

Abstract
We investigated a family in which the index subject presented with severe congenital lactic acidosis and dysmorphic features associated with a cytochrome c oxidase (COX)-assembly defect and a specific decrease in the synthesis of COX I, the subunit that nucleates COX assembly. Using a combination of microcell-mediated chromosome transfer, homozygosity mapping, and transcript profiling, we mapped the gene defect to chromosome 12 and identified a homozygous missense mutation (c.88G>A) in C12orf62. C12orf62 was not detectable by immunoblot analysis in subject fibroblasts, and retroviral expression of the wild-type C12orf62 cDNA rescued the biochemical phenotype. Furthermore, siRNA-mediated knockdown of C12orf 62 recapitulated the biochemical defect in control cells and exacerbated it in subject cells. C12orf62 is apparently restricted to the vertebrate lineage. It codes for a very small (6 kDa), uncharacterized, single-transmembrane protein that localizes to mitochondria and elutes in a complex of ∼110 kDa by gel filtration. COX I, II, and IV coimmunoprecipated with an epitope-tagged version of C12orf62, and 2D blue-native-polyacrylamide-gel-electrophoresis analysis of newly synthesized mitochondrial COX subunits in subject fibroblasts showed that COX assembly was impaired and that the nascent enzyme complex was unstable. We conclude that C12orf62 is required for coordination of the early steps of COX assembly with the synthesis of COX I.
Main Text
Complex IV, or cytochrome c oxidase (COX), is the terminal enzyme of the respiratory chain and catalyzes the oxidation of cytochrome c by molecular oxygen. It is composed of 13 subunits, three of which form the catalytic core of the enzyme and are encoded in mtDNA. Although several mutations in the mtDNA-encoded structural subunits have been reported,1 most isolated COX deficiencies (MIM 220110) are inherited as autosomal-recessive disorders and manifest with an early age of onset and a wide range of clinical phenotypes. These phenotypes include classical Leigh syndrome, the French-Canadian form of Leigh syndrome, fatal infantile COX deficiency, cardiomyopathy, myopathy, fatal infantile lactic acidosis, and a reversible COX deficiency in muscles1, 2. Mutations in several nuclear-encoded factors involved in COX biogenesis,3, 4, 5, 6, 7 as well as in two nuclear-encoded structural subunits, have been reported,8, 9 but the genetic basis for many COX deficiencies remains unknown. Independent of the genetic cause, nearly all COX deficiencies result from a failure to assemble an adequate amount of functional holoenzyme.
Here, we investigate the genetic basis of a mitochondrial disorder in a subject who has a COX-assembly defect and who presented with an unusually severe phenotype. The index subject (II:1) is the first child of healthy consanguineous Portuguese parents. Two subsequent siblings (II:3, II:4) later presented with an identical disease, whereas a third sibling is healthy. Oligoamnios and septum-lucidum cysts were noted at the 28th and 37th weeks of gestation. The subject was born at full term; her birth weight was 3,250 g (50th percentile), and her head circumference was 37 cm (95th percentile). She presented with neurological and respiratory distress immediately after birth. She was dysmorphic with hypotelorism, microphthalmia, an ogival palate, and a single palmar crease on the left hand. Four hours after birth, she had severe metabolic acidosis (pH = 7.03, pCO2 = 30, base excess = −22), which was associated with ketonuria, thus confirming the abnormal intermediary metabolism. Lactate concentration was 23.4 mM in the blood and 26.2 mM in the cerebrospinal fluid 16 hr after birth. Death occurred 24 hr after birth. An autopsy revealed brain hypertrophy, diffuse alteration of the white-matter myelination, and numerous cavities in the parieto-occipital region, brainstem, and cerebellum, as well as hepatomegaly, hypertrophic cardiomyopathy, renal hypoplasia, and adrenal-gland hyperplasia. Her karyotype was normal (46, XX). Informed consent was obtained from the subject's parents, and the research study was approved by the institutional review board of the Montreal Neurological Institute.
Spectrophotometric assays of whole-cell extracts showed that COX activity in immortalized subject fibroblasts was reduced to 30%–40% of that in control fibroblasts.10 Blue native polyacrylamide gel electrophoresis (BN-PAGE) analysis11 showed that the reduced activity was associated with a specific reduction in the amount of fully assembled COX, but did not identify subassemblies of the COX complex (Figure S1A). Immunoblot analysis showed that the steady-state levels of COX I and COX II, two mitochondrially encoded COX subunits, and of COX IV, a nuclear-encoded subunit, had decreased, which is typical for a defect in COX assembly (Figure S1B). The specific reduction in the levels of fully assembled COX and COX subunits prompted us to assess the rate of synthesis and the stability of the mtDNA-encoded subunits of the OXPHOS complexes. To this end, mitochondrial translation products in fibroblasts from the subject and two controls were pulse labeled with a mixture of [35S] methionine and [35S] cysteine as previously described11 and were then chased either for 10 min so that the rate of synthesis of the individual polypeptides could be measured (we refer to this as the PULSE experiment) or for 17 hr so that their stability could be assessed (we refer to this as the CHASE experiment). For the CHASE studies, we incubated cells for 23 hr in 40 μg/ml chloramphenicol to inhibit mitochondrial translation prior to labeling. A specific decrease in the amount of pulse labeling of the COX I subunit was observed (approximately 50% compared to that of the lowest control), whereas the other mtDNA-encoded polypeptides were apparently synthesized at levels similar to those in the control cells (Figure 1A). Newly synthesized COX I appeared much more stable in subject fibroblasts during the CHASE experiment. To ensure that the decrease in the synthesis of the COX I polypeptide was not due to a reduction in the levels of its mRNA, we performed northern-blot analysis for all mtDNA-encoded COX subunits, as well as for a subunit of complex I (ND1) and both ribosomal RNA subunits (Figure 1B). The levels of the COX mRNAs in the subject were not reduced relative to those of the controls when they were normalized to the rRNA levels, suggesting the presence of a mutation in a factor specifically involved with COX I translation.
