

Abstract
Pterygium syndromes are complex congenital disorders that encompass several distinct clinical conditions characterized by multiple skin webs affecting the flexural surfaces often accompanied by craniofacial anomalies. In severe forms, such as in the autosomal-recessive Bartsocas-Papas syndrome, early lethality is common, complicating the identification of causative mutations. Using exome sequencing in a consanguineous family, we identified the homozygous mutation c.1127C>A in exon 7 of RIPK4 that resulted in the introduction of the nonsense mutation p.Ser376X into the encoded ankyrin repeat-containing kinase, a protein that is essential for keratinocyte differentiation. Subsequently, we identified a second mutation in exon 2 of RIPK4 (c.242T>A) that resulted in the missense variant p.Ile81Asn in the kinase domain of the protein. We have further demonstrated that RIPK4 is a direct transcriptional target of the protein p63, a master regulator of stratified epithelial development, which acts as a nodal point in the cascade of molecular events that prevent pterygium syndromes.
Main Text
The epidermis is a highly organized, stratified, self-renewing epithelium that functions as a barrier to protect the organism from dehydration, mechanical trauma, and microbial invasion.1, 2 During development, the immature ectoderm initially consists of a single layer of mitotically active, undifferentiated, cuboidal epithelial cells that progress through a defined series of stratification and differentiation events to produce the mature epidermis that consists of four distinct cellular layers maintained by a constant process of displacement and renewal. Development and subsequent maintenance of the epidermis are therefore critically dependent on the intricate balance between proliferation and differentiation of a mitotically-active basal cell population.1, 2 Disruption of this balance results in a wide variety of congenital anomalies of epidermal development that have profound effects on affected individuals and their families.
Pterygium syndromes are complex, congenital disorders that include several different clinical conditions characterized by multiple skin webs affecting the flexural surfaces and associated craniofacial anomalies. To date, Van der Woude syndrome (MIM 119300) and popliteal pterygium syndrome (MIM 119500) have been demonstrated to result from mutations in the transcription factor IRF6 (MIM 607199),3 whereas mutation of the gene encoding the NF-κB pathway component IκB kinase-α (IKKA [MIM 600664]) underlies an autosomal-recessive lethal syndrome termed severe fetal encasement malformation or Cocoon syndrome (MIM 613630).4 Despite these advances, the molecular basis of other pterygium syndromes remains elusive. Bartsocas-Papas syndrome (MIM 263650) is a devastating autosomal-recessive disorder characterized by multiple popliteal pterygia, ankyloblepharon, filiform bands between the jaws, cleft lip and palate, and syndactyly.5 Early lethality is common, though survival into childhood and beyond has been reported.5
To determine the molecular basis of Bartsocas-Papas syndrome, we performed exome sequencing on a single individual affected by this condition. The proband was the second child of consanguineous parents; no other significant previous family history was identified. Routine anomaly scanning during pregnancy revealed multiple congenital abnormalities. Further investigation during pregnancy was declined. Fetal bradycardia was noted on monitoring 1 week preterm, and an elective Cesarean section was performed. Birth weight was 2.95 kg and the occipitofrontal circumference was 34 cm. Multiple congenital abnormalities, including alopecia totalis with only sparse scalp hair, partial ankyloblepharon, oral synechia resulting in partial occlusion of the oral cavity, hypertelorism, cloudy corneas, absent thumbs, finger oligosyndactyly, hypoplastic genitalia, extensive popliteal pterygia, toe oligosyndactyly, multiple skin tags, and unusual fibrous tethers between the feet and suprapubic region were noted (Figures 1A and 1B). On the basis of these clinical features a diagnosis of Bartsocas-Papas syndrome was made. The patient is alive at 6 years of age and intellectual development appears normal. DNA samples were obtained from three members of the family (Figure 1C) via standard procedures and with informed consent under the relevant institutional review boards and the UK National Research Ethics Service.
Figure 1.

Homozygous Mutations in RIPK4 Underlie Bartsocas-Papas Syndrome
(A and B) Clinical pictures of the affected child whose DNA was subject to exome sequencing. The alopecia, partial ankyloblepharon, oral synechia resulting in partial occlusion of the oral cavity, oligosyndactyly, popliteal pterygia, and fibrous tethers between feet and suprapubic region are apparent.
(C) Partial pedigree of the family. The shaded symbol represents the affected child.
(D) Sequence chromatograms showing segregation of the homozygous NM_020639.2:c.1127C>A mutation, which resulted in the nonsense change p.S376X and the affected phenotype.
