

Abstract
Picoplatin, a third-generation platinum agent, is efficacious against lung cancers that are otherwise resistant or become refractory during platinum treatment. This effort was aimed at the determination of the influence of organic cation transporters 1, 2, and 3 (OCT1, OCT2, and OCT3) and their genetic variants on cellular uptake of picoplatin and on the individual components of the ensuing cytotoxicity such as DNA adduct formation. The effect of OCT1 on picoplatin pharmacokinetics and antitumor efficacy was determined using OCT knockout mice and HEK293 xenografts stably expressing OCT1. The uptake and DNA adduct formation of picoplatin were found to be significantly enhanced by the expression of the OCTs. Expression of OCT1 and OCT2, but not OCT3, significantly enhanced picoplatin cytotoxicity, which was reduced in the presence of an OCT inhibitor. Common reduced functional variants of OCT1 and OCT2 led to reduction in uptake and DNA adduct formation of picoplatin in comparison with the reference OCT1 and OCT2. Pharmacokinetic parameters of picoplatin in Oct1−/− and Oct1+/+ mice were not significantly different, suggesting that the transporters do not influence the disposition of the drug. In contrast, the volume of OCT1-expressing xenografts in mice was significantly reduced by picoplatin treatment, suggesting that OCT1 may enhance the antitumor efficacy of picoplatin. These studies provide a basis for follow-up clinical studies that would seek to examine the relationship between the anticancer efficacy of picoplatin and expression levels of OCTs and their genetic variants in tumors.
Introduction
Lung cancer is the major cause of cancer-related deaths worldwide (1). Among lung cancers, non-small cell lung cancer (NSCLC) is the most common form, accounting for 80% of all lung cancers. Cytotoxic chemotherapeutic agents remain the most effective drugs in the treatment of NSCLC, either as postsurgical adjuvant therapy or in the setting of advanced NSCLC (2). The most widely used chemotherapeutic regimen consists of a combination of cisplatin or carboplatin with a nonplatinum cytotoxic agent (“doublets”; refs. 2, 3). Although effective, such regimens are associated with toxicities, particularly hematologic toxicity, nephrotoxicity, ototoxicity, nausea, and vomiting, which stem from the toxicity of the platinum agent (3, 4). Such toxicities are dose limiting and often result in undertreatment, leading to failure in maximizing the therapeutic benefit. Further, resistance to platinum agents may be present at the initiation of therapy or may develop over time. Clearly, a specific need exists for newer platinum compounds with distinct antitumor and toxicity profiles.
Recent studies suggest that newer platinum agents hold much promise in the treatment of NSCLC (5). In particular, new platinum agents have been developed to overcome resistance to cisplatin and carboplatin and to improve safety, particularly with respect to reducing the toxicities associated with cisplatin. Picoplatin, a new-generation platinum compound, which is currently in phase III clinical trials for treatment of lung cancer (6), has displayed a spectrum of activity distinct from its predecessors in addition to an improved safety profile. To date, it has shown no indication of significant nephrotoxicity, ototoxicity, or neurotoxicity and promising activity in platinum-sensitive, platinum-resistant, and refractory disease. This was attributed to its significantly reduced reactivity to intracellular thiols due to the presence of a sterically hindered amine substituent (using 2-methylpyridine) on the platinum center. There are reports in the literature on unique DNA adducts formed by picoplatin, which helps it to retain its anticancer activity in in vitro human ovarian carcinoma cell lines that have acquired cisplatin resistance (7).
Because of their polarity, platinum compounds require influx transporters to permeate cell membranes. For example, the copper transporter Ctr1 has been previously associated with the uptake of cisplatin (8, 9). We and others have recently established that oxaliplatin is a substrate of the human organic cation transporters 1, 2, and 3 (OCT1, OCT2 and OCT3), which are electrical potential-driven polyspecific transporters that transport several small molecular weight organic cations across the cell membrane (10, 11). Seemingly distinct in their chemical structures, both oxaliplatin and picoplatin have been found active in cisplatin-resistant cancer models and possess only one chemical feature in common (i.e., the presence of an organic amine group at their platinum center). This led us to believe that both share a common transport mechanism to gain entry into the cell.