Figure 1.
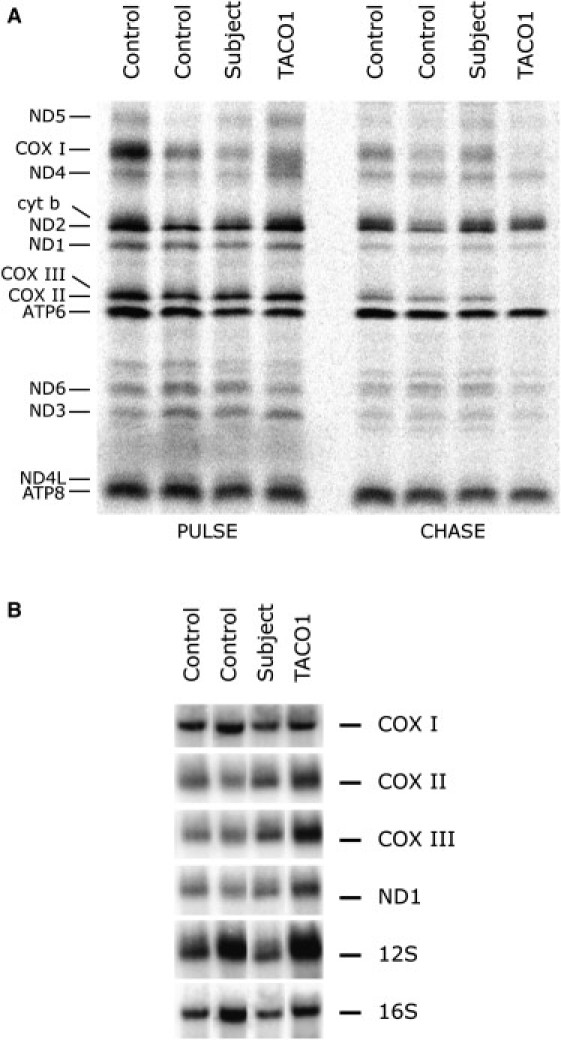
Defect in COX I Translation and Normal Levels of Mitochondrial mRNAs in Subject Fibroblasts
(A) Pulse-labeled mitochondrial polypeptides were chased for 10 min (PULSE) or overnight (CHASE). The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure.
(B) Northern blot analysis of fibroblasts from the subject, TACO1 patient, and controls. Ten micrograms of total RNA was separated on a denaturing formaldehyde agarose gel (MOPS buffer) and transferred to a nylon membrane. We labeled the PCR products (300–500 bp) of individual mitochondrial genes (COX I, COX II, COX III, ND 1, 12S, and 16S) with [α-32P]-dCTP by using the MegaPrime DNA labeling kit (GE Healthcare). The 12S and 16S mitochondrial rRNAs and actin were used as loading controls.
To determine whether the defect was caused by a mutation in nuclear or mitochondrial DNA, we fused patient and control cells with 143 rho0 cells, which are devoid of mtDNA. COX activity in the hybrids obtained with subject fibroblasts was 20% greater than that obtained with control fibroblasts, demonstrating that the subject's biochemical defect is most likely an autosomal-recessive trait. To test whether known COX-assembly factors could suppress the defect, we transduced fibroblasts from the subject with retroviral vectors that express the cDNAs for several known COX-assembly factors, i.e., COX11, COX16, COX17, OXA1, SCO2, PET191, SURF1, OXA2, COX10, COX15.1, COX19, COX15.2, SCO1, and COX23. None of these factors restored COX activity.
We next attempted to identify the defective gene by microcell-mediated monochromosomal transfer.3 A panel of monochromosomal human-mouse hybrid cells containing single human chromosomes tagged with the chimeric selection marker HyTK was used as the donor cell line for human chromosomes.12 All 22 autosomes were transferred one at a time into subject fibroblasts, and the resulting clones were tested for complementation of the COX defect. Rescued clones were considered to be those with a specific COX activity higher than 60% of control activity, a percentage that is about double the COX activity in the subject fibroblasts. Instead of the expected rescue, in which a single chromosome carries the wild-type version of the disease-associated gene, we observed several chromosomes that could apparently restore COX activity (Table S1). Several of the rescued clones were analyzed with BN-PAGE, and all displayed control levels of fully assembled COX (data not shown). One possible explanation for this result was that a mouse chromosome carrying the murine homolog of the gene mutated in the subject was transferred along with the individual human chromosomes. Fluorescence in situ hybridization (FISH) analysis confirmed the presence of mouse chromosomes in the rescued clones (data not shown), and genotyping with mouse microsatellite markers in the rescued and nonrescued clones also showed several mouse chromosomal elements in the tested clones.
