

Abstract
We used trio-based whole-exome sequencing to analyze two families affected by Weaver syndrome, including one of the original families reported in 1974. Filtering of rare variants in the affected probands against the parental variants identified two different de novo mutations in the enhancer of zeste homolog 2 (EZH2). Sanger sequencing of EZH2 in a third classically-affected proband identified a third de novo mutation in this gene. These data show that mutations in EZH2 cause Weaver syndrome.
Main Text
Weaver syndrome [MIM 277590] is a rare congenital anomaly syndrome first described in two families in 1974.1 It consists of generalized overgrowth, advanced bone age, marked macrocephaly, hypertelorism, and characteristic facial features. Intellectual disability is common. Approximately 40 cases are known from the literature. Typically, Weaver syndrome occurs as a sporadic condition, though cases of parent-to-child transmission have been documented.2, 3 Some patients thought to have Weaver syndrome have mutations in NSD1, which is mutated or deleted in most patients with classic Sotos syndrome [MIM 117550].4, 5, 6 This molecular finding has fuelled debate among clinical geneticists regarding whether the Sotos and Weaver syndromes represent variable expressivity of a single locus with allelic heterogeneity or whether they represent distinct disorders caused by mutations in different genes. Clinical features shared by both syndromes include developmental delay, overgrowth, and prominent macrocephaly, and features distinguishing Weaver syndrome from Sotos syndrome are retrognathia with a prominent chin crease (sometimes described as a stuck-on chin), increased prenatal growth, and a carpal bone age that is greatly advanced compared to metacarpal and phalangeal bone age.
Whole-exome sequencing (WES) facilitates the identification of sequence changes in the protein-coding genome in small families and has enabled identification of rare and ultrarare Mendelian disorders that have hitherto been refractory to linkage mapping.7, 8 To search for disease-causing alleles in Weaver syndrome, we collected saliva-derived DNA by using kits (Oragene) from one of the probands included in the original report describing Weaver syndrome,1 from two other unrelated probands with classical features of the condition, and from all six unaffected parents. We obtained informed consent from the parents, assent from the affected individuals, and ethical review and approval according to the Finding of Rare Disease Genes (FORGE) Canada Consortium. Probands 1 and 2 were personally examined by D.D.W., who also reviewed photos and clinical details on proband 3 (examined in childhood; records of examination and referral provided by D.C.; proband was reexamined in adulthood by W.T.G.); probands were confirmed in childhood to have classical features of Weaver syndrome and not Sotos syndrome (Figure 1 and Table 1), and parents were confirmed to be unaffected. None of the three probands had submicroscopic abnormalities detectable by microarray analysis (Illumina Human Omni2.5-Quad chip, analyzed with CNVPartition from GenomeStudio V2010.3). None of the probands had expanded FMR1 alleles [MIM 309550] or abnormalities on clinical karyotyping. We also ruled out rare variants in NSD1 [MIM 606681] by using Sanger sequencing on saliva-derived DNA in all three probands (data not shown, primers available on request).
Figure 1.

Proband from Weaver et al.1 and Two Additional Probands Described in This Study
Proband 1 is shown at 18 months (A), 6 years (B), 11 years (C), 17 years (D), 21 years (E), and 30 years (F, G, and H). Proband 2 is shown at age 7 years (I) and 13 years (J, K, and L). Proband 3 is shown at birth (M), 12 months (N), 24 months (O), 42 months (P), 6 years (Q), 10 years (R), 11 years (S), 16 years (T), and 19 years (U and V). Proband 3 at 8 years in a stance that shows elbow and knee contractures (W). Photos are published with the proxy consent of the parents and assent of the probands.
Table 1.
