfected control hamster (A) are compared with lungs of a hamster intraperitoneally infected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun and euthanatized at 2 days p.i. (B) or 4 days p.i. (C). The arrows show isolated (B) or confluent (C) hemorrhagic areas.
Significant relative Leptospira burden is observed in hamsters compared to rapid clearance from mice.
Live leptospires in tissues were quantified as previously published (55) (Fig. 2 A to D). The normalized expression ratios were compared to the ratio at 3 h p.i. arbitrarily chosen to be equal to 1.0. The results showed high relative Leptospira burdens in the blood of hamsters at day 1 p.i. (ratio = 3.93), with the increase reaching a maximum at day 3 p.i. (ratio = 55.63; Fig. 2A). Opposite that finding, this relative burden continuously decreased in mice, resulting in a total clearance at 15 days p.i. (Fig. 2A). This relative burden was found to be elevated in the livers of the hamsters from 12 h p.i. on (ratio = 3.06), followed by a dramatic increase at day 3 p.i. to a very high level (ratio = 2,577.59) that was maintained at day 4 p.i. (ratio = 1,234.17), while mice presented lower ratios from 24 h p.i. on (ratio = 8.45), rising at day 3 p.i. (ratio = 109.16) and then diminishing remarkably at day 5 p.i. to the initial level (ratio = 0.96) until it was not detected at 15 days p.i. (Fig. 2B). In kidneys, this relative burden was slightly raised in both hamsters and mice from day 1 p.i. on (ratios = 1.74 and 2.54 for hamsters and mice, respectively) to reach a similarly high level at day 3 p.i. (ratios = 35.19 and 44.37 for hamsters and mice, respectively). While it was kept elevated in the kidneys of hamsters at 4 days p.i. (ratio = 52.69), this relative burden decreased to the initial level in mice at day 5 p.i. (ratio = 0.62), and clearance was reached at the end of the experiments at day 15 (Fig. 2C). As observed in liver, this relative load in the lungs of hamsters increased from 12 h p.i. on (ratio = 2.48) to reach a maximum at day 4 p.i. (ratio = 49.51), while it was augmented only slightly in the lungs of mice at day 3 p.i. (ratio = 5.42), before decreasing at day 5 p.i. (ratio = 0.35) and being undetectable at 15 days p.i. (Fig. 2D). Silver impregnation by WS staining showed numerous clusters of spirochetes in the sinusoids of the liver and individual bacteria in the glomeruli and renal tubules in dead and moribund hamsters at day 4.5 p.i. (Fig. 2E and F). In contrast, leptospires were hardly visible by WS staining in the lungs and found only in hemorrhagic foci (data not shown).
Fig. 2.

Leptospire detection in tissues of hamsters or mice infected with virulent L. interrogans serovar Icterohaemorrhagiae. (A to D) Quantification of 16S rRNA by RT-qPCR as described in Materials and Methods for blood (A), liver (B), kidneys (C), and lungs (D) of hamsters (filled bars) or mice (open bars) injected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun. Quantification at day 4 (D4) p.i. for hamsters was compared to quantification at day 5 p.i. in mice (*). As kinetics ended after 4 days p.i. for hamsters, no quantification was established at day 15 p.i. (NE), whereas no detection of 16S rRNA was possible for mice at day 15 p.i. (ND). For clarity, results are expressed as the relative normalized expression ratio of 16S rRNA using results for individuals at 3 h p.i. as calibrators (dotted lines). Values are means ± SEMs (n = 3 animals). (E and F) Warthin-Starry silver stain for spirochetes observation in liver (E) and kidneys (F) of hamsters moribund or deceased at 4.5 days after injection. The arrows indicate large clusters of leptospires in liver sinusoids and individual spirochetes in a renal glomerulus. Magnification, ×1,000.
Severe damage in organs of hamsters contrast with few lesions in mice.
Lesion grading was established using histological criteria reported in previous studies (Table 2). Pathological changes in the livers of hamsters were seen from day 1 p.i. on, with a considerable edema and sinusoidal congestion increasing in severity until the death of the animals. Lymphoplasmacytic inflammation of the portal tract started at day 1 p.i. and evolved from a moderate state at 2 days p.i. to a severe state at the end of the experiment. Hepatocyte necrosis, inflammation, and regeneration were also observed in some individuals from day 1 p.i. on, degenerating to more severe lesions concomitantly with hyperplasia of Kupffer cells (Fig. 3 A and B). In contrast the liver of mice showed limited alterations (i.e., moderate portal inflammation and hepatocyte necrosis) from day 3 p.i. on to day 15 p.i., corroborated by a moderate regeneration. As in the liver, the histological analysis of hamster kidneys evidenced diffuse congestion starting at day 3 p.i. (Fig. 3C and D). A dramatic evolution was associated with severe hemorrhage in renal tissues at the time of death of the animals. However, no interstitial nephritis was found in hamsters, in contrast to a limited interstitial nephritis observed in mice. The histological findings in lungs are consistent with macroscopic observations in hamsters. Indeed, moderate edema and alveolar hemorrhages were observed at day 1 and 2 p.i., respectively, becoming severe from day 3 on concomitantly with the appearance of a severe inflammation of alveolar septa. A generalized deterioration was noticed, with numerous hemorrhagic foci becoming confluent and degenerating until the death of the animals (Fig. 3E and F). Conversely, only moderate lesions were seen late in the lungs of mice at day 5 p.i., but none were detected in mice at the end of the experiment.
Fig. 3.

Histological lesions in hamsters infected with virulent L. interrogans serovar Icterohaemorrhagiae. Shown are sections of liver (A and B), kidneys (C and D), and lungs (E and F) of controls (A, C, and E) or hamsters infected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun (B, D, and F). HES stain. (A and B) Regular hepatocytes in liver of noninfected hamsters compared with necrotic hepatocytes associated with numerous inflammatory elements in liver of leptospiremic animals at 3 days p.i. Magnification, ×400. (C and D) Normal glomerulus with typical renal tubules in control hamsters, in contrast to vascular congestion observed in infected hamsters at day 3 p.i. with infiltration of erythrocytes in glomeruli and tubule. Magnification, ×400. (E and F) Normal alveoli in lungs of noninfected hamsters compared with intense edema and confluent hemorrhagic sites in infected animals at day 3 p.i. Magnification, ×200.