We reasoned that identifying the mouse chromosomal region responsible for the rescue of COX activity could lead to the identification of the syntenic human chromosomal region containing the defective gene. Incorporating DNA isolated from several rescued and nonrescued clones with different transferred human chromosomes, Illumina SNP genotyping (mouse BeadArray technology) identified parts of mouse chromosomes 1, 11, and 15 exclusively in rescued clones. However, because the DNA samples contained both mouse and human DNA, the genotype calling was sometimes ambiguous. Given that the subject is the child of consanguineous parents, we tried homozygosity mapping by using an Affymetrix SNP array (GeneChip 250K Nsp I) to identify subject regions of homozygosity that might be syntenic with the regions identified on the mouse chromosomes in the rescued clones.
The largest region of LOH (loss of heterozygosity) was on chromosome 12 at position 43.5–54.2 Mbp. A region spanning 90–103.4 Mbp on mouse chromosome 15 is syntenic with a large region spanning 38.6–55.1 Mbp on human chromosome 12, and this focused our attention on this region. Using the HumanWG-6 Expression Bead Chip (Illumina), we searched this region for transcripts whose level of expression was low in fibroblast RNA from the subject compared to 11 normal and disease-positive controls (data not shown). This analysis identified C12orf62 as the top candidate for the gene that was mutated in the subject.
We used total RNA isolated from subject fibroblasts with the RNeasy Kit (QIAGEN) to amplify the C12orf62 cDNA by using the OneStep RT-PCR kit (QIAGEN), and we used the gel-purified PCR fragments for direct sequencing. This analysis revealed a homozygous missense mutation (c.88G>A) (Figure 2A), predicting an amino acid change from methionine to isoleucine (p.Met19Ile) (Figure 2D). The mutation was confirmed by restriction fragment-length polymorphism (RFLP) analysis in subject genomic DNA after amplification of C12orf62 exon 2. The mutation abolishes an NlaIII site, and, as predicted, the subject's PCR product remained undigested (Figure 2B).
Figure 2.
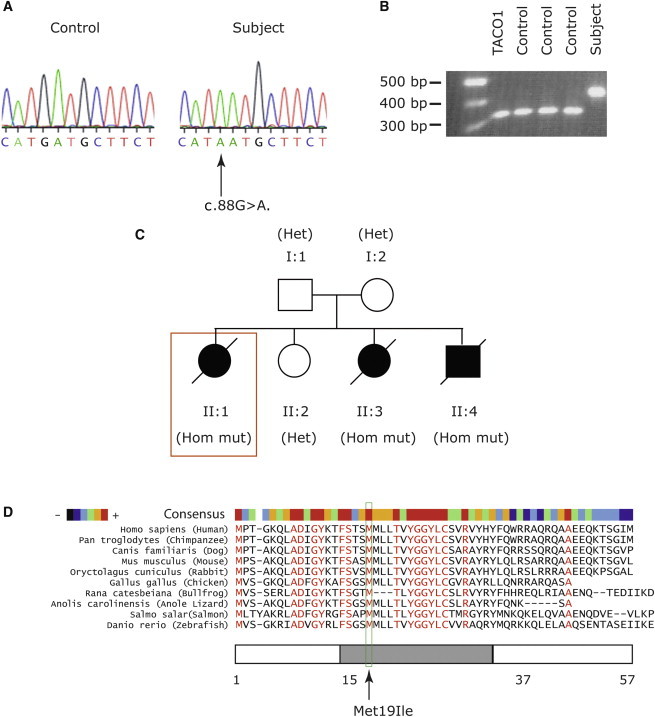
Mutation Analysis of C12orf62 in the Index Subject
(A) Sequence analysis of C12orf62 exon 2 from genomic DNA shows a homozygous c.88G>A missense mutation in the subject fibroblasts.
(B) RFLP analysis shows NlaIII of C12orf62 exon 2 amplified from genomic DNA from the subject, TACO1 patient, and three controls fibroblasts.
(C) Pedigree of the index subject (box) indicates affected and unaffected individuals. The genotype of individuals is shown in parentheses as follows: Hom mut, homozygous for c.88G>A; and Het, heterozygous for c.88G>A.
(D) Protein alignment (MultAlin) of C12orf62 in different species. An arrow indicates the conserved methionine mutated in the subject. The position of the predicted transmembrane domain (predicted with TMHMM Server v. 2.0) is indicated by a gray bar. The color bar reflects the degree of amino acid conservation between species.
The mutation segregated with the disease phenotype in the subject's family (Figure 2C). The two other affected siblings had the same homozygous mutation, whereas the unaffected sibling and the parents were heterozygous carriers of the mutation. C12orf62 contains two exons, only one of which is coding, and it is predicted to produce a 57 amino acid protein of about 6 kDa. Orthologs of the protein appear to be largely confined to the vertebrates, and the mutated methionine, which is predicted to be in a transmembrane helix, is conserved in all C12orf62 orthologs. Analysis of more than 80 other patients with COX deficiency and/or early-onset lactic acidosis failed to identify additional C12orf62 mutations, suggesting that they are a rare cause of COX deficiency.