Phenotypic Manifestations of Weaver Syndrome in Patients with EZH2 Mutations
| Phenotypic Manifestation | Proband 1 c.457_459del (p.Tyr153del) | Proband 2 c.2080C>T (p.His694Tyr) | Proband 3 c.394C>T (p.Pro132Ser) |
|---|---|---|---|
| Gestational age at delivery (weeks) | ∼36.5 | 32 | 42 |
| Birth weight (kg) | 4.82 | 3.26 | 4.50 |
| Birth length (cm) | 55.0 | 50.8 | 55 |
| Birth head circumference (cm) | 36.5 | ∼34 | Nk |
| Recent weight (kg) [age measured] | 118 [30 years] | 64.1 [10 years, 11 months] | 103.3 [19 years, 9 months] |
| Recent height (cm) [age measured] | 190.5 [30 years] | 179 [10 years, 11 months] | 177.7 [19 years, 9 months] |
| Excessive growth of prenatal onset | +++ | +++ | +++ |
| Accelerated osseous maturation | ++++ | ++++ | ++++ |
| Neurological Features | |||
| Hypertonia | ++ | – | – |
| Hypotonia | – | ++ | ++ |
| Hoarse, low-pitched cry | ++ | ++ | ++ |
| Intellectual disability | mild | borderline-mild | mild |
| Excessive appetite | ++ | ++ | – |
| Ventriculomegaly | ++ | Nk | – |
| Delayed myelination | Nk | Nk | + |
| Cerebellar hypoplasia (mild) | – | Nk | + |
| Seizures (age of onset) | tonic-clonic [13 years] | – | brief absence [15 years] |
| Poor fine motor coordination | ++ | + | ++ |
| Poor balance and gravitational insecurity | ++ | ++ | ++ |
| Fatty filum terminale | – | – | ++ |
| Craniofacial | |||
| Macrocephaly | +++ | +++ | +++ |
| Large bifrontal diameter | +++ | +++ | +++ |
| Flat occiput | + | – | + |
| Large ears | +++ | + | + |
| Ocular hypertelorism | ++ | ++ | – |
| Downslanted palpebral fissures | + | + | + |
| Long philtrum | ++ | ++ | ++ |
| Retrognathia | + | + | + |
| Cardiovascular | |||
| Patent ductus arteriosus | – | – | + |
| Hands | |||
| Prominent digit pads | ++ | ++ | ++ |
| Single transverse palmar crease | – | – | – |
| Camptodactyly | ++ | + | – |
| Broad thumbs | ++ | ++ | – |
| Thin, deep-set nails | ++ | ++ | ++ |
| Feet | |||
| Clinodactyly, toes | + | + | – |
| Talipes equinovarus | ++ | – | – |
| Short fourth metatarsals | + | – | – |
| Hind foot valgus | – | – | + |
| Limited elbow and knee extension in early life | + | – | + |
| Limited elbow and knee extension after puberty | + | – | + |
| Widened distal femurs and ulnas | ++ | Nk | – |
| Skin | |||
| Excessive loose skin | ++ | ++ | – |
| Inverted nipples | + | – | – |
| Thin hair | + | – | – |
| Increased pigmented nevi | – | ++ | ++ |
| Connective Tissue | |||
| Umbilical hernia | ++ | + | + |
| Inguinal hernia | ++ | – | – |
| Scoliosis (degrees) | 20 | 10 | 16 |
| Endocrine | |||
| Hypothyroidism [age of onset] | ∼25 years | – | – |
| Growth hormone deficiency [age of onset] | ∼27 years | – | – |
Key: + = minimally present, ++ = obviously present, +++ = very prominent, ++++ = severe, – = assessed and found to be absent, Nk = not known.
We performed exome sequencing on samples from six individuals (probands 1 and 2 and the parents of both), and quantified the DNA concentration by using a Quant-iT dsDNA HS assay kit and a Qubit fluorometer (Invitrogen). We sheared approximately 500 ng DNA for 75 s at a duty cycle of 20% and an intensity of 5 with a Covaris E210 and size fractionated the DNA on an 8% polyacrylamide gel. We excised the 200–250 bp size fraction, eluted it from the gel slice, and ligated it to Illumina paired-end adapters following a standard protocol as previously described.9 Adaptor-ligated DNA was amplified for 10 cycles with the PE primer set (Illumina) and purified. The pre-exome capture library DNA was assessed with an Agilent DNA 1000 Series II assay and subsequently hybridized 500 ng to the 50 Mb exon probe with the Human All Exon Kit (G3370) following Agilent's SureSelect Target Enrichment protocol. The captured DNA was purified with a QIAGEN MiniElute column, and amplified for 12 cycles with the standard Illumina PE primer set. PCR products were separated by size on an 8% PAGE gel before gel extraction at the desired size range (320–370 bp). The samples were then assessed with an Agilent DNA 1000 series II assay. The final library was diluted to a concentration of 10 nM, which was confirmed via a Quant-iT dsDNA HS assay kit and a Qubit fluorometer as above, prior to cluster generation and exome sequencing.
