

Abstract
Cone-rod dystrophy (CRD) and retinitis pigmentosa (RP) are clinically and genetically overlapping heterogeneous retinal dystrophies. By using homozygosity mapping in an individual with autosomal-recessive (ar) RP from a consanguineous family, we identified three sizeable homozygous regions, together encompassing 46 Mb. Next-generation sequencing of all exons, flanking intron sequences, microRNAs, and other highly conserved genomic elements in these three regions revealed a homozygous nonsense mutation (c.497T>A [p.Leu166∗]) in C8orf37, located on chromosome 8q22.1. This mutation was not present in 150 ethnically matched control individuals, single-nucleotide polymorphism databases, or the 1000 Genomes database. Immunohistochemical studies revealed C8orf37 localization at the base of the primary cilium of human retinal pigment epithelium cells and at the base of connecting cilia of mouse photoreceptors. C8orf37 sequence analysis of individuals who had retinal dystrophy and carried conspicuously large homozygous regions encompassing C8orf37 revealed a homozygous splice-site mutation (c.156−2A>G) in two siblings of a consanguineous family and homozygous missense mutations (c.529C>T [p.Arg177Trp]; c.545A>G [p.Gln182Arg]) in siblings of two other consanguineous families. The missense mutations affect highly conserved amino acids, and in silico analyses predicted that both variants are probably pathogenic. Clinical assessment revealed CRD in four individuals and RP with early macular involvement in two individuals. The two CRD siblings with the c.156−2A>G mutation also showed unilateral postaxial polydactyly. These results underline the importance of disrupted ciliary processes in the pathogenesis of retinal dystrophies.
Main Text
Retinitis pigmentosa (RP [MIM 268000]) is the most common inherited retinal degeneration and has an estimated worldwide prevalence of 1/4,000 individuals.1 RP is initially characterized by rod photoreceptor dysfunction, giving rise to night blindness, which is followed by progressive rod and cone photoreceptor dystrophy, resulting in midperipheral vision loss, tunnel vision, and sometimes blindness. The disease is genetically highly heterogeneous and displays all Mendelian patterns of inheritance. In addition, there are some cases with mitochondrial mutations and digenic inheritance.2, 3 Thus far, mutations in 34 genes have been associated with nonsyndromic autosomal-recessive (ar) RP (RetNet).3
In contrast to RP, cone-rod dystrophy (CRD [MIM 120970]) is characterized by a primary loss of cone photoreceptors and subsequent or simultaneous loss of rod photoreceptors.4, 5 The disease in most cases becomes apparent during primary-school years. The symptoms include photoaversion, a decrease in visual acuity with or without nystagmus, color-vision defects, and decreased sensitivity of the central visual field. Because rods are also involved, night blindness and peripheral vision loss can occur. The diagnosis of CRD is mainly based on electroretinogram (ERG) recordings, in which cone (photopic) responses are more severely reduced than, or equally as reduced as, rod (scotopic) responses.5, 6 CRD occurs in 1/40,000 individuals4, 5 and also displays all types of Mendelian inheritance. Mutations in five genes i.e., ABCA4 (MIM 601691), ADAM9 (MIM 602713), CDHR1 (MIM 609502), CERKL (MIM 608381), and RPGRIP1 (MIM 605446) have thus far been implicated in nonsyndromic arCRD.7, 8, 9, 10, 11
Genes harboring arCRD- and arRP-associated mutations encode proteins that are involved in phototransduction, vitamin A (retinoid) metabolism, transport along the connecting cilium, cell-to-cell signaling or synaptic interaction, gene regulation, and phagocytosis.3 Mutations in these genes are estimated to underlie ∼50% of the cases.
We aimed to identify the genetic defects associated with retinal dystrophies and to clinically investigate individuals with RP and CRD. The tenets of the Declaration of Helsinki were followed, and, in accordance with approvals gathered from the appropriate institutional review boards, informed consent was obtained from all participating individuals prior to the donation of blood samples.