Polyclonal antibodies directed against N- and C-terminal C12orf62 peptides (MPTGKQLADIGYKT and CFQWRRAQRQAAEEQKT, respectively) were prepared by 21st Century Biochemicals (Marlboro, MA). Immunoblot analysis was performed on mitochondrial extracts with a mixture of these two antibodies, and it showed that C12orf62 was undetectable in subject fibroblasts but was present in control cells and also in an individual with TACO1 mutations (Figures 3A and 3C), suggesting that the missense mutation destabilizes the protein. To investigate whether the lack of functional C12orf62 was responsible for the COX defect, we expressed wild-type C12orf62 in subject fibroblasts by using a retroviral expression vector. We amplified the C12orf62 cDNA by using OneStep RT-PCR (QIAGEN) with specific primers modified so that they could be cloned into Gateway vectors, and we cloned the cDNA into a Gateway-compatible pBABE vector. Retroviral constructs were transiently transfected into the Phoenix packaging cell line with the HBS/Ca3(PO4)2 method, and fibroblasts were infected 48 hr later by being exposed to virus-containing medium in the presence of 4 μg/ml polybrene.13 Expression of C12orf62 in the controls (Figure 3A) had no measureable effect on the assembly of the OXPHOS complexes or mitochondrial translation, but it completely rescued the COX-assembly defect (Figure 3A), COX I translation in the PULSE assay (Figure 3B), and COX activity (92% of control) in subject fibroblasts.
Figure 3.
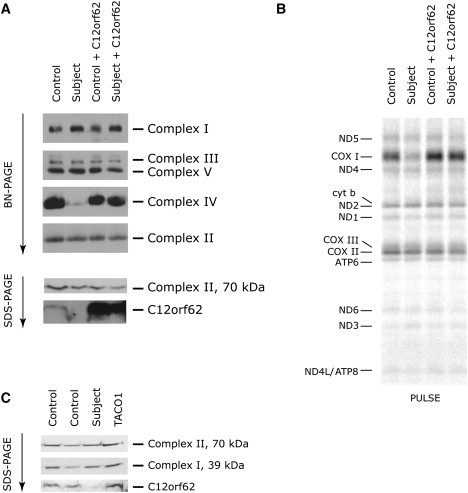
Rescue of COX Assembly and COX I Translation Defect in Subject Fibroblasts
(A) BN-PAGE analysis of the assembly of individual OXPHOS complexes in control and subject fibroblasts expressing C12orf62 from a retroviral vector (upper panel). With complex II subunit 70 kDa as a loading control, SDS-PAGE analysis of C12orf62 indicates its level of expression (lower panel).
(B) Pulse labeling with [35S] methionine and [35S] cysteine of mitochondrial polypeptides and a 10 min chase (PULSE) in control and subject fibroblasts expressing C12orf62 from a retroviral vector. The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure.
(C) SDS-PAGE analysis of C12orf62 steady-state levels in two controls, TACO1 patient and subject fibroblasts. Complex II-70kDa subunit and complexI-39kDa subunit were used as loading controls.
None of the mitochondrial-targeting prediction programs (Predotar, Target P, MitoPred, MitoProt, or MitoPWolf) predict C12orf62 to be targeted to mitochondria, although it appears in the MitoCarta database in multiple tissues, suggesting that C12orf62 is ubiquitously expressed.14 C12orf62 was also one of the highest-scoring genes in a screen for OXPHOS gene expression.15 Immunocytochemistry demonstrated the mitochondrial localization of C12orf62 in cells expressing a C-terminal, Myc-tagged version of C12orf62 (Figure S2A); this version was found to completely rescue the biochemical defect in subject fibroblasts (data not shown). C12orf62 was present exclusively in the pellet obtained after alkaline sodium-carbonate extraction of mitochondria from human embryonic kidney (HEK) 293 cells, indicating that C12orf62, like COX I, is an integral membrane protein, and is not membrane associated like the 70 kDa subunit of complex II (Figure S2B).
To further investigate the function of C12orf62, we used siRNA-mediated knockdown of the protein. Two Stealth RNAi duplexes were designed with Block-iT RNAi Express (Invitrogen). Stealth KD1 (5′-AACAGGACUAGAGCGUUGAUGGUUU-3′), stealth KD2 (5′-GACAUUGGCUAUAAGACCUUCUCUA-3′), and the fluorescent oligo control (Block-iT Alexa FluorRed) were transiently transfected with Lipofectamine RNAiMAX (Invitrogen) into subject and control fibroblasts at a final concentration of 12 nM. The transfections were repeated on days 3 and 6, and the cells were harvested and analyzed on day 8. Using both siRNAs for transient knockdown of C12orf62 in control fibroblasts resulted in both a COX-assembly defect similar to that seen in the subject (Figure 4A) and reduced COX activity (38%–53% of control activity) that correlated with the severity of the assembly defect. It also resulted in a decreased intensity of pulse labeling of COX I in the translation assay (Figure 4B). In the subject fibroblasts, the same knockdown exacerbated the biochemical phenotype, implying that the mutant protein, although not detectable by immunoblot analysis, retained some residual function. The knockdown also reduced the stability of COX I in the subject cells to levels that were indistinguishable from those of the controls (Figure 4C). Interestingly, although siRNA KD2 reduced COX activity to near background levels, it did not reduce the labeling intensity of COX I below that seen in KD1 (Figure 4B), implying that C12orf62 functions in some other aspect of COX biogenesis. We conclude that C12orf62 is specifically involved in the synthesis and subsequent assembly of COX I and that the presence of a small amount of the mutant protein in subject cells stabilizes newly synthesized COX I.
Figure 4.