The goals of our present study were to determine the interaction of picoplatin with OCT1, OCT2, and OCT3 and to elucidate the role of OCTs in picoplatin cytotoxicity in NSCLC cell lines and in vivo in mouse models harboring OCT-expressing xenografts. In addition, we determined the effect of genetic variants in OCT1 and OCT2 on picoplatin uptake and DNA adduct formation. These studies are, in essence, aimed at investigating the mechanistic basis for the observed clinical advantages of picoplatin over older-generation platinums.
Materials and Methods
Drugs and reagents
Cisplatin, carboplatin, oxaliplatin, cimetidine, disopyramide, corticosterone, glutathione, and MTT were purchased from Sigma. Stock solutions of cisplatin and carboplatin (3 mmol/L) were freshly prepared in PBS for every experiment. Oxaliplatin solution (10 mmol/L) was made in water, stored frozen at −20°C, and discarded after 1 mo of preparation. Picoplatin [synthesized according to the method of Battle et. al. (12)] stock solution (20 mmol/L) in DMSO was diluted with PBS to attain the required concentration immediately before use. Stock solution of the MTT reagent was prepared in PBS at 5 mg/mL concentration and was stored at 4°C up to 2 wk from preparation under protection from light. Hygromycin B was obtained from Invitrogen. The cell culture medium DMEM H-21, RPMI 1640, and fetal bovine serum were obtained from the Cell Culture Facility of the University of California (San Francisco, CA).
Cell lines and transfection
Human embryonic kidney (HEK293) cells stably transfected with the full-length reference human OCT1 cDNA (HEK-hOCT1), OCT2 cDNA (HEK-hOCT2), mutant OCT1 and OCT2 cDNA inserts, and the empty vector (HEK-EV or HEK-MOCK) were established previously in our laboratory (10, 13). The stably transfected HEK-hOCT3 cell line was established by transfection of pcDNA5/FRT vector (Invitrogen) containing the full-length human OCT3 cDNA (HEK-hOCT3) into HEK293 Flp-In-293 cells using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions. The stable clones were selected with 75 μg/mL hygromycin B. All the lung cancer cell lines (A549, H460, H838, and H1703) used in the present study were from the American Type Culture Collection.
Cell culture
The stably transfected HEK293 cells were maintained in DMEM H-21 medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 units/mL streptomycin, and 75 μg/mL hygromycin B. The culture medium for all the lung cancer cell lines was RPMI 1640 containing 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. All cell lines were grown at 37°C in a humidified atmosphere with 5% CO2/95% air.
Cytotoxicity studies
The cytotoxicity of picoplatin was measured by standard MTT assays in 96-well plates as previously described (10). Drug exposure time was 7 h followed by 65-h incubation in drug-free medium.
Cellular accumulation of platinum
The cellular accumulation of platinum was determined after a 2-h exposure to platinum drugs as previously described (10). In some experiments, phosphate buffer [PB; 1.06 mmol/L KH2PO4, 2.97 mmol/L Na2HPO4 (pH 7.4)] containing 155 mmol/L NaCl (PB-Cl buffer) or 103 mmol/L Na2SO4 (PB-SO4 buffer) was used instead of the culture medium as specified. The platinum content was then determined by inductively coupled plasma mass spectrometry (ICP-MS) in the Analytical Facility at University of California at Santa Cruz. Cellular platinum accumulation was normalized to the protein content determined by bicinchoninic acid protein assay.
Platinum-DNA adduct formation
The platinum content associated with genomic DNA was determined after a 2-h exposure to picoplatin or other platinum analogues as previously described (10). Genomic DNA was isolated from the cell pellets using Wizard Genomic DNA Purification kit (Promega) according to the manufacturer’s protocol. The DNA-bound platinum was determined by ICP-MS, which was normalized to total DNA content (absorption spectrometry at 260 nm).
RNA isolation
Total RNA was isolated from each cell line on six-well plates using the Invitrogen Micro-to-Midi Total RNA Purification System according to the manufacturer’s protocol. Extracted RNA was stored at −80°C until use. Similarly, total RNA was isolated from the matched samples of tumor and normal lung tissues using Trizol solution (Invitrogen). Patients were consented to tissue specimen collection prospectively, and the study was approved by the University of California, San Francisco, institutional review board (CHR #H8714-11647-14).