Homozygosity mapping has proven to be a fruitful method of identifying mutations underlying autosomal-recessive retinal diseases12, 13, 14, 15, 16 and of establishing genotype-phenotype correlations.17, 18 To identify the genetic defect in a consanguineous family with RP (family 1; Figure 1A), we analyzed the DNA of individual IV:1 by using an Affymetrix GeneChip Human Mapping 250K SNP array (Affymetrix, Santa Clara, CA, USA) and analyzed the SNP data by using Partek Genomic Suite software (Partek, St. Louis, MO, USA). The analyses showed three large homozygous regions of 7.7 Mb (4q34.3-q35.1, rs2128423–rs59156350), 31.6 Mb (8q22.1-q24.13, rs279475–rs7013593), and 7.0 Mb (11p11.2-q11, rs11039487–rs17494990). Because more than 261 genes were present in these three chromosomal regions, a targeted next-generation sequencing (NGS) approach was used. Sequence capture was done on a 385K sequence-capture array (Roche NimbleGen, Madison, WI, USA). The array design comprised all coding and noncoding exons of these regions, including surrounding sequences that covered the splice sites. The array design harbored additional targeted regions used for similar analyses of homozygous regions in two other families. In total, the design included 4,952 targets, comprising 1,903,789 bp. Sequence capture was done according to the manufacturer's (Roche NimbleGen's) instructions with the Titanium optimized protocol as described by Hoischen et al.19 The enriched DNA regions of individual IV:1 from family 1 were sequenced on one of four lanes of a Roche 454 sequencing run, yielding 86 Mb of sequence data. Approximately 86% of the sequences were mapped back to unique regions of the human genome (hg18, NCBI build 36.1) with the use of the Roche Newbler software (version 2.3). Of all mapped reads, 91% were located on or near the targeted regions (i.e., within 500 bp). This was sufficient to reach an average of 19.3-fold coverage for all target regions. For the regions of interest, fewer than 2.6% of all targeted sequences were not covered, and only 22% of the target sequence was covered fewer than ten times. The Roche 454 software detected a total of 2,755 high-confidence variants, i.e., it identified the variants in at least three reads. We used a custom-made data-analysis pipeline as described elsewhere19 to annotate detected variants with various types of information, including known SNPs, amino acid substitutions, genomic location, and evolutionary conservation. A total of 2,573 variants either were found to represent known SNPs or overlapped with a known polymorphic region (dbSNP129); they were therefore not considered to be likely disease-causing variants.
Figure 1.
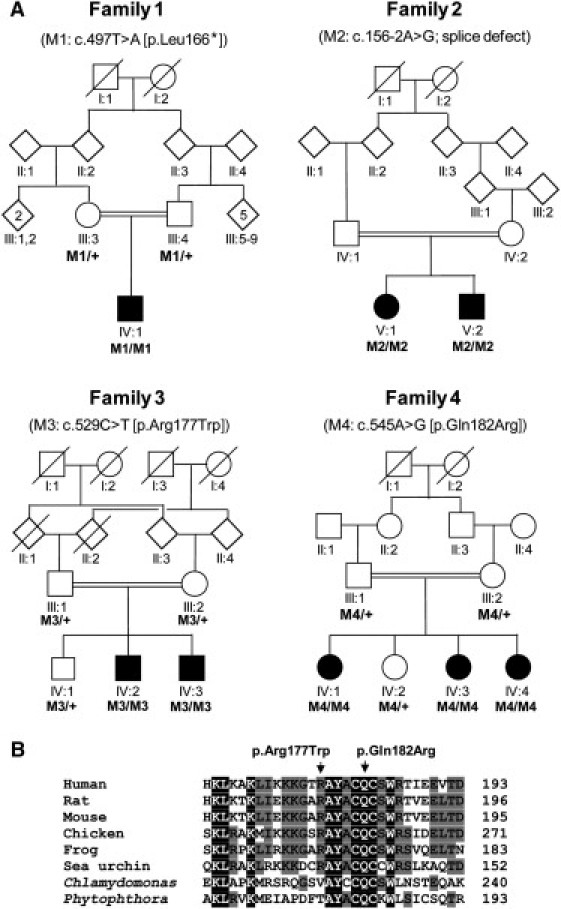
Pedigrees with C8orf37 Mutations and Partial Alignment of C8orf37 Orthologs
(A) Schematic representation of the four consanguineous families in which homozygous C8orf37 variants were identified. The respective mutations (M1–M4) are indicated above the pedigrees. In family 1, a protein-truncating mutation (p.Leu166∗) was identified; in family 2, the c.156−2A>G variant affects one of the canonical nucleotides of a splice-acceptor site. Affected individuals in families 3 and 4 carry C8orf37 missense mutations that are located in close proximity (see B). The + symbol represents wild-type. Diamond-shaped symbols represent male(s) or female(s); the numbers in the symbols indicate the numbers of individuals. Blackened symbols represent affected individuals. Symbols with slashes depict deceased individuals.
(B) Evolutionary conservation of the part of the C8orf37 polypeptide that contains the missense mutations p.Arg177Trp and p.Gln182Arg, which are identified in families 3 and 4, respectively. The alignment was performed via ClustalW2 with the following protein sequences: human (NP_808880.1), rat (NP_001007747.1), mouse (NP_680281.3), chicken (Xp_418346.2), frog (XP_002931575.1), sea urchin (XP_001194284), Phytophtora (XP_002904228.1), and Chlamydomonas (XP_001697126.1). Amino acid residues that are identical in all sequences are white on a black background, whereas amino acids that are similar but not identical are black on a light gray background. Nonconservative changes are indicated in black on a white background.