Early regulation of the proinflammatory mediators in mouse blood and liver contrasts with delayed and massive induction in hamster.
The expression of target genes was assessed using RT-qPCR, and the effect of EMJH medium was first evaluated. Considering that target genes are induced when a 2-fold change in mRNA levels is observed between samples and control (55), the injection of the EMJH medium alone induced the expression of several genes in blood and tissues at 3 h p.i. but not at later time points (data not shown). Therefore, data for this 3-h-p.i. time point were excluded from our analysis, and the expression of immune mediators was studied only from 6 h on (Table 3 and Fig. 4 to 6).
Table 3.
Relative normalized expression ratio of target cytokines, chemokines, and COX-2 inflammatory mediatorsa
| Geneb | Timec | Blood |
Liver |
Kidneys |
Lungs |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Relative normalized expression ratiod |
m/h ratioe | Relative normalized expression ratio |
m/h ratio | Relative normalized expression ratio |
m/h ratio | Relative normalized expression ratio |
m/h ratio | ||||||
| Hamster | Mouse | Hamster | Mouse | Hamster | Mouse | Hamster | Mouse | ||||||
| IL-1β | 0.25 | 0.87 ± 0.22 | 2.21 ± 0.22 | 2.53 | 2.61 ± 2.03 | 3.53 ± 1.62 | 1.36 | 3.19 ± 0.80 | 1.21 ± 0.98 | 0.38 | 1.44 ± 0.35 | 2.49 ± 1.15 | 1.73 |
| 0.5 | 0.82 ± 0.47 | 1.76 ± 0.80 | 2.15 | 6.14 ± 1.22 | 2.04 ± 0.52 | 0.33 | 1.66 ± 0.42 | 2.16 ± 1.03 | 1.30 | 1.96 ± 0.82 | 1.72 ± 0.58 | 0.87 | |
| 1 | 1.38 ± 0.81 | 1.30 ± 0.45 | 0.94 | 3.32 ± 0.95 | 1.26 ± 0.05 | 0.38 | 1.90 ± 0.45 | 1.08 ± 0.24 | 0.57 | 2.37 ± 0.24 | 3.60 ± 0.89 | 1.52 | |
| 2 | 1.25 ± 0.43 | 1.59 ± 0.38 | 1.27 | 11.68 ± 2.23 | 1.79 ± 0.13 | 0.15 | 2.86 ± 1.17 | 1.78 ± 0.62 | 0.62 | 2.79 ± 0.22 | 1.13 ± 0.26 | 0.41 | |
| 3 | 5.15 ± 1.51 | 1.41 ± 0.14 | 0.27 | 24.37 ± 12.17 | 4.22 ± 2.46 | 0.17 | 5.16 ± 0.57 | 2.05 ± 0.28 | 0.40 | 3.59 ± 0.46 | 3.41 ± 1.85 | 0.95 | |
| 4–5 | 2.36 ± 0.64 | 1.51 ± 0.52 | 0.64 | 30.93 ± 7.75 | 3.22 ± 0.67 | 0.10 | 7.62 ± 3.50 | 2.73 ± 1.07 | 0.36 | 1.71 ± 0.26 | 3.38 ± 1.04 | 1.97 | |
| IL-6 | 0.25 | 0.17 ± 0.11 | 2.02 ± 1.72 | 12.07 | 5.58 ± 5.32 | 10.98 ± 8.49 | 1.97 | 1.48 ± 0.30 | 22.54 ± 36.22 | 15.21 | 9.33 ± 6.17 | 1.48 ± 1.10 | 0.16 |
| 0.5 | 2.58 ± 2.23 | 2.18 ± 1.28 | 0.84 | 4.61 ± 1.42 | 3.28 ± 0.95 | 0.71 | 0.78 ± 0.35 | 6.13 ± 5.09 | 7.87 | 6.00 ± 2.36 | 0.52 ± 0.00 | 0.09 | |
| 1 | 7.03 ± 0.00 | 3.02 ± 0.95 | 0.43 | 1.00 ± 0.30 | 0.47 ± 0.23 | 0.47 | 1.02 ± 0.02 | 5.82 ± 1.80 | 5.72 | 6.55 ± 6.26 | 2.98 ± 1.56 | 0.45 | |
| 2 | 0.53 ± 0.45 | 2.36 ± 0.76 | 4.40 | 6.07 ± 1.96 | 1.87 ± 1.48 | 0.31 | 3.31 ± 3.43 | 2.67 ± 1.83 | 0.81 | 34.35 ± 4.69 | 0.75 ± 0.36 | 0.02 | |
| 3 | 6.33 ± 4.87 | 1.04 ± 1.00 | 0.16 | 46.95 ± 46.34 | 1.76 ± 0.77 | 0.04 | 5.19 ± 0.76 | 13.66 ± 0.48 | 2.63 | 10.80 ± 4.29 | 2.04 ± 0.48 | 0.19 | |
| 4–5 | 18.96 ± 25.99 | 0.66 ± 0.43 | 0.03 | 13.27 ± 12.59 | 0.35 ± 0.03 | 0.03 | 7.88 ± 4.30 | 9.80 ± 7.78 | 1.24 | 10.46 ± 4.68 | 2.30 ± 0.58 | 0.22 | |
| TNF-α | 0.25 | 2.23 ± 0.78 | 6.13 ± 3.96 | 2.75 | 6.75 ± 8.93 | 13.29 ± 12.32 | 1.97 | 1.86 ± 0.27 | 4.84 ± 4.20 | 2.61 | 3.96 ± 0.60 | 2.65 ± 0.70 | 0.67 |
| 0.5 | 0.81 ± 0.26 | 1.95 ± 1.31 | 2.40 | 11.02 ± 9.54 | 2.94 ± 0.18 | 0.27 | 1.85 ± 1.30 | 3.47 ± 1.55 | 1.88 | 5.77 ± 1.63 | 4.41 ± 3.31 | 0.76 | |
| 1 | 0.80 ± 0.56 | 1.40 ± 0.42 | 1.75 | 3.48 ± 5.22 | 6.26 ± 1.87 | 1.80 | 1.55 ± 0.71 | 4.55 ± 0.62 | 2.94 | 15.28 ± 1.61 | 3.25 ± 0.24 | 0.21 | |
| 2 | 1.78 ± 0.07 | 3.28 ± 0.85 | 1.84 | 2.28 ± 0.62 | 0.82 ± 0.39 | 0.36 | 2.71 ± 1.30 | 1.97 ± 0.65 | 0.73 | 10.51 ± 5.41 | 8.57 ± 2.47 | 0.82 | |
| 3 | 2.09 ± 0.23 | 2.55 ± 3.48 | 1.22 | 2.41 ± 1.97 | 11.27 ± 7.39 | 4.68 | 4.38 ± 1.84 | 5.04 ± 1.02 | 1.15 | 7.85 ± 1.37 | 2.82 ± 1.44 | 0.36 | |
| 4–5 | 4.10 ± 0.42 | 0.14 ± 0.14 | 0.03 | 6.28 ± 1.37 | 4.71 ± 0.35 | 0.75 | 6.09 ± 2.81 | 1.84 ± 1.00 | 0.30 | 5.47 ± 0.47 | 5.24 ± 0.78 | 0.96 | |
| COX-2 | 0.25 | 0.93 ± 0.09 | 1.18 ± 0.55 | 1.28 | 0.73 ± 0.18 | 47.62 ± 68.94 | 65.66 | 2.57 ± 0.16 | 1.06 ± 0.71 | 0.41 | 1.48 ± 0.38 | 1.83 ± 1.11 | 1.24 |
| 0.5 | 0.85 ± 0.67 | 0.97 ± 0.49 | 1.15 | 2.31 ± 1.05 | 5.77 ± 4.80 | 2.50 | 0.47 ± 0.45 | 4.98 ± 3.32 | 10.54 | 1.33 ± 0.14 | 0.74 ± 0.32 | 0.56 | |
| 1 | 1.44 ± 1.06 | 0.59 ± 0.35 | 0.41 | 0.73 ± 0.67 | 1.93 ± 1.60 | 2.65 | 1.85 ± 1.26 | 1.14 ± 0.76 | 0.62 | 1.08 ± 0.23 | 6.91 ± 2.22 | 6.37 | |