Quantitative real-time PCR
Total RNA (2 μg) was reverse transcribed using High Capacity cDNA Reverse Transcription kit (Applied Biosystems) in a 20 μL volume reaction according to the manufacturer’s protocol. The resulting cDNA was used as template for quantitative real-time PCR using Taqman Gene Expression Assays for human transporter OCT1 (assay ID: Hs00427550_m1), OCT2 (assay ID: Hs00533907_m1), OCT3 (assay ID: Hs01009568_m1), and human glyceraldehyde-3-phosphate dehydrogenase (assay ID: Hs99999905_m1). Quantitative real-time PCR was carried out in 96-well reaction plates in a volume of 10 μL using the Taqman Fast Universal Master Mix (Applied Biosystems). Reactions were run on the Applied Biosystems 7500 Fast Real-Time PCR System with the following profile: 95°C for 20 s followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. The relative expression of each mRNA was calculated by the comparative method (ΔΔCt method). Firstly, the ΔCt values for each samples were obtained by subtracting the Ct value of glyceraldehyde-3-phosphate dehydrogenase mRNA from the Ct value of the target mRNA. Then, the ΔΔCt values for each sample were obtained by subtracting the highest ΔCt value obtained for the sample (which in this case is sample “T8”) from the ΔCt value of each sample. The mRNA expression reported as relative difference was calculated using the arithmetic formula 2-ΔΔCt (ABI Prism 7700 Sequence Detection system User Bulletin No. 2, P/N 4303859).
Western blot analysis
The total protein extracts from HEK293 and lung cancer cells were prepared using CelLytic M cell lysis buffer (Sigma) containing protease inhibitor cocktail at 4°C for 10 min. The cell homogenate was spun for 10 min at 14,000 rpm at 4°C, and the protein concentration in the supernatant was determined by bicinchoninic acid protein assay. Approximately 50 μg of the supernatant were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membrane (Bio-Rad). The blots were incubated for 1 h in blocking buffer containing 5% nonfat dry milk in TBS and then incubated overnight at 4°C with anti-human OCT1 antibody (Abcam) diluted (1:500) in 2% milk in TBS buffer containing 0.1% Tween 20 (TBS-T). After washing with TBS-T, the blots were incubated for 1 h at room temperature with the goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (1:2,500; Santa Cruz Biotechnology) in TBS-T buffer containing 5% milk. The blots were washed again thrice with TBS-T for 10 min and then developed with the enhanced chemiluminescence Western blotting detection reagent (GE Healthcare).
Animals
Oct1+/+ and Oct1−/− mice were housed in a virus-free, temperature-controlled facility on a 12-h light-dark cycle. They were allowed standard mouse food and water ad libitum. Oct1−/− mice were generated as described elsewhere (13). For picoplatin efficacy experiment, 7-wk-old female athymic mice (nu/nu genotype, BALB/c background) were purchased from Taconic, Inc. through a contract with the University of California at San Francisco Helen Diller Comprehensive Cancer Center and housed under aseptic conditions, which included filtered air and sterilized food, water, bedding, and cages. The experiments on mice, which were approved by the Institutional Animal Care and Use Committee, were done in the Preclinical Core of the Cancer Center.
Picoplatin pharmacokinetics in mice
Nine-week-old male Oct1+/+ and Oct1−/− mice were given 20 mg/kg picoplatin in PBS with 5% DMSO via tail vein injection. The animals weighed 25.8 ± 0.75 g and 25.3 ± 0.92 g for Oct1+/+ and Oct1−/− mice, respectively, at the time of treatment. Blood samples (20 μL) were collected at 20 min and 1, 2, 4, 8, 12, 24, 48, 72, 96, 120, and 144 h after picoplatin treatment by tail vein bleeding into heparinized micro-hematocrit capillary tubes (Fisher). Blood was centrifuged for 5 min using Microhematocrit Centrifuge (Thermo Fisher Scientific, Inc.), and the plasma was decanted and frozen at −80°C until analysis. Plasma (5 μL) was used for total platinum measurement by ICP-MS. The pharmacokinetic parameters were obtained by two-compartmental analysis using WinNonlin 4.0 (Pharsight Corp.).