We performed paired-end tag (PE100) sequencing with an Illumina HiSeq2000 machine. Sequencing reads that failed chastity filtering were removed with Illumina's GA Pipeline (1.12.0 RTA 1.12.4.2), and the remaining reads were mapped to the reference genome sequence (hg18) with BWA 0.5.7;10 duplicate reads and reads with a mapping score of 0 were removed. The aligned reads were exported to pileup format and called with SAMtools 0.1.13.11 We filtered single nucleotide variants and retained those with a minimum SNP quality of 20 at varFilter parameter −D 1000. Small insertions and deletions (indels) were processed similarly with varFilter parameters −D 1000, −d 2 and −l 30. We then imported all the variants into a local PostgreSQL database used to store and process human variation data.12 We annotated the filtered variants as known or novel depending on whether they had been previously reported in a public database such as dbSNP13 or the 1000 Genomes Project14 or previously observed in the in-house database of normal germline genomes sequenced at the British Columbia Cancer Agency, Genome Sciences Centre (BCGSC). Currently, this database contains over 1.47 billion observed sequence variants mapping to 63.9 million unique base substitutions derived from over 1,360 individuals. Specifically, we sought to identify variations that cause nonsynonymous changes in protein-coding regions and those that fell within two bases of exon boundaries (such that they might interfere with intron splicing; Table S1, available online).
In proband 1, we identified a heterozygous c.457_459del (p.Tyr153del) variant in isoform A of EZH2 [MIM 601573] (RefSeq NM_004456.4). This was not seen in either of his parents, indicating that this was a de novo mutation. We also identified a heterozygous de novo missense variant c.2080C>T (p.His694Tyr) of the same gene in proband 2. These variants were seen at high coverage in both probands (in 121 out of 239 reads and in 153 out of 304 reads, respectively) but were not seen in the parental reads. In our hands, coverage at this level has a positive predictive value of 100% for subsequent Sanger verification. We went on to validate the presence of both of these mutations and their de novo status by using Sanger sequencing (Figure 2, primers available on request). After filtering out parental variants, no other gene demonstrated private mutations in both of the probands (Table S1), where “private” is defined as not found in dbSNP, 1000 Genomes Project data, or among normal genomes (including the parents of the probands) sequenced in-house at the BCGSC. We then analyzed EZH2 by Sanger sequencing in a third trio (proband 3 and her parents) in whom we had not performed exome sequencing. We identified a c.394C>T (p.Pro132Ser) mutation in Proband 3 and confirmed that it was de novo (i.e., absent in both parents).
Figure 2.
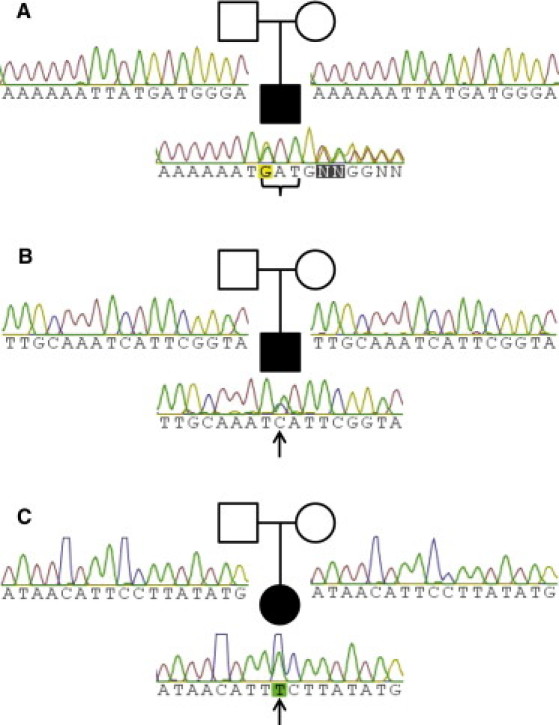
Sanger Confirmation of Sequence Variants
(A) The c.457_459del mutation in Proband 1 (curly bracket) is de novo.
(B) The c.2080C>T mutation in Proband 2 (arrow) is de novo.
(C) The c.394C>T mutation in Proband 3 (arrow) is de novo.