The remaining 182 variants included 39 nongenic variants, 32 untranslated-region variants, 74 intronic variants, two potential splice-site variants, and 35 exonic variants. The exonic variants consisted of three synonymous coding variants and 32 nonsynonymous coding variants. Of the latter 32 variants, only two were called as homozygous variants (i.e., >80% variant reads), whereas the remaining 30 variants appeared heterozygous. The finding of heterozygous variants in homozygous regions can possibly be explained by the existence of pseudogenes that are changing the ratio of variant reads to reference reads.20
The two homozygous nonsynonymous variants (c.2483T>C in C4orf41 [NM_199053.1] and c.497T>A in C8orf37 [NM_177965.2]) were chosen for further candidate analysis. SIFT predicted that the c.2483T>C (p.Val828Ala) variant in C4orf41 would be tolerated, and PolyPhen predicted that it would be benign. In addition, this variant was found in the 1000 Genomes database and in a heterozygous state in 3 out of 133 individuals, who were analyzed via exome NGS. On the basis of the latter finding, this variant would be present in a homozygous state in ∼1/8,000 individuals, a ratio that is 2 orders of magnitude higher than that of other RP-associated variants. Together, these data strongly suggest that the C4orf41 variant is not associated with RP in family 1. The second candidate variant was a homozygous c.497T>A variant in C8orf37 in the 31.6 Mb region of 8q22.1 (Figures 2A and 2B) and resulted in a premature stop codon (p.Leu166∗) (Figures 2C and 2D). Sanger sequencing confirmed the homozygous presence of this variant in IV:1 (Figure 2D) and its presence as a heterozygous change in both parents (Figure 1A). This mutation was not detected in 300 chromosomes of ethnically matched control individuals or in the 1000 Genomes database.
Figure 2.
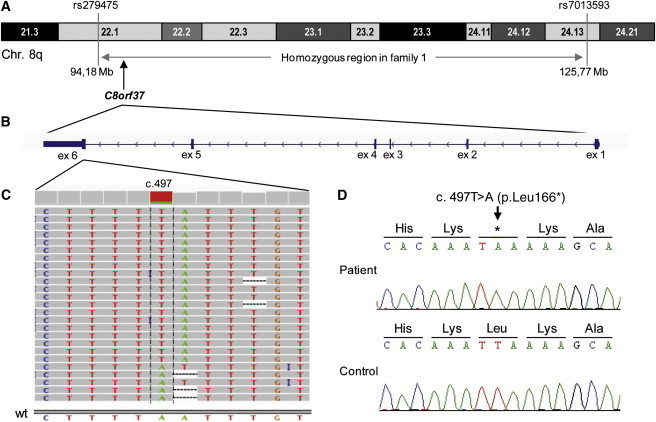
Homozygous Region in Individual IV:1 of Family 1, C8orf37 Exon-Intron Structure, and Sequence Variant
(A) The 31.6 Mb homozygous region encompassing the flanking SNPs and C8orf37 on part of chromosome 8q in IV:1 of family 1.
(B) Genomic structure of C8orf37, which consists of six exons, all of which are protein coding.
(C) Next-generation sequence traces for the reads covering the C8orf37 c.497T>A variant (minus strand), which results in a premature stop codon (p.Leu166∗). Twenty five reads, all of which show the c.497T>A change (visible in antisense direction as c.497A>T), encompass cDNA nucleotide 497. The visibility of this mutation is hampered in the five reads at the bottom, probably because of the mononucleotide stretches around the mutation. These stretches most likely lead to either sequencing or mapping problems when the Roche 454 sequencing technology is used. The homozygosity of this variant was confirmed by traditional Sanger sequencing (see D). The abbreviation wt represents the wild-type anti-sense sequence.
(D) Sanger sequencing confirmation of the c.497T>A variant in individual IV:1 of family 1.
C8orf37 spans 23,203 nucleotides of genomic DNA and consists of six exons that encode a polypeptide that is 207 amino acids long (Figure 2B). The function of C8orf37 is unknown, and because it lacks known functional protein domains, it is difficult to predict its role in the retina. In adult human tissues, C8orf37 is expressed ubiquitously and there are high levels of mRNA expression in the brain, heart, and retinae (Figure 3A). Immunohistochemical studies on hTERT-RPE1 cells with anti-C8orf37 (rabbit polyclonal, Sigma-Aldrich, MO, USA), which was tested for specificity in cow retinal extracts (Figure S1, available online), suggested that C8orf37 is localized in the cytoplasm (data not shown). However, after cilia formation was induced with serum starvation,21 C8orf37 was localized at the base of the primary cilium, as indicated by partial colocalization with polyglutamylated tubulin antibody GT335 (mouse monoclonal, Abcam, Cambridge, UK), a ciliary marker (Figure 3B). Similar results were observed when the ciliary marker antiacetylated α-tubulin (mouse monoclonal, Sigma-Aldrich, MO, USA) and anti-γ-tubulin (mouse monoclonal, Sigma-Aldrich, MO, USA) were used in combination with anti-C8orf37 (Figures 3C and 3D). Immunolocalization studies with the same antibodies in postnatal day 30 mouse retinal sections (Figures 3E–3G) also revealed an intense C8orf37 staining at the base of the photoreceptor connecting cilia. In addition, staining was observed in the photoreceptor inner segments; this staining possibly represents the rootlets.