(E) Sequence chromatogram of the homozygous NM_020639.2:c.242T>A mutation, which resulted in the missense change p.Ile81Asn.
(F) RIPK4 expression in E18.5 mouse epidermis. Cytoplasmic immunofluorescence (red) is observed in basal and suprabasal layers with the nuclei counterstained with DAPI (blue). The dotted line indicates the position of the basement membrane. The scale bar represents 25 μm.
Targeted enrichment and sequencing were performed on 3 μg of DNA extracted from peripheral blood of the affected child. Enrichment was performed with the SureSelect Human All Exon 50 MB Kit (Agilent, Santa Clara, CA, USA) for the ABI SOLiD system following the manufacturer's protocols. ePCR was conducted on the resulting sample library that was then sequenced and indexed with two unrelated samples, on a SOLiD 4 sequencer (Life Technologies, Carlsbad, CA, USA) following the manufacturer's protocols.
Sequence data were mapped with SOLiD Bioscope software (Life Technologies) and hg19 human genome as a reference. An average of 5.92 gigabases of sequence mapped uniquely to the human genome; 71.3% of the targeted exome had coverage at 20-fold or higher. Variants were called with a combination of the diBayes tool in the Bioscope software suite with the medium stringency and Samtools and then filtered for those SNPs with 5-fold or greater coverage. SNPs were initially annotated using Ensembl v61 and Ensembl's defined consequence hierarchically system, the highest impacting consequence for a variant in a gene being retained. Variants were filtered out if they were nonfunctional in dbSNP132 (unless seen in the Human Gene Mutation Database) or in our in-house variant database. At the time of the comparison, the latter database consisted of 23 exomes. For the recessive-mutation model, homozygous variants were filtered, and there was a further 20-fold filtering at novel allele depth and a mapping quality value of 20.
Among the 75 unique homozygous sequence variants identified across the exome of the child affected with Bartsocas-Papas syndrome, a C>A transversion was noted in exon 7 of RIPK4 (MIM 605706) (genomic DNA: chromosome 21[NCBI 36]:g.43164110; cDNA: NM_020639.2:c.1127C>A), which resulted in the homozygous nonsense mutation p.Ser376X (Figure 1D and Figure S1, available online). This variant lies in a 9.3 MB region of homozygosity between rs1053808 and rs1044998. Sanger sequencing confirmed that the parents were heterozygous for the mutation (Figure 1D). In light of these findings, we used Sanger sequencing to analyze a DNA sample from an additional unrelated affected individual who has been reported previously6 and identified a homozygous T>A transversion in exon 2 of RIPK4 (genomic DNA: chromosome 17[NCBI 36]:g.43176917; cDNA: NM_020639.2:c.242T>A), which resulted in the homozygous missense change p.Ile81Asn in the kinase domain of the protein (Figure 1E).7 Although DNA samples from the parents are not available, bioinformatics analysis confirmed that neither of these mutations was present in either dbSNP, the 1000 Genomes Database, or in over 4,000 chromosomes for the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project with over 20× coverage. Moreover, the isoleucine amino acid residue at position 81 is completely conserved in mammals, fish, chickens, and frogs (Figure S2).
To provide further support for RIPK4 mutations underlying the Bartsocas-Papas syndrome phenotype, we performed immunolocalization studies of neonatal mouse epidermis. Following dissection and fixation, samples were dehydrated through a graded ethanol series, cleared in chloroform, embedded in paraffin wax, and sectioned. Immunofluorescence analysis was performed with an antibody raised against RIPK4 (Abcam, Cambridge, UK). The primary antibody was detected with an Alexafluor 488-conjugated secondary antibody (Invitrogen, Grand Island, NY, USA) and mounted in fluorescence mountant containing 4’,6-diamidino-2-phenylindole ([DAPI] Vector Laboratories, Burlingame, CA, USA). Sections were examined with a Digital Module RB (DMRB) microscope (Leica) with a Spot digital camera and associated software (RTKE/SE, Diagnostic Instruments, Sterling Heights, MI, USA). In agreement with previous reports that used mouse tail skin that closely resembles the multilayered human epidermis,8 RIPK4 immunoreactivity was detected in the cytoplasm of the basal and suprabasal epidermal keratinocytes (Figure 1F).