Preparation of xenografts and picoplatin treatment
In this study, nude mice were divided into two treatment groups (five mice per group): xenografts consisting of HEK293 cells stably expressing OCT1 and xenografts consisting of HEK293 cells expressing the vector alone. Tumors were induced by s.c. injection of □ 10 × 106 HEK293 cells into the flanks of mice. Animals were maintained in pathogen-free conditions inside autoclaved microisolator cages. Serial tumor measurements were obtained every 3 to 4 d by caliper in three dimensions. Tumor volumes were calculated according to the following formula: volume = height × weight × length × 0.5236. About 14 d later, when the tumor sizes reached □ 100 to 150 mm3 in both tumor types, mice (n = 4) were randomized into picoplatin and saline treatment groups, and tumor volume was ascribed to day 0. Mice were then injected i.v. via tail vein with picoplatin (20 mg/kg) or saline containing 5% DMSO once a week for 3 wk. Tumor and body weight measurements were done twice a week. Comparison of the relative change in tumor volume with respect to day 0 in picoplatin- and saline-treated groups for OCT1 and EV tumors was considered an indicator of picoplatin efficacy.
Data analysis
Data were analyzed statistically by unpaired or paired Student’s t tests, as appropriate. Probability values of <0.05 were considered statistically significant. For inhibition studies, concentration response graphs were generated for each drug using GraphPad Prism software (GraphPad Software, Inc.). These graphs were analyzed using a curve fit for sigmoid dose response, and IC50 values were derived. Results are expressed as mean IC50 with the SEM. For xenograft studies in mice, changes in tumor volumes were recorded at various time points and are represented as the relative change in tumor volume at that time point with respect to day 0 (before initiation of treatment). ANOVA was used to compare within and between treatment groups to derive statistical significance.
Results
The cytotoxicity of picoplatin was enhanced in cells expressing OCT1 and OCT2
To study the effect of the expression of the OCTs on the cytotoxicity of picoplatin, we did the standard MTT assay in HEK293 cells transfected with the OCTs and the empty vector. These transfected cell lines have previously been characterized for the mRNA overexpression level of the desired transporter (10). The IC50 of picoplatin in HEK-hOCT1 after 7 hours of exposure was 0.44 ± 0.01 μmol/L, whereas in the corresponding empty vector-transfected cell line, HEK-EV, it was 2.97 ± 0.10 μmol/L (Fig. 1; Table 1). The modification factor, which is the ratio of IC50 value in EV cells to that in the corresponding OCT-transfected cells, was 6.74 (Table 1). Similarly, when picoplatin cytotoxicity was determined in hOCT2-transfected HEK cells, a modification factor of 10.7-fold was observed (P < 0.001; Table 1; Fig. 1C). Coincubation of an OCT1 inhibitor, disopyramide (150 μmol/L), and OCT2 inhibitor, cimetidine (1.5 mmol/L), substantially increased the IC50 of picoplatin in HEK-hOCT1 cells by 5.9-fold and in HEK-hOCT2 cells by 10.9-fold (P < 0.01; Table 1; Fig. 1B and D). There was a small effect of disopyramide and cimetidine on picoplatin IC50 in HEK-EV cells (P < 0.05; Table 1; Fig. 1B and D). Under the same experimental conditions, disopyramide and cimetidine themselves did not exhibit any cytotoxicity up to 400 μmol/L and 5 mmol/L, respectively (data not shown). In contrast to OCT1 and OCT2, the overexpression of hOCT3 did not have any effect on picoplatin cytotoxicity (Table 1). Because OCT1 and OCT2 enhanced picoplatin cytotoxicity, whereas OCT3 did not, we focused mostly on these two transporters.
Figure 1.