Figure 3.
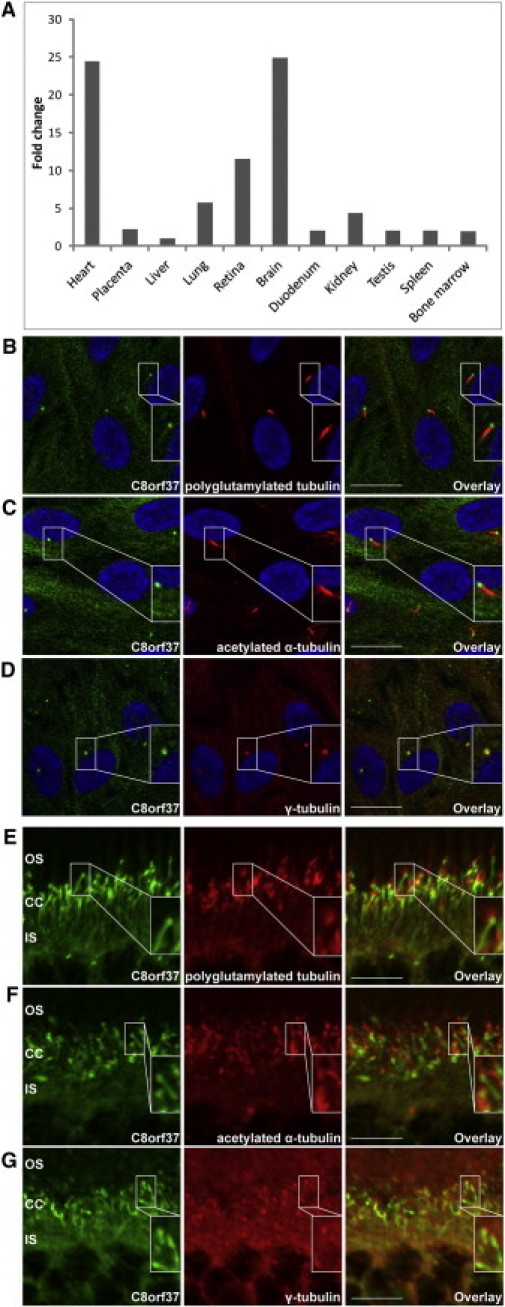
mRNA and Protein-Expression Characteristics of C8orf37
(A) mRNA expression of C8orf37 in adult human tissues is the highest in the heart and the brain, followed by the retinae. The y axis shows the relative expression of C8orf37 in comparison to the lowest expression detected in the liver. For liver expression, the ΔΔCt value was set at 0, resulting in an arbitrary expression of 1.33, 34 The x axis shows the adult human tissues that were tested for C8orf37 expression.
(B–D) Immunohistochemical staining of ciliated hTERT-RPE1 cells with antibodies against human C8orf37 (in green), and (in red) the ciliary markers GT335 (antipolyglutamylated tubulin) (B), antiacetylated α-tubulin (C), and anti-γ-tubulin (D) revealed that endogenous C8orf37 localizes to the base of the primary cilia. Nuclei were stained with DAPI (in blue). Insets show selected magnifications.
(E–G) Immunohistochemical staining in mouse retinal sections (P30). The most intense labeling for C8orf37 (in green) was noted at the base of the photoreceptor connecting cilia which partially colocalized with (in red) the connecting cilium markers GT335 (antipolyglutamylated tubulin) (E), antiacetylated α-tubulin (F), and anti-γ-tubulin (G). C8orf37 also stains structures that extend from the base of the cilium toward the inner segments, suggestive of ciliary rootlets. Abbreviations are as follows: CC, connecting cilia; IS, photoreceptor inner segments; and OS, photoreceptor outer segments. The scale bars represent the following: (B–D), 20 μm and (E–G), 10 μm.