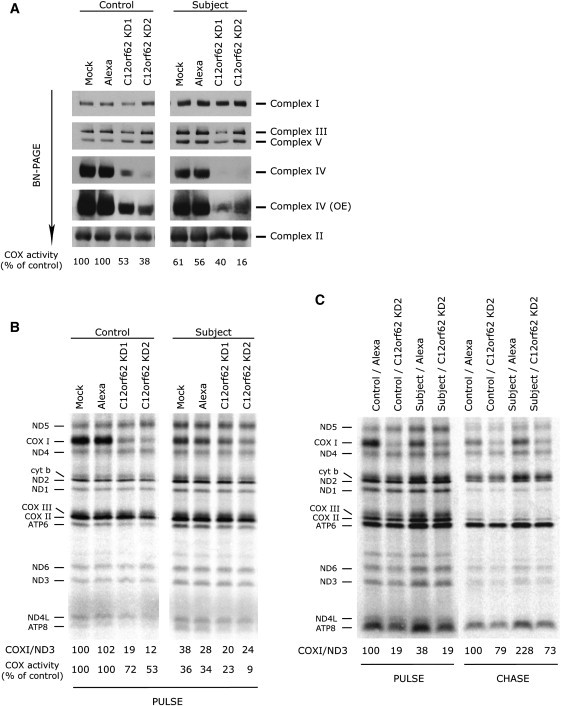
Knockdown of C12orf62 in Control and Subject Fibroblasts Results in a Specific Defect in COX Assembly and COX I Translation
(A) BN-PAGE analysis of the assembly of individual OXPHOS complexes in control and subject fibroblasts transiently transfected with two different siRNA constructs specific to C12orf62 (KD1 and KD2), with a fluorescent control siRNA (Alexa), and without siRNA (Mock). Complex IV (OE) is a longer exposure of the same blot.
(B) Pulse labeling with [35S] methionine and [35S] cysteine of mitochondrial polypeptides of the samples in panel (A). The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure. For quantification of COX I synthesis, COX I labeling was normalized to the labeling of ND3. COX activity was also measured in these samples. The values are shown under the translation gel and are expressed as percentages of the mock control.
(C) The mitochondrial translation products were pulse labeled and chased for 10 min (PULSE) or 17 hr (CHASE) in control and subject fibroblasts and in cells that were transfected with either a siRNA construct specific to C12orf62 (KD2) or a fluorescent control siRNA (Alexa). COX I labeling was normalized to the labeling of ND3.
To investigate whether C12orf62 interacts or associates with COX I, we performed a size-exclusion experiment in HEK 293 cells. The majority of C12orf62 appeared in the fraction corresponding to a molecular weight (MW) of about 110 kDa and containing COX I and a small amount of COX IV (Figure S3), suggesting that C12orf62 interacts with newly synthesized COX I at an early step in holoenzyme assembly. (Fully assembled COX has a MW of about 220 kDa). A minority of C12orf62 also appeared in the same fraction that contained COX II and COX IV, both of which are involved in early COX-assembly steps. To confirm the interaction of C12orf62 and COX 1, we used the FLAG antibody to perform immunoprecipitation of the control fibroblasts expressing C12orf62-FLAG. Mitochondria (400 μg) isolated from fibroblasts expressing C12orf62-FLAG were extracted on ice for 45 min in 100 μl of 50 mM HEPES buffer (pH7.6), 150 mM NaCl, 1% taurodeoxycholate, and complete protease inhibitors (Roche). The extract was centrifuged at 25,000 × g at 4°C for 40 min, and we precleared the supernatant overnight with noncoated agarose beads (EZ view Red Protein G Affinity Gel, Sigma) to reduce nonspecific protein binding. The precleared extract was used for immunoprecipitation with FLAG-coated agarose beads (EZ view Red anti-FLAG M2 Affinity Gel, Sigma). The proteins were eluted from the beads with 0.1 M glycine (pH 2.5), 0.08% Tween 20, and 1% DDM. The immunoprecipitated fractions were analyzed via immunoblotting, and the eluates were precipitated with trichloroacetic acid and analyzed with mass spectrometry (Orbitrap, Thermo Scientific, Waltham, MA) at the Institute de Recherches Cliniques de Montreal. C12orf62 coimmunoprecipitated with a small fraction of COX I, COX II, and COX IV (Figure 5A). Reciprocal immunoprecipitation with the COX I and COX II antibodies verified the interactions (Figure 5B).
Figure 5.
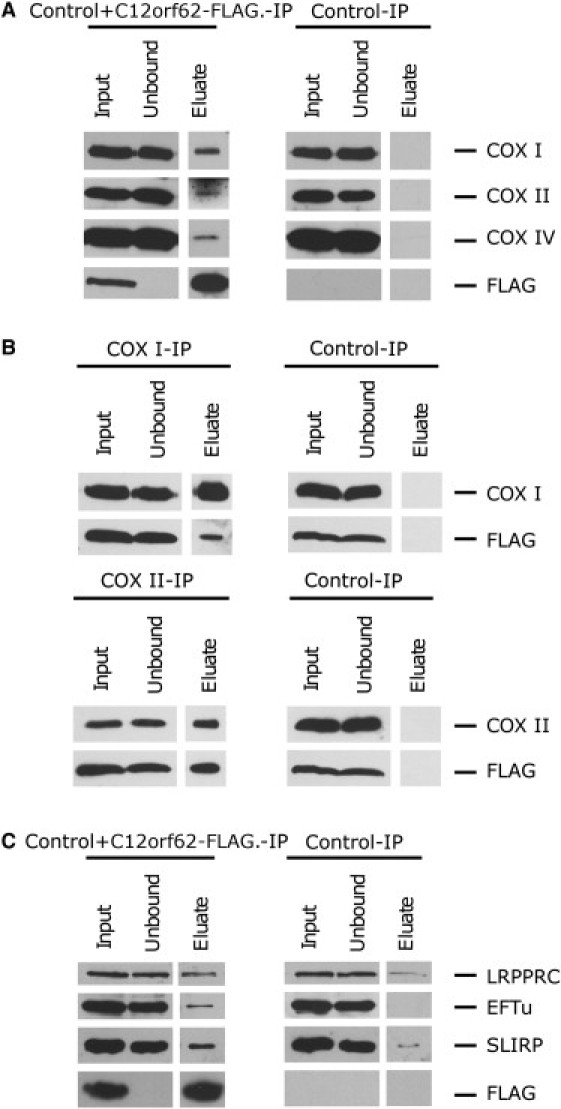
C12orf62-FLAG Coimmunoprecipates with COX I, II, and IV and with LRPPRC, EFTu, and SLIRP
(A) Mitochondria from either fibroblasts expressing C12orf62-FLAG or control fibroblasts were extracted and incubated with anti-FLAG agarose beads. Immunoblotting with antibodies against FLAG, COX I, COX II, and COX IV was performed on each fraction.