Previous results have demonstrated that RIPK4 plays a key role in controlling keratinocyte differentiation,8, 9, 10, 11 Ripk4-null mice exhibiting severe epidermal adhesions that phenocopy Bartsocas-Papas syndrome.9 A highly similar phenotype is also observed in mice carrying loss-of-function mutations in either the transcription factor Interferon Regulatory Factor 6 (Irf6), the NF-κB pathway component IκB kinase-α (Ikka), or the cell-cycle regulator protein stratifin (Sfn),12, 13, 14, 15, 16, 17, 18, 19 proteins that are under the direct control of the transcription factor p63.20, 21, 22, 23 TP63 (MIM 603273) encodes at least six protein variants resulting from the usage of two different transcription start sites and alternative splicing. Although all isoforms contain a DNA binding domain, different promoters give rise to two alternative N termini; TA isoforms that contain a transactivation sequence and ΔN isoforms that contain a shorter activation domain.24, 25 Alternative splicing toward the carboxy terminus generates three subtypes; α, β, and γ.25 ΔNp63α is the major isoform expressed in basal epithelial cells24, 26, 27, 28 and is essential for epidermal development with mutations in TP63 resulting in a series of developmental disorders characterized by varying combinations of ectodermal dysplasia, facial clefting, and limb abnormalities including ectrodactyly-ectodermal dysplasia-clefting syndrome (MIM 604292) and ankyloblepharon-ectodermal dysplasia-clefting syndrome (MIM 106260).26, 29 Although these features are partially recapitulated in Trp63 mutant mice that exhibit truncations of the limbs and craniofacial anomalies, the epidermis of Trp63-null mice is thin, fails to stratify, and lacks ectodermal appendages such as hairs, whiskers, teeth, and several glands, including mammary, salivary, and lacrimal glands.30, 31
As p63 is required for development and maintenance of epidermal keratinocytes,32 we tested whether RIPK4 is a direct transcriptional target of p63 by analyzing a genome-wide p63 binding profile in human primary keratinocytes generated with ChIP-seq analysis.33 We identified four high-fidelity peaks within 56 kb of the RIPK4 transcription start site; two peaks resided upstream of RIPK4 (+16.5 kb and +11 kb), one peak resided in RIPK4 intron 2 (−15 kb), and the final peak was located downstream of the gene (−39 kb). All four peaks fell in conserved regions of the genome (Figure 2A and Figure S3). In addition, all four regions were strongly enriched for monomethylated histone H3 lysine 4, dimethylated histone H3 lysine 4, acetylated histone H3 lysine 9, and acetylated histone H3 lysine 27 in normal human epidermal keratinocytes (Figure 2A), chromatin signatures that mark sites of enhancer activity.34, 35 The p53scan algorithm, which is suitable for binding motif searches for p53 family members,36 identified consensus binding motifs in all four binding sites; however, only binding sites 3 and 4 were conserved in the mouse (Figure S3).
Figure 2.
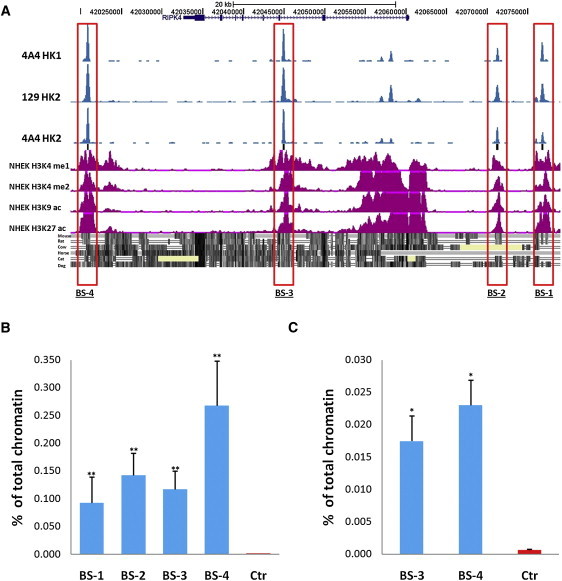
p63 Binds in to Genomic Regions in Close Proximity to RIPK4
(A) The position of the four p63 binding regions (BS-1, BS-2, BS-3, and BS-4) around RIPK4 previously identified by ChIP-seq analysis of human primary keratinocytes with the p63 antibodies 4A4 and H129 (blue peaks)33 correlate with highly conserved regions of the genome that are enriched for regulatory marks in normal human epidermal keratinocytes (NHEK) that are strongly associated with enhancer regions. The following abbreviations are used: H3K4 me1, monomethylated histone H3 lysine 4; H3K4 me2 dimethylated histone H3 lysine 4; H3K9 ac, acetylated histone H3 lysine 9; H3K27 ac, acetylated histone H3 lysine 27.