To determine whether mutations in C8orf37 are more widely involved in retinal dystrophies, we assessed ∼400 unrelated individuals who had arCRD, arRP, or Leber congenital amaurosis (LCA [MIM 204000]) and who had significant homozygous regions, i.e., >2 Mb for individuals in nonconsanguineous families and >4 Mb for individuals in consanguineous families.13, 14, 16, 20, 22 In 15 consanguineous families and in none of the nonconsanguineous families, C8orf37 was located in conspicuously large homozygous regions, and sequence analysis of the C8orf37 coding exons revealed DNA variants in three additional consanguineous families (Figure 1A). Two affected siblings in family 2 carried a homozygous splice-site mutation (c.156−2A>G), whereas in families 3 and 4, all affected siblings carried the missense mutations p.Arg177Trp (c.529C>T) and p.Gln182Arg (c.545A>G), respectively. When available, additional affected and unaffected family members were analyzed, revealing that in each family, mutations fully cosegregated with the disease if one assumed an autosomal-recessive mode of inheritance (Figure 1A). The c.156−2A>G and c.529C>T variants were not found in 179 ethnically matched control individuals, dbSNP129, or the 1000 Genomes database. The c.545A>G mutation that was detected in a family of Israeli origin was found to be heterozygous in 2 out of 91 ethnically matched control individuals of Druze origin; this finding is in line with previous carrier-frequency data reported for other mutations that were identified in this population.23 The splice mutation c.156−2A>G affects one of two canonical nucleotides of the 3′ splice site and hence potentially leads to the skipping of exon 2, introducing a frameshift and a premature truncation of the protein. Alignment of the C8orf37 amino-acid sequences of various orthologs showed that the substituted amino acids, i.e., arginine at position 177 and glutamine at position 182, are highly conserved (Figure 1B). Gln-182 is fully conserved up to Chlamydomonas and Phytophtora, whereas Arg-177 is conserved up to sea urchins. PolyPhen predicted that p.Arg177Trp is probably damaging, and SIFT analysis revealed that it is not tolerated. PolyPhen predicted that p.Gln182Arg is probably damaging, and SIFT analysis revealed that it is tolerated.
The clinical characteristics of six individuals with C8orf37 variants are summarized in Table 1. The age of onset varied from infancy to the end of the second decade. All affected individuals showed early involvement of the macula, resulting in a loss of central vision at an early age. Visual acuity in these individuals did not exceed 20/60 and was as low as light perception in one individual. Figure 4 shows a representative fundus image of V:2 of family 2. It shows atrophy with a beaten-bronze aspect at the center of the macula. The optic disc shows temporal pallor, and there is a gliosis over the posterior pole. The retinal arterioles are mildly attenuated. The optical coherence tomography of this person shows a section of the central retina that includes the fovea; the retina is atrophic and much thinner than a normal retina.
Table 1.
Summary of the Clinical Data of Six Individuals with C8orf37-Associated Retinal Dystrophies
| Family | Person | Gender | bAge of Onset |
Visual Acuitya |
Ophthalmoscopy | Full-Field ERG (ODS) | Diagnosis | Homozygous Mutation | |
|---|---|---|---|---|---|---|---|---|---|
| OD | OS | ||||||||
| 1 | IV:1 | M | infancy | LP | LP | Macular atrophy, bone spicule pigmentations, attenuated retinal vessels. | nonrecordable | RP (with early macular involvement) | c.497T>A (p.Leu166∗) |
| 2 | V:1 | F | 17 | CF | CF | Atrophy of the central macula with gliosis. Temporal pallor of optic disc. Moderate attenuation of retinal arterioles. Some peripheral pigmentations. | nonrecordable | CRD | c.156−2A>G |
| 2 | V:2 | M | 10 | 20/125 | 20/125 | Severe atrophy of the central macula with gliosis. Temporal pallor of optic disc. Mild attenuation of retinal arterioles. | nonrecordable | CRD | c.156−2A>G |
| 3 | IV:2 | M | infancy | 20/250 | 20/400 | Peripapillar atrophy and temporal optic disc pallor, central isolated macular RPE atrophy, and pigment clumping. Attenuation of retinal vessels. | nonrecordable cone pattern, severe reduced rod recording | CRD | c.529C>T (p.Arg177Trp) |
| 3 | IV:3 | M | infancy | 20/60 | 20/400 | Central isolated macular atrophy. Attenuation of retinal vessels. No pigment clumping associated with the retinal dystrophy. | nonrecordable cone pattern, severe reduced rod recording | CRD | c.529C>T (p.Arg177Trp) |
| 4 | IV:1 | F | 18 | HM | HM | Waxy optic disc; grayish atrophic changes in maculas with pigmentation. Bone spicule-like pigmentation and heavy pigment in mid-periphery. | nonrecordable | RP (with early macular involvement) | c.545A>G (p.Gln182Arg) |
Abbreviations are as follows: CF, counting fingers; CRD, cone-rod dystrophy; F, female; HM, hand movements; LP, light perception; M, male; OD, right eye; OS, left eye; ODS, both eyes; and RP, retinitis pigmentosa.
Fraction measured at 20 feet.
Age at which the affected individuals of families 2 and 4 first noticed visual abnormalities; retinal abnormalities could have been present earlier.
Figure 4.