| 2 | 0.61 ± 0.40 | 0.67 ± 0.37 | 1.10 | 3.79 ± 1.24 | 1.31 ± 0.05 | 0.35 | 1.17 ± 0.95 | 3.56 ± 1.11 | 3.05 | 1.65 ± 0.36 | 0.82 ± 0.19 | 0.50 | |
| 3 | 3.79 ± 1.31 | 1.62 ± 0.56 | 0.43 | 4.22 ± 4.04 | 2.28 ± 2.38 | 0.54 | 4.41 ± 3.11 | 0.38 ± 0.20 | 0.09 | 1.36 ± 0.18 | 2.55 ± 0.55 | 1.88 | |
| 4–5 | 2.65 ± 1.29 | 1.41 ± 0.88 | 0.53 | 5.58 ± 2.86 | 0.90 ± 0.06 | 0.16 | 3.68 ± 3.28 | 1.65 ± 1.14 | 0.45 | 1.82 ± 0.29 | 4.20 ± 1.34 | 2.31 | |
Hamsters and mice were intraperitoneally infected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun, and expression of target genes was quantified in each tissue by RT-qPCR as described in Materials and Methods.
Name of target genes.
Time p.i. is expressed in days. Quantification at day 4 p.i. in hamsters was compared to quantification at day 5 p.i. in mice.
Relative normalized expression ratios were calculated after normalization on reference genes (β-actin and GAPDH) and using noninfected animals as calibrators. Values are means ± standard errors of the means (n = 3).
The m/h ratio represents the ratio of relative normalized expression in mouse to relative normalized expression in hamsters in each tissue. An m/h ratio of >1 represents higher gene expression in mice, and a ratio of <1 represents higher gene expression in hamsters.
Fig. 4.

Modulation of proinflammatory mediators in tissues after injection with virulent L. interrogans serovar Icterohaemorrhagiae. Shown is the m/h ratio of relative normalized expression calculated as defined in the text for IL-1β (filled bars), IL-6 (open bars), TNF-α (hatched bars), and COX-2 (gray bars) in blood (A), liver (B), kidneys (C), and lungs (D) from animals (n = 3) injected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun. An m/h ratio of >1 represents higher target gene expression in mice than hamsters, and conversely, a ratio of <1 represents higher target gene expression in hamsters.
Fig. 6.

Time course of expression of the chemokines MIP-1α/CCL3 and IP-10/CXCL10 in tissues after infection with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun. Quantification of MIP-1α/CCL3 (A) and IP-10/CXCL10 (B) mRNA in blood, liver, kidneys, and lungs from hamsters (filled bars) and mice (open bars) infected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun. Kinetics are time points ranging from 6 h p.i. to day 4 p.i. in hamsters and to day 15 p.i. in mice (see details in Table 3). Results are expressed as relative normalized expression ratios using noninfected animals as calibrators. Values are means ± SEMs (n = 3 animals).
Soon after infection (6 h p.i.), the proinflammatory cytokines IL-1β and IL-6 (ratios > 2) and TNF-α (ratio = 6.13) were overexpressed in mouse blood. In contrast, they were either not induced (IL-1β and IL-6; ratios < 1) or less induced (TNF-α; ratio = 2.23) in hamsters. COX-2 was expressed at the basal level in both animals (Table 3). Along the time course of the kinetics, IL-1β, IL-6, and TNF-α mRNA levels decreased in mouse blood and went down to or under the constitutive level, but, in contrast, they increased in hamsters until 4 days p.i. (ratios = 2.36, 18.96, and 4.10, respectively). This was notably evidenced from an initial value of the m/h ratio of >2 for these cytokines in blood at 6 h p.i., reflecting a higher expression level in mice (Fig. 4A). This ratio then decreased moderately for IL-1β (m/h ratio = 0.64) and strongly for IL-6 and TNF-α (m/h ratios = 0.03 for both) at later time points. COX-2 mRNA levels did not vary in mouse blood but increased significantly in hamster blood and was responsible for low m/h ratios at days 3 and 4 p.i. (m/h ratio < 0.6). In the liver, all four proinflammatory mediators were highly induced in mouse as soon as 6 h p.i., with relative normalized expression ratios of >3.5 in a higher proportion of mice than hamsters (Table 3). However, while IL-6 and COX-2 mRNA levels decreased in mouse liver, they increased to high expression levels in hamsters (ratios = 46.95 and 5.58 for IL-6 and COX-2, respectively). This was evidenced by m/h ratios decreasing to values under 1 for proinflammatory cytokines (Fig. 4B). Interestingly, TNF-α presented a biphasic expression profile in the liver of both animals (Table 3) but was delayed in hamsters compared to mice. Moreover, the minimum ratio reached at day 2 p.i. was higher in hamsters (ratio = 2.28) than in mice (ratio = 0.82).