(B) ChIP-qPCR analysis using the p63 antibody H129 in human HaCaT cells confirms specific binding of p63 to all four binding sites but not to the negative control myoglobin exon 2 (Control [Ctr]) (∗p = 0.0079).
(C) ChIP-qPCR analysis using the p63 antibody H129 confirms specific binding of p63 to the conserved binding sites BS-3 and BS-4 but not to the negative control myoglobin exon 2 (Ctr) in E18.5 mouse epidermis (∗p = 0.03 Mann; Whitney U test).
The error bars in B and C indicate standard error of the mean.
To confirm the p63 binding sites identified in ChIP-seq analysis, we performed independent ChIP-qPCR experiments. Human HaCaT cells were crosslinked with 1% formaldehyde for 10 min, and chromatin was collected as described previously.37 Chromatin was sonicated with a Vibracell sonicator (Sonics, Newtown, CT, USA) for eight pulses, each of 10 s, at an amplitude setting of 40 and precipitated with the H129 antibody (SC-8344; Santa Cruz Biotechnology, Santa Cruz, CA, USA) raised against the α-tail of p63 (amino acids 513–641). ChIP efficiencies expressed as a percentage of total chromatin confirmed enrichment from HaCaT cells at all four peaks compared to the myoglobin negative control (p = 0.0079 Mann-Whitney U; Figure 2B). As enhancer elements have been shown to exhibit tissue- and species-specific activities,38 we performed ChIP-qPCR on epidermis dissected from embryonic day (E)18.5 wild-type mice and, in agreement with the conservation data (Figure S3), demonstrated that enrichment was achieved at binding sites 3 and 4 (Figure 2C). ChIP-qPCR further demonstrated that binding site 3, but not binding site 4, was enriched in chromatin precipitated from facial processes dissected from E11.5 mouse embryos (Figure S4).
p63 can act as either an activator or a repressor depending on the target gene.39, 40 To investigate the specific effects of p63 on RIPK4 transcription, the genomic regions encompassing the four p63 binding sites identified in human keratinocytes were amplified by PCR (Table S2) and cloned into a firefly luciferase reporter gene. Two hundred nanograms each of the firefly luciferase reporter, the Renilla luciferase control pRL-CMV, and a ΔNp63α wild-type expression plasmid that has been described previously27 were transfected into Sarcoma osteogenic cell line (SAOS2) cells for 24 hr with Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). The cells were then lysed and luciferase activity measured with a dual luciferase reporter assay according to the manufacturer's instructions (Promega). All transfections were performed in quadruplicate and standard errors calculated. In these transient transfection assays, wild-type ΔNp63α strongly activated transcription of the luciferase reporter through the p63 binding sites as follows: binding site 1, to 3-fold; binding site 2, to 5-fold; binding site 3, to 2-fold; and binding site 4, >5-fold (p = 0.03 Mann-Whitney U test; Figure 3A).
Figure 3.
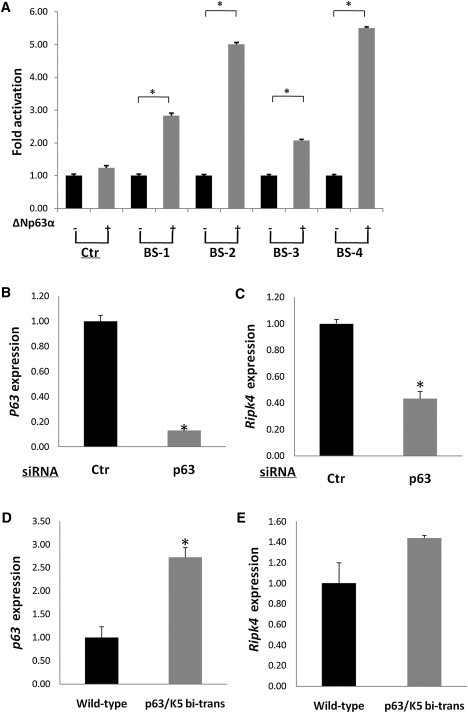
p63 Transactivates RIPK4
(A) Luciferase reporter assays demonstrate that wild-type ΔNp63α strongly activates transcription through the four binding sites (BS) located within 56 kb of RIPK4 (∗p = 0.03, Mann-Whitney U test).