(B) Mitochondria from control fibroblasts expressing C12orf62-FLAG were extracted and incubated with magnetic beads that either were uncoated or were coated with COX I or COX II antibodies. Immunoblotting with antibodies against FLAG, COX I, and COX II was performed on the input, unbound, and eluate fractions.
(C) The immunoprecipitation was performed with FLAG beads as described in (A), and immunoblotting with antibodies against FLAG, LRPPRC, EFTu, and SLIRP was performed on each fraction.
To further investigate the function of C12orf62 in the synthesis of COX I, we searched for binding partners of C12orf62 by using the FLAG-tagged construct, which we had previously shown to be functional. C12orf62 coimmunoprecipitated with the translation elongation factors EFTu, LRPPRC, and SLIRP, the latter two of which form a ribonucleoprotein complex that regulates the stability and handling of mature mRNAs.15, 16 EFTu was not detected in the control, and LRPPRC and SLIRP were enriched more than 10-fold (Figure 5C).
The above data suggest that C12orf62 plays a role in the assembly or stability of the COX complex. To test this, we pulse labeled the mitochondrial translation products in the presence of a reversible cytosolic translation inhibitor (anisomycin) and assessed the stability of the newly synthesized COX subunits after 8, 17, and 32 hr chases (Figure 6). The labeling intensity of the newly synthesized COX subunits was normalized to ATP6. In control cells, the labeling intensity of all three COX subunits decreased to about 20%–30% of that observed in the pulse, even in the shortest chase (8 hr), indicating that they are apparently synthesized at levels in excess of that required for new assembly of the COX holoenzyme (Figure 6B). In the subject cells, although COX I was synthesized at ∼25% of control levels, the newly synthesized protein was completely stable. This contrasted with COX II and COX III, whose turnover was much faster than in control cells (Figure 6B).
Figure 6.
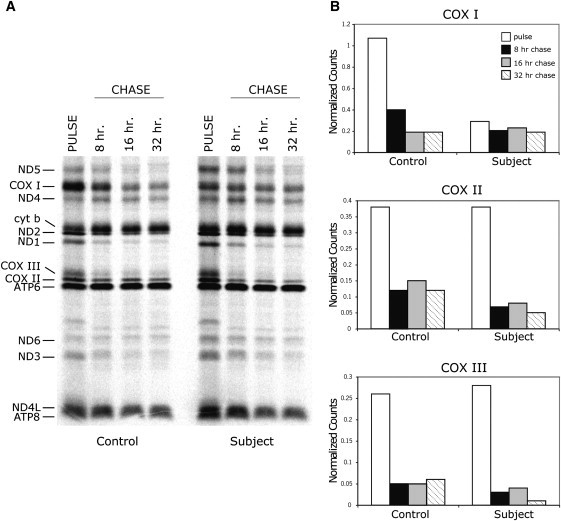
Synthesis and Turnover of COX Subunits in Control and Subject Fibroblasts
(A) Pulse labeling with [35S] methionine and [35S] cysteine of mitochondrial polypeptides and a 10 min chase (PULSE), as well as the time course of degradation via chases for 8, 17, and 32 hr (CHASE) are shown in control and subject fibroblasts. The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure.
(B) Histograms in which the level of the three COX subunits is normalized to the level of ATP6 in the PULSE and CHASE experiments in (A).
Incorporation of the newly synthesized mtDNA-encoded COX subunits into the holoenzyme complex was assessed with 2D BN-PAGE (Figure 7). For this analysis, 30 μg of 35S-labeled mitochondrial translation products was run in the first dimension on 8%–12% nondenaturing polyacrylamide gradient gels, followed by a separation on 10%–16% Tricine-SDS gradient gels.17 In control cells, COX I is incorporated into two complexes, one corresponding to the fully assembled holoenzyme (S4) and another lower-molecular-weight complex corresponding to an intermediate assembly complex (S3).18 A similar pattern was observed in the subject cells: COX I stability appeared normal in the fully assembled complex but slightly decreased in the assembly intermediate. By contrast, newly synthesized COX II and III were less efficiently incorporated into the COX complex, and they were rapidly turned over after a long chase.
Figure 7.