The modulation of proinflammatory mediators is different between mice and hamsters in kidneys and lungs.
A higher induction of IL-1β was observed in kidneys of hamsters than in those of mice, evidenced by low m/h ratios ranging from 0.36 to 1.30 (Fig. 4C). The expression of COX-2 was maintained at the basal level in mouse kidneys during the experiment (except for a few time points with high interindividual variability), in contrast to a continuous increase in hamsters (Table 3). The opposite was found for IL-6 and TNF-α, which were intensively overexpressed in mouse kidneys from 6 h p.i. on (ratios = 22.54 and 4.84, respectively), and although their expression decreased during the time course, expression was maintained at higher levels in mouse kidneys than in hamster kidneys until 3 days p.i., with the m/h ratio being higher than 1 along the kinetics (Fig. 4C). This tendency was then reversed for TNF-α at day 4 p.i., with a noteworthy decrease in mouse compared with an increasing level in hamster (m/h ratio = 0.30). The expression profiles of proinflammatory cytokines were different in lungs, with higher induction of IL-6 and TNF-α in hamsters than in mice. Indeed, although both IL-6 and TNF-α were overexpressed in mouse lungs from early time points (ratio > 2.5), their expression levels in hamster lungs were higher along the kinetics, reflected by low m/h ratios (Fig. 4D). The results for IL-1β in lungs were quite discontinuous, with poor regulation in both animal models. No overexpression of COX-2 was observed in hamster lungs, which is in contrast to a sustained induction in mouse lungs until day 5 p.i. (ratio = 4.20).
The induction of the anti-inflammatory IL-10 is higher in mice than in hamsters.
IL-10 was slightly induced in hamster blood at day 1 p.i. (ratio = 2.95), with a little increase until a maximum value was reached at day 3 p.i. (ratio = 7.57). However, it was induced earlier and at higher levels from 6 h p.i. on (ratio = 22.00) in mouse blood and maintained from day 1 to day 5 (ratios = 13.93 and 22.72, respectively), before returning to its basal expression level (ratio = 1.34) at the end of the experiment at day 15 p.i. (Fig. 5A). In the liver, hamsters and mice presented similar levels of overexpression of IL-10 from 6 h p.i. on (ratios = 4.70 and 3.80, respectively), increasing until day 3 p.i. (ratios = 31.52 and 39.79, respectively). It then decreased faster in hamsters (ratio = 12.95) than in mice (ratio = 27.76) at 4 days p.i. (Fig. 5B) and returned to the basal level at day 15 p.i. in mice (ratio = 1.68). Similar to the findings for liver, IL-10 was induced in kidneys of both mice and hamsters from 6 h p.i. on (ratios = 4.09 and 4.77, respectively). However, it was then constant during the kinetics in hamster kidneys, while it increased remarkably in mouse kidneys from 12 h p.i. on, reaching maximum expression at day 5 p.i. (ratio = 34.40). In the lungs, IL-10 was induced from 6 h p.i. on in mice (ratio = 4.05) but not in hamsters (ratio = 1.24). The mRNA level of this cytokine then greatly increased in mouse lungs, with high expression ratios at day 3 p.i. (ratio = 11.07) and day 4 p.i. (ratio = 13.70), while it was only weakly induced in hamster lungs, with a maximum ratio at day 3 p.i. (ratio = 4.00). It is noticeable that, as was observed in the blood and liver, IL-10 expression also almost regained its basal level in kidneys and lungs of mice at the end of the experiment (ratios = 3.64 and 0.49, respectively).
Fig. 5.

Modulation of IL-10 in tissues after injection with virulent L. interrogans serovar Icterohaemorrhagiae. Quantification of IL-10 mRNA was conducted by RT-qPCR as described in Materials and Methods for blood (A), liver (B), kidneys (C), and lungs (D) from hamsters (filled bars) and mice (open bars) infected with virulent L. interrogans serovar Icterohaemorrhagiae strain Verdun. Quantification at day 4 p.i. for hamsters was compared to quantification at day 5 p.i. in mice (*). As kinetics ended at day 4 p.i. for hamsters, no quantification was established at day 15 p.i. (NE). Results are expressed as the relative normalized expression ratio of IL-10 mRNA using expression levels in noninfected animals as calibrators (dotted line). Values are means ± SEMs (n = 3 animals).
Late induction of the chemokines MIP-1α/CCL3 and IP-10/CXCL10 is observed in hamsters, in contrast to early chemokine regulation in mice.
The expression of MIP-1α/CCL3 continuously increased in the blood, liver, and kidneys of hamsters, with significant levels from the earliest time point onwards in the kidneys (ratio = 5.60) and from day 2 p.i. on in the blood and liver (ratios = 2.60 and 5.14, respectively), until high expression levels at days 3 and 4 p.i. (Fig. 6A to C). In contrast, MIP-1α/CCL3 was induced at lower levels in the same tissues of mice. Differences were less evident for MIP-1α/CCL3 expression in lungs between mice and hamsters (Fig. 6D). An early induction of IP-10/CXCL10 was observed in all mouse tissues as soon as 6 h p.i. at the highest expression ratio (ranging from 7.09 to 22.37, depending on the tissue) and then diminished along the kinetics to its basal level, except in the lungs, where the ratio was maintained above 2 from days 3 to 5 p.i. (Fig. 6D). This is in contrast to a later induction of IP-10/CXCL10 in hamster tissues.