The p.Tyr153 del mutation in Proband 1 lies six amino acid residues from the N terminus of the Simple Modular Architecture Research Tool (SMART)15-predicted SANT (switching-defective protein 3 [Swi3]), adaptor 2 [Ada2], nuclear receptor corepressor [N-CoR], transcription factor [TF]IIIB′) domain (Figure 3, annotated with InterPro16). The deletion of an entire amino acid (p.Tyr153del) in EZH2 removes a bulky polar residue near the putative SANT DNA-binding domain, which is suggestive of functional consequences for the protein. Also, the deleted codon is evolutionarily conserved according to phyloP.17 The placental mammalian genome-wide alignment-based phyloP score averaged across the three codon sites (chromosome 7:148,157,778–148,157,780) is 1.59 (taken from the UCSC Genome Browser18 Conservation track for the hg18 assembly). A positive phyloP score is interpreted as a signature of evolutionary conservation, which is consistent with functional importance.
Figure 3.
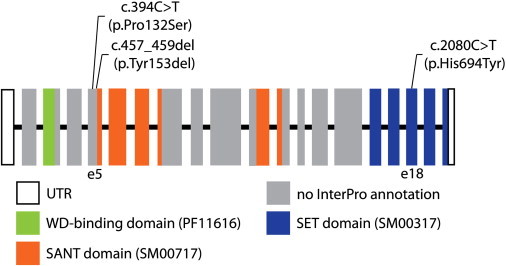
Schematic of Human EZH2
Coding exons are indicated by rectangles and noncoding exons by open rectangles. The exons are numbered starting from the exon containing the 5′ untranslated region (UTR). The putative SANT DNA-binding domain is shown in orange, the SET domain in blue, and the WD-binding domain in green. The SMART or Pfam domain identifier is presented in parentheses. Exons with no InterPro annotation are indicated in gray.
The Sorting Intolerant From Tolerant (SIFT)19, 20 scores for p.Pro132Ser and p.His694Tyr were 0.00 (with values ≤ 0.05 interpreted as damaging). With PolyPhen221 trained on the HumDiv data set, p.Pro132Ser and p.His694Tyr were both predicted to be probably damaging. Additionally, the nucleotide site (chromosome 7:148,157,843) where Pro132 occurs has a placental mammalian phyloP score of 3.17, and the nucleotide site (chromosome 7:148,137,365) where His694 occurs has a score of 2.90. The positive phyloP scores suggest that these nucleotide sites are evolutionarily conserved, whereas a score near 0 would have suggested neutral selection. Perhaps the most suggestive evidence for the pathogenicity of p.His694Tyr comes from the specific location of this histidine residue—it is located in the Su(var)3,9, Enhancer of zeste, Trithorax (SET) domain, within the knot substructure of the active site, and is predicted to form part of the binding domain for the enzymatic cofactor S-adenosyl-L-methionine (AdoMet).22 We made a 3D model of human EZH2 p.His694Tyr by using SWISS-MODEL23 and ICM software24 (Molsoft) based on the structure of the related protein euchromatic histone-lysine N-methyltransferase 1 ([MIM 607001] selected because of its lack of gaps and high-resolution crystal structure [1.6 Å]; see Figure 4).25 The conformation of this conserved histidine is highly similar across known crystal structures for the SET domain. Because of its proximity to the AdoMet-binding site, replacement of this histidine with a bulkier tyrosine side chain could well interfere with cofactor binding and methyltransferase activity of the mutant molecule. Mutation of the histidine residue that occupies a similar position in SUV39H1 abolished its methyltransferase activity in an in vitro assay,26 and a mutation of this specific histidine residue to arginine (with functional effects) has been reported in a 41-year-old male with chronic myelomonocytic leukemia.27 Furthermore, mutations in nearby residues at positions 690 and 693 were also reported in other hematological malignancies.27
Figure 4.
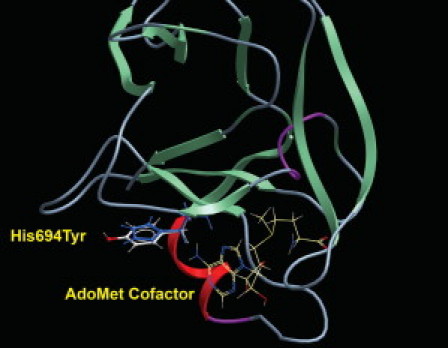
Ribbon Model of the EZH2 SET Domain
The wild-type histidine residue is shown in blue and the bulkier tyrosine in white and red. The nearby binding site of the S-Adenosylmethionine cofactor is also indicated.