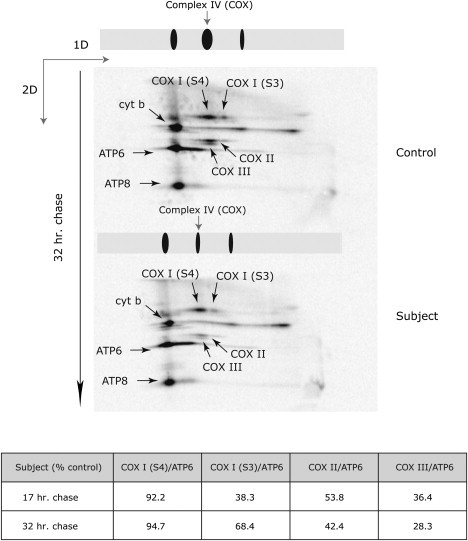
COX Subunits II and III Are Not Properly Assembled into the Nascent COX Complex in Subject Fibroblasts
2D BN-PAGE analysis of the sample pulse that was labeled with [35S] methionine and [35S] cysteine and chased for 32 hr. The gels were dried and the labeled mitochondrial translation products were detected through direct autoradiography. The arrows show different mtDNA-encoded polypeptides: one complex III subunit (cyt b), two complex V subunits (ATP6 and ATP8), COX subunit 1 (COX I-S4) in fully assembled complex IV, COX subunit 1 (COX I-S3) in the S3 intermediate of complex IV, COX subunit 2 (COX II), and COX subunit 3 (COX III) in fully assembled COX. The amount of label in each of the identified COX subunits was normalized to the label in ATP6 and is shown beneath the figure for both the 17 and 32 hr chases.
Together, the results of this study suggest that the role of C12orf62 is to couple the synthesis of COX I with subsequent assembly of the nascent subunits into the holoenzyme complex. This function is reminiscent of S. cerevisiae Mss51p, a protein that acts both as a translator activator for COX1 mRNA and as an early assembly factor in a heteroligomeric complex. Mss51p binds to the 5′ UTR of the COX1 mRNA to control translation and also associates with several other proteins in a high-molecular-weight complex to assist in early assembly; such proteins include Cox14p, Surf1p, Coa1p, and Coa3p.19 The current model proposes that Mss51p exists in two pools, one that can activate COX1 translation and another that assists in chaperoning Cox1p in the assembly process. This, then, would serve to couple the supply of newly synthesized Cox1p subunits with demand.
It is still not clear exactly how translation initiation of the mitochondrial mRNAs occurs in mammals because most mammalian mitochondrial mRNAs lack significant 5′ UTRs; however, the existence of a family of translational activator proteins analogous to those in yeast seems probable. Only one such protein, TACO1, has been identified to date in mammals. It is essential for the efficient translation of full-length COX I, but the molecular mechanism remains unknown.7 Deletion mutants of the yeast ortholog of TACO1 (YGR021w) have normal Cox1p synthesis and respire normally, indicating that the protein has evolved a different function in mammals. C12orf62 is essential for the normal level of COX I labeling in a pulse-translation experiment, and the labeling intensity of newly synthesized COX I correlates with the severity of the COX-assembly defect up to a point. Although we cannot be certain that this represents a true synthesis defect rather than rapid proteolysis of the newly synthesized COX I polypeptide, a number of observations argue against the latter. First, in fibroblasts that are null for TACO1, prematurely truncated forms of COX I accumulate in the pulse and are only turned over in a longer chase,7 suggesting that the quality-control system does not immediately recognize unassembled COX I subunits. Second, both LRPPRC and SLIRP, which form a ribonucleoprotein complex with COX I mRNA and are essential for its stability,16 are enriched more than 10-fold in the immunoprecipitate of C12orf62, suggesting that C12orf62 plays a direct role in the synthesis of COX I. Finally, although newly synthesized COX I turns over rapidly in the context of other reported COX-assembly defects such as those related to SCO111 or TACO1,7 it is completely stable in the subject fibroblasts.
The immunoprecipitation data suggest that the majority of C12orf62 interacts with the newly synthesized core subunits of COX. It also coelutes on a size-exclusion column with a fraction of COX I and COX IV, two subunits that nucleate the earliest step of COX assembly.18 Many small single-transmembrane proteins are structural components of the OXPHOS complexes, and it has been proposed that they might function to organize the hydrophobic domains of these complexes.20 One possibility is that C12orf62 acts to stabilize COX I in the inner membrane, allows COX I to assemble with COX IV and COX Va, and permits the addition of heme A and the two copper atoms that are necessary to mature this subunit. This activity might make C12orf62 unavailable for a role in translation.
The translation factor EFTu also coimmunoprecipitated with C12orf62-FLAG. Although its major role is delivering aminoacyl-tRNAs to the acceptor (A) site on the ribosome during the elongation stage of translation, it also appears to have a chaperone function.21 It interacts with misfolded, newly synthesized polypeptides from the mitochondrial ribosome, prevents thermal aggregation of proteins in vitro, and enhances protein refolding in vitro. Thus, EFTu could have a role in quality control of the nascent COX enzyme complexes through the binding of C12orf62.
Surprisingly, despite the fact that COX activity in the patient fibroblasts was 30%–40% of that in the control fibroblasts, the phenotypic presentation and clinical outcome in the subject investigated here are much more severe than that of SURF1 3- or TACO1-7 mutation-positive patients, whose fibroblasts have even less fully assembled COX. This most likely reflects tissue-specific requirements for the individual COX-assembly factors;22, 23 however, because all of these factors are ubiquitously expressed, the molecular basis for this specificity remains unclear. A resolution of this question awaits the development of appropriate cellular or animal models of disease.