DISCUSSION
Leptospirosis is known to cause severe disease in humans, but the processes involved in the response to pathogenic Leptospira are not fully understood. Several animal models are available to study leptospirosis. We used the recognized hamster model injected with a dose of a virulent L. interrogans serovar Icterohaemorrhagiae strain known to induce severe fatal leptospirosis. This severity is related to the infecting species, strain, and dose, as shown in hamsters, with different LD50s for several Leptospira strains (47). Because severe human cases are frequently, though not exclusively, caused by the Icterohaemorrhagiae serogroup (18), we have selected the virulent strain L. interrogans serogroup Icterohaemorrhagiae Verdun. Our results validated the pathogenicity of this strain and again highlighted the usefulness of the hamster as a susceptible model for leptospirosis (16). In contrast, mice injected with the same inoculum were asymptomatic with limited histological changes. Interstitial nephritis was noticed in mice but not in hamsters. The same pathological changes were observed in an asymptomatic reservoir model of rat experimentally infected with L. interrogans serovar Copenhageni but were associated with persistent leptospires in renal tubules (4, 53). Herein, asymptomatic mice presented resolution of the histological lesion 15 days after infection, and a rapid decrease in relative leptospiral loads ended in complete clearance in all tissues, even in kidneys. The moderate virulence of our model strain Verdun might notably account for this unexpected result. These results confirmed that the OF1 mouse is valuable as a resistant but not reservoir model of leptospirosis after infection with a high dose of virulent L. interrogans Icterohaemorrhagiae strain Verdun.
The mechanisms by which leptospires cause host tissue damage or, conversely, the pathways used by infected hosts to avoid organ failure are not well-defined yet. In this study, we aimed to put the lesions observed in infected animals in parallel with relative bacterial burdens and expression profiles of some relevant cytokines. We focused on the proinflammatory cytokines IL-1β, IL-6, and TNF-α, the chemokines MIP-1α/CCL3 and IP-10/CXCL10, and the anti-inflammatory cytokine IL-10. Our results showed that the proinflammatory cytokines were rapidly regulated in the blood of mice, compared to a massive and delayed induction in hamsters, as observed for IP-10/CXCL10 and MIP-1α/CCL3. Both chemokines are involved in chemotaxis and transendothelial migration of leukocytes from the bloodstream to tissues, but MIP-1α/CCL2 preferentially attracts T cells, monocytes, and natural killer (NK) cells, whereas IP-10/CXCL10 especially regulates chemotaxis of neutrophils and NK cells (28, 34). Moreover, IL-1β and TNF-α are known to stimulate leukocyte adhesion to endothelial cells by upregulating cellular adhesion molecule (CAM) expression, especially in sepsis (48). Interestingly, high circulating levels of soluble CAMs were evidenced in the serum of leptospirosis patients (38). Thus, the high expression level of proinflammatory cytokines and chemokines in the tissues of infected hamsters could interfere with chemotaxis, activation, and adhesion of circulating leukocytes toward the target organs.
Few studies have focused on gene expression in the liver of animals modeling leptospirosis. The modulation of chemokines was investigated in susceptible C3H/HeJ mice (Toll-like receptor 4 deficient [TLR4−/−]) infected by L. interrogans serovar Copenhageni, showing that MIP-1α/CCL3 was rapidly induced and maintained at a level higher than that observed in the resistant BALB/c mouse (10). This is consistent with a massive induction of MIP-1α/CCL3 in the hamster liver, whereas it was induced at a lower level in mice. This possibly contributes to the inflammation of the portal tract in the liver of hamsters with numerous inflammatory elements, ending in a massive necrosis. Similar lesions were reported in the liver of susceptible gerbils infected with a virulent Leptospira strain (6). An in vitro experiment showed that the incubation of Kupffer cells with L. interrogans Icterohaemorrhagiae led to fast TNF-α overproduction in culture supernatants (24), and our in vivo results showed that it was greatly induced at the early stage of infection in the liver of hamsters compared with mice. Infected guinea pigs also presented hepatocyte apoptosis in vivo, as shown using the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling assay (31). Because TNF-α is known to trigger apoptosis via the caspase pathway (17), the role of this cytokine might be pivotal in establishing the lesions in the liver of susceptible individuals.
An upregulation of TNF-α and IP-10/CXCL10 was evidenced in kidneys of hamsters infected with L. interrogans serovar Pyrogenes (21). A progressive induction of IP-10/CXCL10 and MIP-1α/CCL3 was also observed in kidneys of a mouse model of sepsis-induced acute renal failure associated with an accumulation of neutrophils and monocytes (22). Our results evidenced that chemokines and proinflammatory cytokines were overexpressed in kidneys of both hamsters and mice infected by Leptospira. However, the expression levels of IP-10/CXCL10 and MIP-1α/CCL3 progressively increased in hamsters, whereas their induction, though earlier, rapidly diminished in mice. IL-6 and TNF-α were also induced in higher proportions in mice than in hamsters within the first days of infection. This is consistent with the increasing immunodetection of TNF-α and IL-6 in the kidneys of resistant BALB/c mice infected with a virulent strain of L. interrogans serovar Canicola (25). Mice deficient for the TNF-α receptor exhibited more severe residual kidney inflammation after inoculation with L. interrogans serovar Copenhageni, suggesting that this cytokine might be important in the rapid control of renal lesions during infection (3). Thus, an early regulation of chemokines and proinflammatory cytokines in mouse kidneys might be associated with limited pathological lesions in this resistant model. In contrast, their massive and later upregulation could account for the massive alterations of hamster renal tissues.
The pulmonary lesions, especially edema, congestion, inflammatory infiltration, and hemorrhage, confirmed the severe disease in hamsters. Similar lesions were observed in human leptospirosis or in susceptible animal models (12). Proinflammatory cytokines are involved in the regulation of nitric oxide (NO), a major reactive oxygen species involved in septic shock (37). Interestingly, high overexpression of pulmonary endothelial NO synthase (eNOS) and TNF-α was associated with an infiltration of neutrophils and mononuclear cells in the lungs of hamsters infected with L. interrogans serovar Icterohaemorrhagiae (26), consistent with our results. Moreover, TNF-α and IL-6 were greatly induced in hamster lungs compared to mouse lungs, and this upregulation was faster, as early as 6 h p.i. Biological activities of TNF-α in neutrophil recruitment to lungs and its involvement in ARDS have been previously documented (32). Interestingly, the NO pathway was associated with ARDS in a human case of leptospirosis, and NO production was evidenced in bronchoalveolar fluid and alveolar macrophages of hamsters infected with L. interrogans serogroup Icterohaemorrhagiae (27, 63). Thus, high levels of expression of proinflammatory cytokines in hamster lungs could be related to alterations by acting on diverse pathways, including attraction and activation of immune cells and the NO pathway.