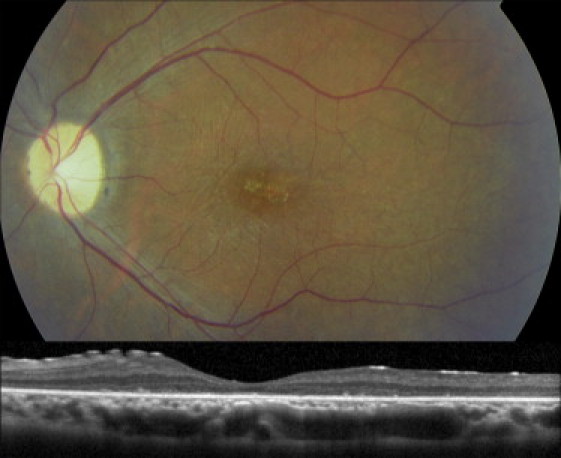
Fundus Photograph and Optical Coherence Tomography of the Left Eye of Individual V:2 of Family 2 at age 27
The upper panel of the fundus photograph shows atrophy with a beaten-bronze aspect at the center of the macula. The optic disc shows temporal pallor, and there is a gliosis over the posterior pole. The retinal arterioles are mildly attenuated. The optical coherence tomography in the lower panel shows a section of the central retina that includes the fovea; the retina is atrophic and much thinner than a normal retina.
The retinal phenotypes of four of the affected individuals (V:1 and V:2 from family 2, IV:2 and IV:3 from family 3) were classified as CRD in view of the initial symptoms, including photophobia and vision loss, ophthalmoscopic abnormalities that involved the macula rather than the periphery, and a cone pattern that was more reduced than the rod pattern on the ERG in IV:2 and IV:3. In the remaining four affected individuals, the deterioration of the rod and cone systems appeared to occur simultaneously. Besides a loss of visual acuity, these individuals also experienced night blindness in the early stage of their disease. Whether the photoreceptor dystrophy in these persons should be classified as CRD or rather as RP with early macular involvement seems largely academic. However, the phenotype in these individuals is more severe than that of the four individuals with classic CRD, considering the greater amount of vision loss and the retinal abnormalities that spread into the far periphery.
Interestingly, two siblings (V:1 and V:2 from family 2) also demonstrated a postaxial polydactyly at birth; they each had an extra finger or toe on the right hand or foot, respectively. Given that polydactyly is one of the features of Bardet-Biedl syndrome (BBS [MIM 20900])24 (a ciliopathy that includes retinal dystrophy), it is unlikely that this is a coincidental finding. Therefore, C8orf37 mutations might also contribute to extraocular abnormalities.
In this study, one nonsense, one splice-site, and two missense mutations were identified in 4 of 15 families with conspicuously large homozygous regions encompassing C8orf37, two of which (p.Leu166∗ and c.156−2A>G) have an unequivocal pathogenic effect. The two missense mutations (p.Arg177Trp and p.Gln182Arg) have been subjected to in silico prediction analysis, which suggested a pathogenic effect. Further functional studies are warranted to confirm an impact of these missense mutations on the function of C8orf37.
On the basis of the data presented above, it is difficult to deduce a genotype-phenotype correlation. The individuals with CRD and postaxial polydactyly from family 2 carry a splice-site mutation that most likely results in the skipping of exon 2, yielding a frameshift and a truncation of the 154 carboxy-terminal amino acids. Moreover, nonsense-mediated decay (NMD) could reduce the amount of mutant RNA, effectively resulting in a null allele. On the other hand, the p.Leu166∗ mutation identified in family 1 resides in exon 6, the last exon of C8orf37, and thereby is not predicted to give rise to NMD. In theory, only the 42 carboxy-terminal amino acids would be truncated, which could result in a partially functional protein. The precise effect of the missense mutations p.Arg177Trp and p.Gln182Arg on the function of the C8orf37 protein in families 3 and 4 cannot be predicted.
Given the extraocular clinical features of the siblings in family 2, one could argue that they are the most severely affected. This, however, is not the case for their retinal phenotypes because the age of onset of retinal disease in these individuals was in their teens (that of affected individuals from families 1 and 3 was in infancy). Postaxial polydactyly is one of the cardinal clinical features of BBS.24 To date, 15 genes have been associated with BBS (RetNet),25 but DNA variants in BBS1 (MIM 209901)25, 26 (A.E.-C., A.I.d.H., F.P.M.C., unpublished data), ARL6 (MIM 608845),27 BBS12 (MIM 610683),28 CEP290 (MIM 610142),29, 30 and TTC8 (MIM 608132)31 have also been associated with mild BBS or nonsyndromic retinal dystrophies. Therefore, it is very possible that C8orf37 is also implicated in full-blown BBS or in other ciliopathies. In view of the recent identification of a genetic modifier that was enriched in ciliopathies with an ocular phenotype,32 it is possible that the affected persons in family 2 carry a genetic modifier that, together with the C8orf37 variants, influenced hand and foot development.