The EZH2 protein partners with SUZ12 and EED to form the polycomb repressive complex (PRC2). This complex catalyzes the trimethylation of lysine 27 of histone H3 (H3K27), and EZH2 itself forms the catalytic subunit for this reaction. Thus, EZH2 forms a key component of molecular machinery that shuts off transcription of loci to which trimethylated H3K27 is bound. EZH2 is known to be mutated somatically in lymphoid and myeloid cancers.9, 27 Mutation of arginine 690 to histidine or histidine 694 to arginine appears to block EZH2's ability to facilitate trimethylation of H3K27,27 and mutation of tyrosine at position 641 also alters the affinity of EZH2 for H3K27.28 Residues other than tyrosine at position 641 reduce the preference for unmethylated and monomethylated lysine and favor trimethylation of lysine.28 It is worth noting that some patients with Weaver syndrome have been reported to develop tumors or malignancies, including acute lymphoblastic leukemia.29 The lifetime risk of malignancy in Weaver syndrome patients has been estimated at 11%, though this is likely an overestimate because of reporting bias. Nevertheless, constitutive EZH2 mutations might confer a mild predisposition to malignancy. Mutations that conferred a more profound effect on histone methylation might also confer a stronger selective advantage for cell growth. Such mutations might not be viable in the fully heterozygous state and would be observed in nature only as postzygotic somatic mutations. In vitro studies will be required to determine whether the p.His694Tyr mutation we have observed in this study affects the affinity of EZH2 for AdoMet and whether any of the three mutations affect H3K27 trimethylation.
It is clear from the resulting phenotypes that EZH2 mutations and structural variants also affect developmentally important pathways. Several patients with deletions and duplications encompassing EZH2 are reported in the DECIPHER database (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources),30 and one patient with a duplication (patient 250841) does manifest macrocephaly. The fact that there is little other concordance between the DECIPHER phenotypes (apart from intellectual disability) and those of our patients is likely attributable to the multiple other genes affected by these structural variants. It is also possible that the EZH2 protein variants expressed in our patients act through molecular mechanisms other than haploinsufficiency; subtle but important changes in specific subfunctions of EZH2 are known to occur in association with specific protein variants.28
Animal studies are yielding insights into the specific role of mouse Ezh2 in organ systems other than the hemopoietic system. Ezh2 regulates proximodistal axis elongation and anteroposterior axis specification in the developing mouse limb.31 Limb anomalies in humans with Weaver syndrome are relatively mild, though deep-set nails, joint contractures, and dysharmonic bone age might be consequences of aberrant EZH2 signaling in human limb patterning. Mice with targeted knockout of Ezh2 in beta cells had reduced beta cell proliferation and beta cell mass,32 and mice with targeted knockout of Ezh2 in satellite cells had impaired regeneration of muscle.33 None of our three probands had elevated fasting glucose, though probands 2 and 3 continue to manifest hypotonia (Table 1).
Several patients with Weaver syndrome are reported to have mutations in NSD1, a gene first associated with overgrowth in the Sotos syndrome.4, 5, 6 NSD1 mutations in Weaver syndrome appear to cluster toward the C terminus of the molecule, 5′ of the SET domain, though one frameshift mutation in exon 5 and one mutation within the SET domain itself have been reported.5 Direct protein-protein interactions between EZH2 and NSD1 are not yet known, but the similarity of the human phenotypes caused by rare mutations in these genes suggests interactive links between gene networks containing these two SET-domain-containing proteins.
Our data demonstrate de novo mutations in EZH2 in three families, including one of the original families that led to the definition of the disorder. These results illustrate the power of next-generation sequencing methods to identify rare disease-causing variants with a small number of samples (six individuals in this case, only two of whom are affected), provided that detailed clinical studies first identify phenotypic concordance and additional families are available as a replication set. Because Weaver syndrome is a genetically heterogeneous condition, other genes associated with the condition might yet be identified.