The massive induction of chemokines and proinflammatory cytokines in hamsters can be paralleled to the cytokine storm in sepsis (48). Opposite this, the anti-inflammatory IL-10, though upregulated, was induced later or maintained at lower levels in hamsters than in mice, a finding consistent with previous reports in hamster blood or kidneys (21, 55, 56). In contrast, a rapid and higher induction of IL-10 gene expression was observed in mouse tissues, except the liver. IL-10 is a major regulator of innate immunity and belongs to Th2 cytokines: it interferes with the production of inflammatory mediators and upregulates the expression of other anti-inflammatory molecules (29). Herein, we evidenced an essential difference in the cytokine balance related to specific tissue lesions between a susceptible (hamster) and a resistant (mouse) model of leptospirosis. This correlated with the findings of a previous clinical study on leptospirosis patients (49), evidencing that a low ratio of IL-10/TNF-α levels was associated with a poor outcome in leptospirosis. The regulation of cytokines, especially IL-10, was also found to be modulated in inflammation during arthritis induced by another spirochete, Borrelia burgdorferi in Lyme disease (7). Thus, the therapeutic modulation of the proinflammatory response could be valuable in leptospirosis. Among the immunosuppressive drugs tested, rapamycin actually diminished the renal and pulmonary injuries in a hamster model (41).
Besides its anti-inflammatory activity, IL-10 is known to regulate immunoglobulin (Ig) secretion by Ig switching to IgG in activated B cells (23, 46). Interestingly, a pioneer study presented IgM-to-IgG switching profiles in a rat model of leptospirosis depending on the serovar, Icterohaemorrhagiae or Grippotyphosa, inoculated (51). Moreover, the depletion of antibody production was correlated to mortality in a mouse model of leptospirosis in which mice were rendered susceptible by the injection of cyclophosphamide (2). This study also evidenced that a decrease in leptospiremia coincided with the production of antibodies in mice. Taken together, this suggests that IL-10 overexpression could efficiently regulate the proinflammatory response with limited lesions in mice but also successfully regulate B cells and the production of specific antibodies, in turn leading to the elimination of the pathogen in mouse tissues. Our results, however, showed that the leptospiremia decreased as early as 6 h p.i. in mouse, until it was not detected at the end of the experiment, while it continuously increased in hamsters. This rapid decrease in mouse highlights that other, earlier mechanisms contribute to leptospirosis resistance. Indeed, killing mechanisms with fast recruitment of phagocytic cells as macrophages and neutrophils in the blood and peritoneal cavity could participate in bacterial clearance (13). This is suggested by our results showing a fast and high overexpression of leukocyte-attracting chemokines in mice during the early stage of infection. Interestingly, these particular leukocytes are known to produce antimicrobial peptides, and an interesting in vitro study demonstrated a fast inhibitory and bactericidal effect of cathelicidin-derived peptides from mammals on Leptospira (44).
We evidenced major differences in the expression profiles of cytokines depending on organs and host susceptibility to leptospirosis, and these can be part of an explanation for host tissue damage. This variation in susceptibility between the resistant and susceptible models might be related to host genetic polymorphism, especially for proteins involved in the recognition and/or response to pathogens, as seen in sepsis (9). Indeed, previous studies evidenced some genetic polymorphisms associated with susceptibility to leptospirosis (14, 19). In addition to host genetic determinants, it could be interesting to also consider the gene regulation of leptospires. Indeed, several studies focused on leptospiral proteins responsible for host immune responses such as the outer membrane protein LipL32, which induced TNF-α, inducible NO synthase (iNOS), and NO production in mouse proximal tubular cells, possibly associated with the tubulointerstitial nephritis in mouse kidneys (62). Moreover, microarray approaches evidenced the regulation of the Leptospira transcriptome under environmental conditions mimicking host infection (39, 61); further studies could investigate the regulation of Leptospira genes in different infected hosts.
ACKNOWLEDGMENTS
We are very grateful to Yannick Rougier for his expert assistance and helpful suggestions on the histological part of the study during preliminary experiments. Warm thanks are also addressed to the staff of the Pathological Anatomic Laboratory, especially Evelyne Tuheiava and Daniel Wagino, for their skillful technical assistance.
The postdoctoral fellowship of M.M. was financed by the New Caledonia government.
Footnotes
Published ahead of print on 15 August 2011.
REFERENCES
- 1. Adler B., de la Pena Moctezuma A. 2009. Leptospira and leptospirosis. Vet. Microbiol. 140:287–296 [DOI] [PubMed] [Google Scholar]
- 2. Adler B., Faine S. 1976. Susceptibility of mice treated with cyclophosphamide to lethal infection with Leptospira interrogans serovar pomona. Infect. Immun. 14:703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Athanazio D. A., Santos C. S., Santos A. C., McBride F. W., Reis M. G. 2008. Experimental infection in tumor necrosis factor alpha receptor, interferon gamma and interleukin 4 deficient mice by pathogenic Leptospira interrogans. Acta Trop. 105:95–98 [DOI] [PubMed] [Google Scholar]
- 4. Athanazio D. A., et al. 2008. Rattus norvegicus as a model for persistent renal colonization by pathogenic Leptospira interrogans. Acta Trop. 105:176–180 [DOI] [PubMed] [Google Scholar]
- 5. Bharadwaj R., et al. 2002. An urban outbreak of leptospirosis in Mumbai, India. Jpn. J. Infect. Dis. 55:194–196 [PubMed] [Google Scholar]