In conclusion, we have identified mutations in a ciliary-expressed gene, C8orf37, and have found that these mutations are associated with arCRD and arRP with early macular involvement. Large-scale mutation analysis in the future will reveal the pathologic burden of C8orf37 mutations in these diseases.
Acknowledgments
We thank all participating families, and we thank Emine Bolat, Lisette Hetterschijt, Liliana Mizrahi-Meissonnier, Susanne Roosing, and Theo Peters for their scientific and technical support. We thank Christian Gilissen and Nienke Wieskamp for their excellent bioinformatical analysis and Carsten Janke for antibody GT335. The other members of the European Retinal Disease Consortium are Carmen Ayuso, Sandro Banfi, Tamar Ben-Yosef, Elfride De Baere, Christian Hamel, Chris Inglehearn, Robert K. Koenekoop, Bart P. Leroy, and Carmel Toomes. These studies were supported by the Radboud University Nijmegen Medical Centre (to F.P.M.C. and A.I.d.H.); the European Community's Seventh Framework Programs FP7/2007–2013 under grant agreement number 223143–TECHGENE (to H.S. and J.A.V.) and FP7/2009 under grant agreement number 241955–SYSCILIA (to R.R.); the Netherlands Organization for Health Research and Development ZonMW grants 917-66-363, 911-08-025 (to J.A.V.), and Vidi-917-86-396 (to R.R.); the Foundation Fighting Blindness USA (BR-GE-0510-0489-RAD, to A.I.d.H.; BR-GE-0510-0490-HUJ to D.S.); Algemene Nederlandse Vereniging ter Voorkoming van Blindheid; Gelderse Blinden Stichting; Landelijke Stichting voor Blinden en Slechtzienden; Retina Nederland; Stichting Oogfonds Nederland; Stichting Wetenschappelijk Onderzoek het Oogziekenhuis; Rotterdamse Stichting Blindenbelangen; and Stichting AF Deutman Researchfonds Oogheelkunde (to F.P.M.C. and A.I.d.H.).
Published online: December 15, 2011
Footnotes
Supplemental Data include one figure and can be found with this article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
dbSNP Build 129, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id%BC129
Integrative Genomics Viewer (IGV) Browser, http://www.broadinstitute.org/igv
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2 (Polymorphism Phenotyping v2), http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
Supplemental Data
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol. Scand. Suppl. 2002;233:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Daiger S.P., Bowne S.J., Sullivan L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007;125:151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger W., Kloeckener-Gruissem B., Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010;29:335–375. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Michaelides M., Hunt D.M., Moore A.T. The cone dysfunction syndromes. Br. J. Ophthalmol. 2004;88:291–297. doi: 10.1136/bjo.2003.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamel C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007;2:7. doi: 10.1186/1750-1172-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szlyk J.P., Seiple W., Fishman G.A., Alexander K.R., Grover S., Mahler C.L. Perceived and actual performance of daily tasks: relationship to visual function tests in individuals with retinitis pigmentosa. Ophthalmology. 2001;108:65–75. doi: 10.1016/s0161-6420(00)00413-9. [DOI] [PubMed] [Google Scholar]
- 7.Maugeri A., Klevering B.J., Rohrschneider K., Blankenagel A., Brunner H.G., Deutman A.F., Hoyng C.B., Cremers F.P.M. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000;67:960–966. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostergaard E., Batbayli M., Duno M., Vilhelmsen K., Rosenberg T. Mutations in PCDH21 cause autosomal recessive cone-rod dystrophy. J. Med. Genet. 2010;47:665–669. doi: 10.1136/jmg.2009.069120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parry D.A., Toomes C., Bida L., Danciger M., Towns K.V., McKibbin M., Jacobson S.G., Logan C.V., Ali M., Bond J., et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am. J. Hum. Genet. 2009;84:683–691. doi: 10.1016/j.ajhg.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aleman T.S., Soumittra N., Cideciyan A.V., Sumaroka A.M., Ramprasad V.L., Herrera W., Windsor E.A., Schwartz S.B., Russell R.C., Roman A.J., et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest. Ophthalmol. Vis. Sci. 2009;50:5944–5954. doi: 10.1167/iovs.09-3982. [DOI] [PubMed] [Google Scholar]
- 11.Hameed A., Abid A., Aziz A., Ismail M., Mehdi S.Q., Khaliq S. Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J. Med. Genet. 2003;40:616–619. doi: 10.1136/jmg.40.8.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collin R.W.J., Littink K.W., Klevering B.J., van den Born L.I., Koenekoop R.K., Zonneveld M.N., Blokland E.A., Strom T.M., Hoyng C.B., den Hollander A.I., Cremers F.P. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008;83:594–603. doi: 10.1016/j.ajhg.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collin R.W.J., van den Born L.I., Klevering B.J., de Castro-Miró M., Littink K.W., Arimadyo K., Azam M., Yazar V., Zonneveld M.N., Paun C.C., et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Invest. Ophthalmol. Vis. Sci. 2011;52:2227–2239. doi: 10.1167/iovs.10-6185. [DOI] [PubMed] [Google Scholar]