Finally, it is interesting to note the involvement of SET-domain proteins in molecular networks that, when perturbed, cause intellectual disability syndromes and/or cancer. Mutations in NSD1 cause Sotos syndrome and Weaver syndrome,4, 5, 6 and mutations in MLL2 (which also bears a SET domain) cause Kabuki syndrome.34 Histone-modifying proteins such as NSD1, EZH2, and MLL2 appear repeatedly as targets of somatic mutation in hematological malignancies35 and are emerging as a cause of neurodevelopmental disorders. Detailed studies of larger cohorts of well-phenotyped probands will assist in determination of the prevalence of mutations in EZH2 and other SET-domain proteins in Weaver and other syndromes and of their consequences on metabolism and cancer risk. At this time, the evidence from animal models is insufficient to recommend routine surveillance in Weaver syndrome for diabetes or myopathy, beyond what would ordinarily be performed in pediatric and adult practice. Data from long-term follow-up of adult individuals with Weaver syndrome will assist physicians in deciding the optimal time to screen for potential metabolic and neoplastic complications of this rare disorder. The possibility that dietary supplementation with methionine (or with other methyl donors such as betaine and choline) might improve the activity of certain EZH2 variants is attractive but awaits further investigation.
Acknowledgments
The authors gratefully acknowledge the generosity of the families in providing samples and clinical details for this study. We would like to thank J. Marcadier (Clinical Coordinator) and C. Beaulieu (Project Manager) for their contribution to the infrastructure of the FORGE Canada Consortium. This work was funded by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research (CIHR), and the Ontario Genomics Institute (OGI-049). Additional funding was provided by Genome Québec and Genome British Columbia. W.T.G. is supported by a Clinician Scientist Phase 2 Award from the CIHR Institute of Genetics and a Clinician Scientist Salary Award from the Child and Family Research Institute. K.M.B. is supported by a Clinical Investigatorship Award from the CIHR Institute of Genetics. S.J.M.J. is a Senior Scholar of the Michael Smith Foundation for Health Research. The FORGE Canada Steering Committee includes Kym Boycott (University of Ottawa), Jan Friedman (University of British Columbia), Jacques Michaud (Université de Montréal), Francois Bernier (University of Calgary), Michael Brudno (University of Toronto), Bridget Fernandez (Memorial University), Bartha Knoppers (McGill University), Mark Samuels (Université de Montréal), and Steve Scherer (University of Toronto).
Published online: December 15, 2011
Footnotes
Supplemental Data include one figure and one table and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Burrows-Wheeler Aligner BWA, http://bio-bwa.sourceforge.net/
Database of Genomic Variants, http://projects.tcag.ca/variation/
DECIPHER database, http://decipher.sanger.ac.uk/
Human Variation Database (Vancouver version), http://vancouvershortr.sourceforge.net/
Online Mendelian Inheritance in man (OMIM), http://www.omim.org
Phred, http://www.phrap.com/phred/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
PostgreSQL, http://www.postgresql.org/
SAMtools, http://samtools.sourceforge.net/
SIFT, http://sift.jcvi.org/
Simple Modular Architecture Research Tool, http://smart.embl-heidelberg.de/
SWISS-Model Server, http://swissmodel.expasy.org/
Accession Codes
Reference sequences reported in this study are available from GenBank under the following accession codes: EZH2 longest isoform, NM_004456.4. Local identifiers are as follows: NM_004456.4:c.457_459del (NCBI ss 472336142), NM_004456.4:c.2080C>T (NCBI ss 472336143) and NM_004456.4:c.394C>T (NCBI ss 472336144).