(B and C) siRNA knockdown of Trp63 in mouse primary keratinocytes results in a 7-fold reduction in Trp63 levels (p = 0.02) resulting in a more than 2-fold reduction in Ripk4 levels (p = 0.02).
(D and E) qPCR analysis of epidermis from wild-type and mice overexpressing ΔNp63α indicates that Trp63 transcripts are increased ∼2.6-fold in Krt5-tTA/pTRE-ΔNp63α bitransgenic mice compared to their wild-type littermates (∗p = 0.05) resulting in an increase in Ripk4 transcripts in ΔNp63α overexpressing mice compared to their wild-type littermates. Although the increase in Ripk4 expression levels did not reach the threshold for significance, Pearson's correlation coefficient demonstrated an association between the relative expression of Trp63 and Ripk4 in Krt5-tTA/pTRE-ΔNp63α bitransgenic and wild-type mice that was significant (p = 0.02). Regression analysis of these data confirmed a linear relationship between increased Trp63 and Ripk4 expression levels (r2 = 0.49; p = 0.02) (Figure S5).
Error bars indicate standard error of the mean.
To complement these findings, we knocked down Trp63 in mouse primary keratinocytes by using a pan-Trp63-specific siRNA41 and found a statistically significant decrease in the levels of Ripk4 transcripts (p = 0.05, Mann-Whitney U test; Figures 3B and 3C). Taken together, the reporter and knockdown experiments strongly point to p63, specifically the ΔNp63α isoform, being an activator of RIPK4. To provide an in vivo context for these findings, we analyzed RNA extracted from the epidermis of postnatal day 1 mice in which ΔNp63α overexpression is targeted to the epidermis under the control of the bovine keratin 5 promoter, Krt5-tTA/pTRE-ΔNp63α bitransgenic mice.42 qPCR was performed with Power SYBR Green master mix (Invitrogen) and primers defining Trp63 and Ripk4 (Table S2) according to manufacturer's protocol. β-actin was used as a housekeeping gene to normalize the amount of cDNA used. The results of these analyses confirmed that Trp63 was upregulated 2.6-fold in Krt5-tTA/pTRE-ΔNp63α mice compared to their wild-type littermates (p = 0.05, Mann-Whitney U test; Figure 3D). Although Ripk4 expression levels were also upregulated in the Krt5-tTA/pTRE-ΔNp63α bitransgenic mice, this did not reach the threshold for significance; however, Pearson's correlation coefficient demonstrated an association between the relative expression of Trp63 and Ripk4 in Krt5-tTA/pTRE-ΔNp63α bitransgenic and wild-type mice that was significant (p = 0.02) (Figure 3E). Regression analysis of these data confirmed a linear relationship between increased Trp63 and Ripk4 expression levels (r2 = 0.49; p = 0.02) (Figure S5).
The RIP kinase family is composed of a group of seven proteins characterized by homology within their serine/threonine kinase domain; however, each member is defined by the unique domains linked to this region.7 RIPK4 contains an N-terminal kinase domain that displays ∼40% identity with other RIP kinases attached via an intermediate region that can be cleaved by caspases to a C terminus containing 11 ankyrin repeats.7, 43, 44, 45 Overexpression and inhibition studies have shown that RIPK4 is an activator of the NF-κB and JNK signaling pathways; however, in contrast to RIPK1, these activities can occur in a kinase-independent manner.11, 45, 46 Although the isoleucine residue at amino acid position 81, which has been shown to be mutated to asparagine in this study (Figure 1E), has not been directly implicated in the kinase activity of RIPK4,46 it is highly conserved across evolution suggesting a fundamental role in RIPK4 function (Figure S2). Taken together with the position of the C>A transversion in exon 7 of RIPK4, which results in the introduction of a homozygous termination codon into the protein and which is therefore predicted to result in nonsense-mediated degradation of the mutant transcript,47 it is likely that both mutations abrogate RIPK4 function. Support for this hypothesis is provided by the observation that Ripk4-null mice exhibit abnormalities of the keratinocyte proliferation-differentiation switch that result in multiple epidermal adhesions thereby phenocopying Bartsocas-Papas syndrome.9, 10 It is also notable that mice carrying loss-of-function mutations in either Irf6, Ikka, or Sfn exhibit highly similar defects of stratified, squamous, keratinizing epithelia suggesting that at least a subset of these proteins function in a common pathway with RIPK4.12, 13, 14, 15, 16, 17, 18, 19 In this context, SFN has been shown to be a downstream target of IKKα in regulating the G2/M cell-cycle checkpoint in response to DNA damage in keratinocytes; IKKα shields SFN from hypermethylation to prevent its silencing.48 Similarly, Irf6 and Sfn have been shown to interact epistatically, although the molecular basis of this interaction has yet to be determined.18 Although the full extent of the interactions between these molecules has yet to be elucidated, we have now demonstrated that RIPK4 is a direct transcriptional target of ΔNp63α, a protein that also activates IRF6 and IKKA,20, 21, 22, 23 genes that are mutated in clinical conditions that exhibit marked phenotypic overlap with Bartsocas-Papas syndrome.3, 4 These observations suggest that the transcription factor ΔNp63α acts as a nodal point in the molecular events that prevent pterygium syndromes.