- 6. Bourhy P., et al. 2010. Isolation and characterization of new Leptospira genotypes from patients in Mayotte (Indian Ocean). PLoS Negl. Trop. Dis. 4:e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown J. P., Zachary J. F., Teuscher C., Weis J. J., Wooten R. M. 1999. Dual role of interleukin-10 in murine Lyme disease: regulation of arthritis severity and host defense. Infect. Immun. 67:5142–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chierakul W., et al. 2004. Differential expression of interferon-γ and interferon-γ-inducing cytokines in Thai patients with scrub typhus or leptospirosis. Clin Immunol. 113:140–144 [DOI] [PubMed] [Google Scholar]
- 9. Dahmer M. K., Randolph A., Vitali S., Quasney M. W. 2005. Genetic polymorphisms in sepsis. Pediatr. Crit. Care Med. 6:S61–S73 [DOI] [PubMed] [Google Scholar]
- 10. da Silva J. B., et al. 2009. Chemokines expression during Leptospira interrogans serovar Copenhageni infection in resistant BALB/c and susceptible C3H/HeJ mice. Microb. Pathog. 47:87–93 [DOI] [PubMed] [Google Scholar]
- 11. De Fost M., et al. 2007. Release of granzymes and chemokines in Thai patients with leptospirosis. Clin. Microbiol. Infect. 13:433–436 [DOI] [PubMed] [Google Scholar]
- 12. Dolhnikoff M., Mauad T., Bethlem E. P., Carvalho C. R. 2007. Pathology and pathophysiology of pulmonary manifestations in leptospirosis. Braz. J. Infect. Dis. 11:142–148 [DOI] [PubMed] [Google Scholar]
- 13. Dunn D. L., Barke R. A., Knight N. B., Humphrey E. W., Simmons R. L. 1985. Role of resident macrophages, peripheral neutrophils, and translymphatic absorption in bacterial clearance from the peritoneal cavity. Infect. Immun. 49:257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fialho R. N., et al. 2009. Role of HLA, KIR and cytokine gene polymorphisms in leptospirosis. Hum. Immunol. 70:915–920 [DOI] [PubMed] [Google Scholar]
- 15. Fujishima S., et al. 1996. Serum MIP-1 alpha and IL-8 in septic patients. Intensive Care Med. 22:1169–1175 [DOI] [PubMed] [Google Scholar]
- 16. Haake D. A. 2006. Hamster model of leptospirosis. Curr. Protocols Microbiol. Chapter 12:Unit 12E.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatano E. 2007. Tumor necrosis factor signaling in hepatocyte apoptosis. J. Gastroenterol. Hepatol. 22(Suppl. 1):S43–S44 [DOI] [PubMed] [Google Scholar]
- 18. Levett P. N. 2001. Leptospirosis. Clin. Microbiol. Rev. 14:296–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lingappa J., et al. 2004. HLA-DQ6 and ingestion of contaminated water: possible gene-environment interaction in an outbreak of leptospirosis. Genes Immun. 5:197–202 [DOI] [PubMed] [Google Scholar]
- 20. Lourdault K., Aviat F., Picardeau M. 2009. Use of quantitative real-time PCR for studying the dissemination of Leptospira interrogans in the guinea pig infection model of leptospirosis. J. Med. Microbiol. 58:648–655 [DOI] [PubMed] [Google Scholar]
- 21. Lowanitchapat A., et al. 2010. Expression of TNF-alpha, TGF-beta, IP-10 and IL-10 mRNA in kidneys of hamsters infected with pathogenic Leptospira. Comp. Immunol. Microbiol. Infect. Dis. 33:423–434 [DOI] [PubMed] [Google Scholar]
- 22. Maier S., et al. 2000. Massive chemokine transcription in acute renal failure due to polymicrobial sepsis. Shock 14:187–192 [DOI] [PubMed] [Google Scholar]
- 23. Malisan F., et al. 1996. Interleukin-10 induces immunoglobulin G isotype switch recombination in human CD40-activated naive B lymphocytes. J. Exp. Med. 183:937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marangoni A., et al. 2000. Uptake and killing of Leptospira interrogans and Borrelia burgdorferi, spirochetes pathogenic to humans, by reticuloendothelial cells in perfused rat liver. Infect. Immun. 68:5408–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marinho M., Monteiro C. M. R., Peiro J. R., Machado G. F., Oliveira-Junior I. S. 2008. TNF-α and IL-6 immunohistochemistry in rat renal tissue experimentally infected with Leptospira interrogans serovar Canicola. J. Venom. Anim. Toxins incl. Trop. Dis. 14:533–540 [Google Scholar]
- 26. Marinho M., Oliveira-Junior I. S., Monteiro C. M., Perri S. H., Salomao R. 2009. Pulmonary disease in hamsters infected with Leptospira interrogans: histopathologic findings and cytokine mRNA expressions. Am. J. Trop. Med. Hyg. 80:832–836 [PubMed] [Google Scholar]
- 27. Marinho M., et al. 2008. Response activity of alveolar macrophages in pulmonary dysfunction caused by Leptospira infection. J. Venom. Anim. Toxins incl. Trop. Dis. 14:58–70 [Google Scholar]
- 28. Maurer M., von Stebut E. 2004. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 36:1882–1886 [DOI] [PubMed] [Google Scholar]
- 29. Mege J. L., Meghari S., Honstettre A., Capo C., Raoult D. 2006. The two faces of interleukin 10 in human infectious diseases. Lancet Infect. Dis. 6:557–569 [DOI] [PubMed] [Google Scholar]
- 30. Merien F., Amouriaux P., Perolat P., Baranton G., Saint Girons I. 1992. Polymerase chain reaction for detection of Leptospira spp. in clinical samples. J. Clin. Microbiol. 30:2219–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merien F., Truccolo J., Rougier Y., Baranton G., Perolat P. 1998. In vivo apoptosis of hepatocytes in guinea pigs infected with Leptospira interrogans serovar icterohaemorrhagiae. FEMS Microbiol. Lett. 169:95–102 [DOI] [PubMed] [Google Scholar]
- 32. Mukhopadhyay S., Hoidal J. R., Mukherjee T. K. 2006. Role of TNFalpha in pulmonary pathophysiology. Respir. Res. 7:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nally J. E., et al. 2004. Alveolar septal deposition of immunoglobulin and complement parallels pulmonary hemorrhage in a guinea pig model of severe pulmonary leptospirosis. Am. J. Pathol. 164:1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neville L. F., Mathiak G., Bagasra O. 1997. The immunobiology of interferon-gamma inducible protein 10 kD (IP-10): a novel, pleiotropic member of the C-X-C chemokine superfamily. Cytokine Growth Factor Rev. 8:207–219 [DOI] [PubMed] [Google Scholar]
- 35. Nicodemo A. C., et al. 1997. Lung lesions in human leptospirosis: microscopic, immunohistochemical, and ultrastructural features related to thrombocytopenia. Am. J. Trop. Med. Hyg. 56:181–187 [DOI] [PubMed] [Google Scholar]