Supplemental Data
References
- 1.Weaver D.D., Graham C.B., Thomas I.T., Smith D.W. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J. Pediatr. 1974;84:547–552. doi: 10.1016/s0022-3476(74)80675-x. [DOI] [PubMed] [Google Scholar]
- 2.Fryer A., Smith C., Rosenbloom L., Cole T. Autosomal dominant inheritance of Weaver syndrome. J. Med. Genet. 1997;34:418–419. doi: 10.1136/jmg.34.5.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Proud V.K., Braddock S.R., Cook L., Weaver D.D. Weaver syndrome: Autosomal dominant inheritance of the disorder. Am. J. Med. Genet. 1998;79:305–310. doi: 10.1002/(sici)1096-8628(19981002)79:4<305::aid-ajmg13>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 4.Douglas J., Hanks S., Temple I.K., Davies S., Murray A., Upadhyaya M., Tomkins S., Hughes H.E., Cole T.R., Rahman N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am. J. Hum. Genet. 2003;72:132–143. doi: 10.1086/345647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rio M., Clech L., Amiel J., Faivre L., Lyonnet S., Le Merrer M., Odent S., Lacombe D., Edery P., Brauner R., et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J. Med. Genet. 2003;40:436–440. doi: 10.1136/jmg.40.6.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baujat G., Rio M., Rossignol S., Sanlaville D., Lyonnet S., Le Merrer M., Munnich A., Gicquel C., Colleaux L., Cormier-Daire V. Clinical and molecular overlap in overgrowth syndromes. Am. J. Med. Genet. C. Semin. Med. Genet. 2005;137C:4–11. doi: 10.1002/ajmg.c.30060. [DOI] [PubMed] [Google Scholar]
- 7.Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 8.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 9.Morin R.D., Johnson N.A., Severson T.M., Mungall A.J., An J., Goya R., Paul J.E., Boyle M., Woolcock B.W., Kuchenbauer F., et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fejes A.P., Khodabakhshi A.H., Birol I., Jones S.J. Human variation database: An open-source database template for genomic discovery. Bioinformatics. 2011;27:1155–1156. doi: 10.1093/bioinformatics/btr100. [DOI] [PubMed] [Google Scholar]
- 13.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., et al. the 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letunic I., Doerks T., Bork P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr931. in press. Published online November 3, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter S., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009;37(Database issue):D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita P.A., Rhead B., Zweig A.S., Hinrichs A.S., Karolchik D., Cline M.S., Goldman M., Barber G.P., Clawson H., Coelho A., et al. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res. 2011;39(Database issue):D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 20.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs S.A., Harp J.M., Devarakonda S., Kim Y., Rastinejad F., Khorasanizadeh S. The active site of the SET domain is constructed on a knot. Nat. Struct. Biol. 2002;9:833–838. doi: 10.1038/nsb861. [DOI] [PubMed] [Google Scholar]
- 23.Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 24.Abagyan R.A., Totrov M.M., Kuznetsov D.A. ICM: A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994;15:488–506. [Google Scholar]
- 25.Wu H., Min J., Lunin V.V., Antoshenko T., Dombrovski L., Zeng H., Allali-Hassani A., Campagna-Slater V., Vedadi M., Arrowsmith C.H., et al. Structural biology of human H3K9 methyltransferases. PLoS ONE. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rea S., Eisenhaber F., O'Carroll D., Strahl B.D., Sun Z.W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D., Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 27.Makishima H., Jankowska A.M., Tiu R.V., Szpurka H., Sugimoto Y., Hu Z., Saunthararajah Y., Guinta K., Keddache M.A., Putnam P., et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24:1799–1804. doi: 10.1038/leu.2010.167. [DOI] [PubMed] [Google Scholar]
- 28.Yap D.B., Chu J., Berg T., Schapira M., Cheng S.W., Moradian A., Morin R.D., Mungall A.J., Meissner B., Boyle M., et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basel-Vanagaite L. Acute lymphoblastic leukemia in Weaver syndrome. Am. J. Med. Genet. A. 2010;152A:383–386. doi: 10.1002/ajmg.a.33244. [DOI] [PubMed] [Google Scholar]
- 30.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyngaarden L.A., Delgado-Olguin P., Su I.H., Bruneau B.G., Hopyan S. Ezh2 regulates anteroposterior axis specification and proximodistal axis elongation in the developing limb. Development. 2011;138:3759–3767. doi: 10.1242/dev.063180. [DOI] [PubMed] [Google Scholar]
- 32.Chen H., Gu X., Su I.H., Bottino R., Contreras J.L., Tarakhovsky A., Kim S.K. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23:975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juan A.H., Derfoul A., Feng X., Ryall J.G., Dell'Orso S., Pasut A., Zare H., Simone J.M., Rudnicki M.A., Sartorelli V. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011;25:789–794. doi: 10.1101/gad.2027911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C., et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morin R.D., Mendez-Lago M., Mungall A.J., Goya R., Mungall K.L., Corbett R.D., Johnson N.A., Severson T.M., Chiu R., Field M., et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.