In summary, we report the use of whole-exome sequencing to demonstrate that the autosomal-recessive disorder Bartsocas-Papas syndrome results from loss-of-function mutations in the gene encoding receptor-interacting kinase 4, an ankyrin repeat-containing kinase essential for keratinocyte differentiation.
Acknowledgments
We express our gratitude to the family members for participating in the study. We thank Drs. J. Zhou and Hans Van Bokhoven for kindly expediting access to their ChIP-seq data set. The financial support from the Medical Research Council, UK to M.J.D. and J.D. (G0901539), the National Institute for Health Research-funded Manchester Biomedical Research Centre to G.C.M.B and M.J.D., the Healing Foundation to M.J.D., Telethon Foundation, Italy to C.M (GGP09230), and the National Institutes of Health to J.C.M. (DE08559) and B.C.S. (DE13513) is gratefully acknowledged.
Published online: December 22, 2011
Footnotes
Supplemental Data include five figures and two tables and can be found with this article online at http://www.cell.com/AJHG/.
Contributor Information
Jill Dixon, Email: jill.dixon@manchester.ac.uk.
Michael J. Dixon, Email: mike.dixon@manchester.ac.uk.
Web Resources
The URLs for data presented herein are as follows:
Human Gene Mutation Database, http://www.hgmd.org
NHLBI Exome Sequencing Project (ESP), http://snp.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man, http://www.omim.org
Supplemental Data
References
- 1.Fuchs E., Raghavan S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 2002;3:199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- 2.Watt F.M. Stem cell fate and patterning in mammalian epidermis. Curr. Opin. Genet. Dev. 2001;11:410–417. doi: 10.1016/s0959-437x(00)00211-2. [DOI] [PubMed] [Google Scholar]
- 3.Kondo S., Schutte B.C., Richardson R.J., Bjork B.C., Knight A.S., Watanabe Y., Howard E., Ferreira de Lima R.L.L., Daack-Hirsch S., Sander A., et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lahtela J., Nousiainen H.O., Stefanovic V., Tallila J., Viskari H., Karikoski R., Gentile M., Saloranta C., Varilo T., Salonen R., et al. Mutant CHUK and severe fetal encasement malformation. N. Engl. J. Med. 2010;363:1631–1637. doi: 10.1056/NEJMoa0911698. [DOI] [PubMed] [Google Scholar]
- 5.Veenstra-Knol H.E., Kleibeuker A., Timmer A., ten Kate L.P., van Essen A.J. Unreported manifestations in two Dutch families with Bartsocas-Papas syndrome. Am. J. Med. Genet. 2003;123A:243–248. doi: 10.1002/ajmg.a.20308. [DOI] [PubMed] [Google Scholar]
- 6.Shanske A.L., Hoper S.A., Krahn K., Schutte B.C. Mutations in IRF6 do not cause Bartsocas-Papas syndrome in a family with two affected sibs. Am. J. Med. Genet. 2004;128A:431–433. doi: 10.1002/ajmg.a.30096. [DOI] [PubMed] [Google Scholar]
- 7.Meylan E., Tschopp J. The RIP kinases: Crucial integrators of cellular stress. Trends Biochem. Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Holland P., Willis C., Kanaly S., Glaccum M., Warren A., Charrier K., Murison J., Derry J., Virca G., Bird T., et al. RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr. Biol. 2002;12:1424–1428. doi: 10.1016/s0960-9822(02)01075-8. [DOI] [PubMed] [Google Scholar]
- 9.Rountree R.B., Willis C.R., Dinh H., Blumberg H., Bailey K., Dean C., Jr., Peschon J.J., Holland P.M. RIP4 regulates epidermal differentiation and cutaneous inflammation. J. Invest. Dermatol. 2010;130:102–112. doi: 10.1038/jid.2009.223. [DOI] [PubMed] [Google Scholar]
- 10.Adams S., Munz B. RIP4 is a target of multiple signal transduction pathways in keratinocytes: Implications for epidermal differentiation and cutaneous wound repair. Exp. Cell Res. 2010;316:126–137. doi: 10.1016/j.yexcr.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Adams S., Pankow S., Werner S., Munz B. Regulation of NF-kappaB activity and keratinocyte differentiation by the RIP4 protein: Implications for cutaneous wound repair. J. Invest. Dermatol. 2007;127:538–544. doi: 10.1038/sj.jid.5700588. [DOI] [PubMed] [Google Scholar]