- 36. O'Grady N. P., et al. 1999. Detection of macrophage inflammatory protein (MIP)-1alpha and MIP-1beta during experimental endotoxemia and human sepsis. J. Infect. Dis. 179:136–141 [DOI] [PubMed] [Google Scholar]
- 37. Opal S. M. 2007. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int. J. Med. Microbiol. 297:365–377 [DOI] [PubMed] [Google Scholar]
- 38. Paris D. H., et al. 2008. Differential patterns of endothelial and leucocyte activation in ‘typhus-like’ illnesses in Laos and Thailand. Clin. Exp. Immunol. 153:63–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patarakul K., Lo M., Adler B. 2010. Global transcriptomic response of Leptospira interrogans serovar Copenhageni upon exposure to serum. BMC Microbiol. 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Philippart F., Cavaillon J. M. 2007. Sepsis mediators. Curr. Infect. Dis. Rep. 9:358–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Praditpornsilpa K., et al. 2006. Alleviation of renal and pulmonary injury by immunomodulation in leptospirosis: hamster model. J. Med. Assoc. Thai. 89:S178–S187 [PubMed] [Google Scholar]
- 42. Punyadeera C., et al. 2010. A biomarker panel to discriminate between systemic inflammatory response syndrome and sepsis and sepsis severity. J. Emerg. Trauma Shock 3:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rozen S., Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. (Clifton, NJ) 132:385–386 [DOI] [PubMed] [Google Scholar]
- 44. Sambri V., et al. 2002. Comparative in vitro activity of five cathelicidin-derived synthetic peptides against Leptospira, Borrelia and Treponema pallidum. J. Antimicrob. Chemother. 50:895–902 [DOI] [PubMed] [Google Scholar]
- 45. Segura E. R., et al. 2005. Clinical spectrum of pulmonary involvement in leptospirosis in a region of endemicity, with quantification of leptospiral burden. Clin. Infect. Dis. 40:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shparago N., et al. 1996. IL-10 selectively regulates murine Ig isotype switching. Int. Immunol. 8:781–790 [DOI] [PubMed] [Google Scholar]
- 47. Silva E. F., et al. 2008. Characterization of virulence of Leptospira isolates in a hamster model. Vaccine 26:3892–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sriskandan S., Altmann D. M. 2008. The immunology of sepsis. J. Pathol. 214:211–223 [DOI] [PubMed] [Google Scholar]
- 49. Tajiki M. H., Nakama A. S. Y., Salomão R. 1997. The ratio of plasma levels of Il-10/TNF-alpha and its relationship to disease severity and survival in patients with leptospirosis. Braz. J. Infect. Dis. 1:138–141 [PubMed] [Google Scholar]
- 50. Tajiki M. H., Salomão R. 1996. Association of plasma levels of tumour necrosis factor α with severity of disease and mortality among patients with leptospirosis. Clin. Infect. Dis. 23:1177–1178 [DOI] [PubMed] [Google Scholar]
- 51. Thiermann A. B. 1981. The Norway rat as a selective chronic carrier of Leptospira icterohaemorrhagiae. J. Wildl. Dis. 17:39–43 [DOI] [PubMed] [Google Scholar]
- 52. Truccolo J., Serais O., Merien F., Perolat P. 2001. Following the course of human leptospirosis: evidence of a critical threshold for the vital prognosis using a quantitative PCR assay. FEMS Microbiol. Lett. 204:317–321 [DOI] [PubMed] [Google Scholar]
- 53. Tucunduva de Faria M., et al. 2007. Morphological alterations in the kidney of rats with natural and experimental Leptospira infection. J. Comp. Pathol. 137:231–238 [DOI] [PubMed] [Google Scholar]
- 54. van den Ingh T. S., Hartman E. G. 1986. Pathology of acute Leptospira interrogans serotype icterohaemorrhagiae infection in the Syrian hamster. Vet. Microbiol. 12:367–376 [DOI] [PubMed] [Google Scholar]
- 55. Vernel-Pauillac F., Goarant C. 2010. Differential cytokine gene expression according to outcome in a hamster model of leptospirosis. PLoS Negl. Trop. Dis. 4:e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vernel-Pauillac F., Merien F. 2006. Proinflammatory and immunomodulatory cytokine mRNA time course profiles in hamsters infected with a virulent variant of Leptospira interrogans. Infect. Immun. 74:4172–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vijayachari P., Sehgal S. C., Goris M. G., Terpstra W. J., Hartskeerl R. A. 2003. Leptospira interrogans serovar Valbuzzi: a cause of severe pulmonary haemorrhages in the Andaman Islands. J. Med. Microbiol. 52:913–918 [DOI] [PubMed] [Google Scholar]
- 58. Wagenaar J. F., et al. 2009. Soluble ST2 levels are associated with bleeding in patients with severe leptospirosis. PLoS Negl. Trop. Dis. 3:e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wagenaar J. F., et al. 2009. Long pentraxin PTX3 is associated with mortality and disease severity in severe Leptospirosis. J. Infect. 58:425–432 [DOI] [PubMed] [Google Scholar]
- 60. Wuscher N., Huerre M. 1993. Méthode de Warthin-Starry modifiée au pyrocatechol: intérêt pour la mise en évidence des spirochètes et agents infectieux. (Method of Warthin-Starry modified with pyrocatechol: interest for revealing spirochetes and infectious agents.) Rev. Fr. Histotech. 6:5–8 [Google Scholar]
- 61. Xue F., et al. 2010. Transcriptional responses of Leptospira interrogans to host innate immunity: significant changes in metabolism, oxygen tolerance, and outer membrane. PLoS Negl. Trop. Dis. 4:e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang C. W., et al. 2002. The Leptospira outer membrane protein LipL32 induces tubulointerstitial nephritis-mediated gene expression in mouse proximal tubule cells. J. Am. Soc. Nephrol. 13:2037–2045 [DOI] [PubMed] [Google Scholar]
- 63. Yang G. G., Hsu Y. H. 2005. Nitric oxide production and immunoglobulin deposition in leptospiral hemorrhagic respiratory failure. J. Formos. Med. Assoc. 104:759–763 [PubMed] [Google Scholar]
- 64. Yang H. L., et al. 2006. Thrombocytopenia in the experimental leptospirosis of guinea pig is not related to disseminated intravascular coagulation. BMC Infect. Dis. 6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yersin C., et al. 2000. Pulmonary haemorrhage as a predominant cause of death in leptospirosis in Seychelles. Trans. R. Soc. Trop. Med. Hyg. 94:71–76 [DOI] [PubMed] [Google Scholar]