- 14.Bandah-Rozenfeld D., Collin R.W.J., Banin E., van den Born L.I., Coene K.L.M., Siemiatkowska A.M., Zelinger L., Khan M.I., Lefeber D.J., Erdinest I., et al. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:199–208. doi: 10.1016/j.ajhg.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.den Hollander A.I., Koenekoop R.K., Mohamed M.D., Arts H.H., Boldt K., Towns K.V., Sedmak T., Beer M., Nagel-Wolfrum K., McKibbin M., et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat. Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 16.Littink K.W., Koenekoop R.K., van den Born L.I., Collin R.W.J., Moruz L., Veltman J.A., Roosing S., Zonneveld M.N., Omar A., Darvish M., et al. Homozygosity mapping in patients with cone-rod dystrophy: Novel mutations and clinical characterizations. Invest. Ophthalmol. Vis. Sci. 2010;51:5943–5951. doi: 10.1167/iovs.10-5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Estrada-Cuzcano A., Koenekoop R.K., Coppieters F., Kohl S., Lopez I., Collin R.W.J., De Baere E.B.W., Roeleveld D., Marek J., Bernd A., et al. IQCB1 mutations in patients with leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2011;52:834–839. doi: 10.1167/iovs.10-5221. [DOI] [PubMed] [Google Scholar]
- 18.Khan M.I., Kersten F.F.J., Azam M., Collin R.W.J., Hussain A., Shah S.T., Keunen J.E.E., Kremer H., Cremers F.P.M., Qamar R., den Hollander A.I. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2011;118:1444–1448. doi: 10.1016/j.ophtha.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 19.Hoischen A., Gilissen C., Arts P., Wieskamp N., van der Vliet W., Vermeer S., Steehouwer M., de Vries P., Meijer R., Seiqueros J., et al. Massively parallel sequencing of ataxia genes after array-based enrichment. Hum. Mutat. 2010;31:494–499. doi: 10.1002/humu.21221. [DOI] [PubMed] [Google Scholar]
- 20.Walsh T., Lee M.K., Casadei S., Thornton A.M., Stray S.M., Pennil C., Nord A.S., Mandell J.B., Swisher E.M., King M.C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2010;107:12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graser S., Stierhof Y.D., Lavoie S.B., Gassner O.S., Lamla S., Le Clech M., Nigg E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.den Hollander A.I., Lopez I., Yzer S., Zonneveld M.N., Janssen I.M., Strom T.M., Hehir-Kwa J.Y., Veltman J.A., Arends M.L., Meitinger T., et al. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest. Ophthalmol. Vis. Sci. 2007;48:5690–5698. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 23.Falik-Zaccai T.C., Kfir N., Frenkel P., Cohen C., Tanus M., Mandel H., Shihab S., Morkos S., Aaref S., Summar M.L., Khayat M. Population screening in a Druze community: The challenge and the reward. Genet. Med. 2008;10:903–909. doi: 10.1097/GIM.0b013e31818d0e0f. [DOI] [PubMed] [Google Scholar]
- 24.Beales P.L., Elcioglu N., Woolf A.S., Parker D., Flinter F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 25.Zaghloul N.A., Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon P.S., Clayton-Smith J., Beales P.L., Lloyd I.C. Bardet-biedl syndrome: An atypical phenotype in brothers with a proven BBS1 mutation. Ophthalmic Genet. 2008;29:128–132. doi: 10.1080/13816810802216464. [DOI] [PubMed] [Google Scholar]
- 27.Pretorius P.R., Aldahmesh M.A., Alkuraya F.S., Sheffield V.C., Slusarski D.C. Functional analysis of BBS3 A89V that results in non-syndromic retinal degeneration. Hum. Mol. Genet. 2011;20:1625–1632. doi: 10.1093/hmg/ddr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pawlik B., Mir A., Iqbal H., Li Y., Nürnberg G., Becker C., Qamar R., Nürnberg P., Wollnik B. A novel familial BBS12 mutation associated with a mild phenotype: Implications for clinical and molecular diagnostic strategies. Mol Syndromol. 2010;1:27–34. doi: 10.1159/000276763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E.J., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G., et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Lewis R.A., Eyaid W., Banin E., et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 31.Riazuddin S.A., Iqbal M., Wang Y., Masuda T., Chen Y., Bowne S., Sullivan L.S., Waseem N.H., Bhattacharya S., Daiger S.P., et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010;86:805–812. doi: 10.1016/j.ajhg.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khanna H., Davis E.E., Murga-Zamalloa C.A., Estrada-Cuzcano A., Lopez I., den Hollander A.I., Zonneveld M.N., Othman M.I., Waseem N., Chakarova C.F., et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.