- 12.Guenet J.L., Salzgeber B., Tassin M.T. Repeated epilation: A genetic epidermal syndrome in mice. J. Hered. 1979;70:90–94. doi: 10.1093/oxfordjournals.jhered.a109223. [DOI] [PubMed] [Google Scholar]
- 13.Hu Y., Baud V., Delhase M., Zhang P., Deerinck T., Ellisman M., Johnson R., Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 14.Li Q., Lu Q., Hwang J.Y., Büscher D., Lee K.F., Izpisua-Belmonte J.C., Verma I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda K., Takeuchi O., Tsujimura T., Itami S., Adachi O., Kawai T., Sanjo H., Yoshikawa K., Terada N., Akira S. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 16.Herron B.J., Liddell R.A., Parker A., Grant S., Kinne J., Fisher J.K., Siracusa L.D. A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat. Genet. 2005;37:1210–1212. doi: 10.1038/ng1652. [DOI] [PubMed] [Google Scholar]
- 17.Li Q., Lu Q., Estepa G., Verma I.M. Identification of 14-3-3sigma mutation causing cutaneous abnormality in repeated-epilation mutant mouse. Proc. Natl. Acad. Sci. USA. 2005;102:15977–15982. doi: 10.1073/pnas.0508310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson R.J., Dixon J., Malhotra S., Hardman M.J., Knowles L., Boot-Handford R.P., Shore P., Whitmarsh A., Dixon M.J. IRF6 is a key determinant of the keratinocyte proliferation/differentiation switch. Nat. Genet. 2006;38:1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 19.Ingraham C.R., Kinoshita A., Kondo S., Yang B., Sajan S., Trout K.J., Malik M.I., Dunnwald M., Goudy S.L., Lovett M., et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat. Genet. 2006;38:1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomason H.A., Zhou H., Kouwenhoven E.N., Dotto G.P., Restivo G., Nguyen B.C., Little H., Dixon M.J., van Bokhoven H., Dixon J. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 2010;120:1561–1569. doi: 10.1172/JCI40266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moretti F., Marinari B., Lo Iacono N., Botti E., Giunta A., Spallone G., Garaffo G., Vernersson-Lindahl E., Merlo G., Mills A.A., et al. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. J. Clin. Invest. 2010;120:1570–1577. doi: 10.1172/JCI40267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koster M.I., Dai D., Marinari B., Sano Y., Costanzo A., Karin M., Roop D.R. p63 induces key target genes required for epidermal morphogenesis. Proc. Natl. Acad. Sci. USA. 2007;104:3255–3260. doi: 10.1073/pnas.0611376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marinari B., Ballaro C., Koster M.I., Giustizieri M.L., Moretti F., Crosti F., Papoutsaki M., Karin M., Alema S., Chimenti S., et al. IKKalpha is a p63 transcriptional target involved in the pathogenesis of ectodermal dysplasias. J. Invest. Dermatol. 2009;129:60–69. doi: 10.1038/jid.2008.202. [DOI] [PubMed] [Google Scholar]
- 24.Yang A., Kaghad M., Wang Y., Gillett E., Fleming M.D., Dötsch V., Andrews N.C., Caput D., McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 25.van Bokhoven H., Brunner H.G. Splitting p63. Am. J. Hum. Genet. 2002;71:1–13. doi: 10.1086/341450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laurikkala J., Mikkola M.L., James M., Tummers M., Mills A.A., Thesleff I. p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development. 2006;133:1553–1563. doi: 10.1242/dev.02325. [DOI] [PubMed] [Google Scholar]
- 27.Rinne T., Clements S.E., Lamme E., Duijf P.H., Bolat E., Meijer R., Scheffer H., Rosser E., Tan T.Y., McGrath J.A., et al. A novel translation re-initiation mechanism for the p63 gene revealed by amino-terminal truncating mutations in Rapp-Hodgkin/Hay-Wells-like syndromes. Hum. Mol. Genet. 2008;17:1968–1977. doi: 10.1093/hmg/ddn094. [DOI] [PubMed] [Google Scholar]
- 28.Thomason H.A., Dixon M.J., Dixon J. Facial clefting in Tp63 deficient mice results from altered Bmp4, Fgf8 and Shh signaling. Dev. Biol. 2008;321:273–282. doi: 10.1016/j.ydbio.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 29.Rinne T., Brunner H.G., van Bokhoven H. p63-associated disorders. Cell Cycle. 2007;6:262–268. doi: 10.4161/cc.6.3.3796. [DOI] [PubMed] [Google Scholar]