Acknowledgments
This research was supported by a grant from the Canadian Institutes of Health Research to E.A.S., grants from the Agence Nationale de la Recherche (ANR; MNP 2008 Mitopark: ANR-08-MNPS-020-01) and the Association Française contre les Myopathies to A.L., and a grant from ANR (ANR-08-GENOPAT-043) to A.R. The antibody against EFTu/Ts was a kind gift from Linda Spremulli. E.A.S. is an International Scholar of the Howard Hughes Medical Institute. SNP genotyping, homozygosity mapping, and expression profiling were performed by The Centre for Applied Genomics and the Hospital for Sick Children, Toronto, Canada. We thank Timothy Johns and Neil Webb for technical assistance.
Published online: January 12, 2012
Footnotes
Supplemental Data include three figures, one table, and supplemental references and can be found with this article online at http://www.ajhg.org.
Web Resources
The URLs for data presented herein are as follows:
Block-iT RNAi Designer, http://rnaidesigner.invitrogen.com/rnaiexpress
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
TMHMM Server v. 2.0, http://www.cbs.dtu.dk/services/TMHMM/
Supplemental Data
References
- 1.Shoubridge E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001;106:46–52. doi: 10.1002/ajmg.1378. [DOI] [PubMed] [Google Scholar]
- 2.Robinson B.H. Human cytochrome oxidase deficiency. Pediatr. Res. 2000;48:581–585. doi: 10.1203/00006450-200011000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M., et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 4.Papadopoulou L.C., Sue C.M., Davidson M.M., Tanji K., Nishino I., Sadlock J.E., Krishna S., Walker W., Selby J., Glerum D.M., et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 5.Valnot I., Osmond S., Gigarel N., Mehaye B., Amiel J., Cormier-Daire V., Munnich A., Bonnefont J.P., Rustin P., Rötig A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000;67:1104–1109. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mootha V.K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F., et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weraarpachai W., Antonicka H., Sasarman F., Seeger J., Schrank B., Kolesar J.E., Lochmüller H., Chevrette M., Kaufman B.A., Horvath R., Shoubridge E.A. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 8.Shteyer E., Saada A., Shaag A., Al-Hijawi F.A., Kidess R., Revel-Vilk S., Elpeleg O. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am. J. Hum. Genet. 2009;84:412–417. doi: 10.1016/j.ajhg.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massa V., Fernandez-Vizarra E., Alshahwan S., Bakhsh E., Goffrini P., Ferrero I., Mereghetti P., D'Adamo P., Gasparini P., Zeviani M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008;82:1281–1289. doi: 10.1016/j.ajhg.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao J., Shoubridge E.A. Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum. Mol. Genet. 1999;8:2541–2549. doi: 10.1093/hmg/8.13.2541. [DOI] [PubMed] [Google Scholar]
- 11.Leary S.C., Sasarman F., Nishimura T., Shoubridge E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009;18:2230–2240. doi: 10.1093/hmg/ddp158. [DOI] [PubMed] [Google Scholar]
- 12.Cuthbert A.P., Trott D.A., Ekong R.M., Jezzard S., England N.L., Themis M., Todd C.M., Newbold R.F. Construction and characterization of a highly stable human: Rodent monochromosomal hybrid panel for genetic complementation and genome mapping studies. Cytogenet. Cell Genet. 1995;71:68–76. doi: 10.1159/000134066. [DOI] [PubMed] [Google Scholar]
- 13.Lochmüller H., Johns T., Shoubridge E.A. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 1999;248:186–193. doi: 10.1006/excr.1999.4407. [DOI] [PubMed] [Google Scholar]
- 14.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K., et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baughman J.M., Nilsson R., Gohil V.M., Arlow D.H., Gauhar Z., Mootha V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009;5:e1000590. doi: 10.1371/journal.pgen.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasarman F., Brunel-Guitton C., Antonicka H., Wai T., Shoubridge E.A., LSFC Consortium LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antonicka H., Ogilvie I., Taivassalo T., Anitori R.P., Haller R.G., Vissing J., Kennaway N.G., Shoubridge E.A. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J. Biol. Chem. 2003;278:43081–43088. doi: 10.1074/jbc.M304998200. [DOI] [PubMed] [Google Scholar]
- 18.Nijtmans L.G., Taanman J.W., Muijsers A.O., Speijer D., Van den Bogert C. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 1998;254:389–394. doi: 10.1046/j.1432-1327.1998.2540389.x. [DOI] [PubMed] [Google Scholar]
- 19.Mick D.U., Fox T.D., Rehling P. Inventory control: Cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zickermann V., Angerer H., Ding M.G., Nübel E., Brandt U. Small single transmembrane domain (STMD) proteins organize the hydrophobic subunits of large membrane protein complexes. FEBS Lett. 2010;584:2516–2525. doi: 10.1016/j.febslet.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki H., Ueda T., Taguchi H., Takeuchi N. Chaperone properties of mammalian mitochondrial translation elongation factor Tu. J. Biol. Chem. 2007;282:4076–4084. doi: 10.1074/jbc.M608187200. [DOI] [PubMed] [Google Scholar]
- 22.Antonicka H., Sasarman F., Kennaway N.G., Shoubridge E.A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 23.Leary S.C., Cobine P.A., Kaufman B.A., Guercin G.H., Mattman A., Palaty J., Lockitch G., Winge D.R., Rustin P., Horvath R., Shoubridge E.A. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 2007;5:9–20. doi: 10.1016/j.cmet.2006.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.