- 30.Mills A.A., Zheng B., Wang X.J., Vogel H., Roop D.R., Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 31.Yang A., Schweitzer R., Sun D., Kaghad M., Walker N., Bronson R.T., Tabin C., Sharpe A., Caput D., Crum C., et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 32.Koster M.I., Roop D.R. Mechanisms regulating epithelial stratification. Annu. Rev. Cell Dev. Biol. 2007;23:93–113. doi: 10.1146/annurev.cellbio.23.090506.123357. [DOI] [PubMed] [Google Scholar]
- 33.Kouwenhoven E.N., van Heeringen S.J., Tena J.J., Oti M., Dutilh B.E., Alonso M.E., de la Calle-Mustienes E., Smeenk L., Rinne T., Parsaulian L., et al. Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet. 2010;6:e1001065. doi: 10.1371/journal.pgen.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 36.Smeenk L., van Heeringen S.J., Koeppel M., van Driel M.A., Bartels S.J., Akkers R.C., Denissov S., Stunnenberg H.G., Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639–3654. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denissov S., van Driel M., Voit R., Hekkelman M., Hulsen T., Hernandez N., Grummt I., Wehrens R., Stunnenberg H. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J. 2007;26:944–954. doi: 10.1038/sj.emboj.7601550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F., et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghioni P., Bolognese F., Duijf P.H., Van Bokhoven H., Mantovani R., Guerrini L. Complex transcriptional effects of p63 isoforms: Identification of novel activation and repression domains. Mol. Cell. Biol. 2002;22:8659–8668. doi: 10.1128/MCB.22.24.8659-8668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang A., Zhu Z., Kapranov P., McKeon F., Church G.M., Gingeras T.R., Struhl K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 41.De Rosa L., Antonini D., Ferone G., Russo M.T., Yu P.B., Han R., Missero C. p63 Suppresses non-epidermal lineage markers in a bone morphogenetic protein-dependent manner via repression of Smad7. J. Biol. Chem. 2009;284:30574–30582. doi: 10.1074/jbc.M109.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romano R.A., Smalley K., Liu S., Sinha S. Abnormal hair follicle development and altered cell fate of follicular keratinocytes in transgenic mice expressing DeltaNp63alpha. Development. 2010;137:1431–1439. doi: 10.1242/dev.045427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bähr C., Rohwer A., Stempka L., Rincke G., Marks F., Gschwendt M. DIK, a novel protein kinase that interacts with protein kinase Cdelta. Cloning, characterization, and gene analysis. J. Biol. Chem. 2000;275:36350–36357. doi: 10.1074/jbc.M004771200. [DOI] [PubMed] [Google Scholar]
- 44.Chen L., Haider K., Ponda M., Cariappa A., Rowitch D., Pillai S. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J. Biol. Chem. 2001;276:21737–21744. doi: 10.1074/jbc.M008069200. [DOI] [PubMed] [Google Scholar]
- 45.Meylan E., Martinon F., Thome M., Gschwendt M., Tschopp J. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-kappa B and is processed during apoptosis. EMBO Rep. 2002;30:1201–1208. doi: 10.1093/embo-reports/kvf236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moran S.T., Haider K., Ow Y., Milton P., Chen L., Pillai S. Protein kinase C-associated kinase can activate NFkappaB in both a kinase-dependent and a kinase-independent manner. J. Biol. Chem. 2003;278:21526–21533. doi: 10.1074/jbc.M301575200. [DOI] [PubMed] [Google Scholar]
- 47.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 48.Zhu F., Xia X., Liu B., Shen J., Hu Y., Person M., Hu Y. IKKalpha shields 14-3-3sigma, a G(2)/M cell cycle checkpoint gene, from hypermethylation, preventing its silencing. Mol. Cell. 2007;27:214–227. doi: 10.1016/j.molcel.2007.05.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.