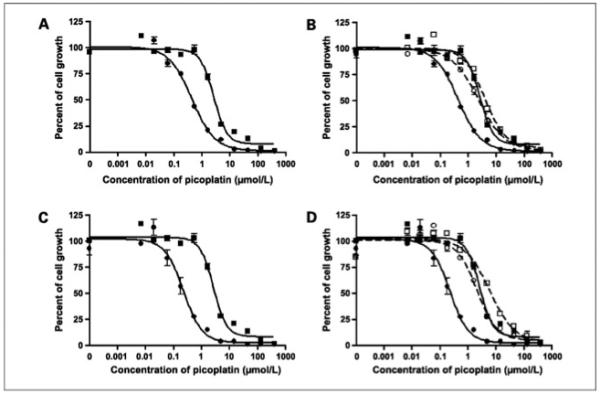
Cytotoxicity of picoplatin in HEK293 cells stably transfected with OCTs. The MTT assays of picoplatin were done in OCT1-transfected (A) and OCT2-transfected (C) HEK cells (•) and in the corresponding EV-transfected (■) cells as described in Materials and Methods. In addition, the cytotoxicity of picoplatin was determined in these cell lines in the absence (• and■) and presence (○ and □) of OCT inhibitors (B and D): disopyramide (150 μmol/L) for OCT1 and cimetidine (1.5 mmol/L) for OCT2. Data represent results from a typical experiment. Four independent experiments were done and similar results were obtained.
Table 1.
Effects of OCT1, OCT2, and OCT3 on picoplatin cytotoxicity
| Transporter | Treatment | IC50 (μmol/L), HEK-EV |
IC50 (μmol/L), HEK-transporter |
Modification factor (IC50–EV/IC50 –transporter) |
|---|---|---|---|---|
| OCT1 | Picoplatin | 2.97 ± 0.105 |
0.440 ±0.011 | 6.74* |
| Picoplatin + disopyramide |
3.72 ±0.15 | 2.60 ± 0.45 | 1.43† | |
| OCT2 | Picoplatin | 2.97 ± 0.105 |
0.277 ± 0.026 | 10.7* |
| Picoplatin + cimetidine |
4.09 ± 0.41 | 2.88 ± 0.55 | 1.42‡ | |
| OCT3 | Picoplatin | 2.64 ± 0.265 |
3.20 ± 0.364 | 0.91 |
NOTE: The IC50 values in OCT-transfected cell lines and the corresponding EV cell line were determined using the MTT assay with drug exposure for 7 h as described in Materials and Methods. Increase in IC50 values of picoplatin was studied in the presence of disopyramide (150 μmol/L) and cimetidine (1.5 mmol/L) as OCT1 and OCT2 inhibitors, respectively. Data are expressed as the mean ± SEM of four independent experiments. The ratio of the mean IC50 value in the EV cells to that in OCT-transfected cells was defined as the modification factor.
P < 0.001.
P < 0.01.
P < 0.05.
OCTs enhanced the cellular uptake and DNA adduct formation rate of picoplatin
The cellular accumulation rate of picoplatin (10 μmol/L) after 2-hour exposure in HEK-hOCT1 cells [0.252 ± 0.017 nmol/(mg protein–hour)] was 6.4-fold higher than in HEK-EV cells [0.039 ± 0.006 nmol/(mg protein–hour); P < 0.001; Fig. 2A]. The 2-hour exposure time period was necessitated by limitations in the capability to detect lower platinum concentrations that would prevail during very early time points at pharmacologically relevant platinum concentrations. Incubation of picoplatin with disopyramide significantly reduced the platinum accumulation in OCT1 cells (P < 0.001; Fig. 2A), with little effect in HEK-EV cells. Similarly, picoplatin accumulation rate in HEK-hOCT2 [0.541 ± 0.026 nmol/(mg protein-hour)] and HEK-hOCT3 cells [0.290 ± 0.006 nmol/(mg protein-hour)] was markedly higher than in HEK-EV cells (P < 0.001; Fig. 2B and C). Coincubation of picoplatin with an OCT2 inhibitor, cimetidine (1.5 mmol/L), and OCT3 inhibitor, corticosterone (10 μmol/L), reduced the platinum accumulation in HEK-hOCT2 and HEK-hOCT3 cells by 6.27- and 3.53-fold, respectively (P < 0.05; Fig. 2B and C), with a significant but small effect in HEK-EV cells (control versus cimetidine treated versus corticosterone treated; P < 0.05).
Figure 2.
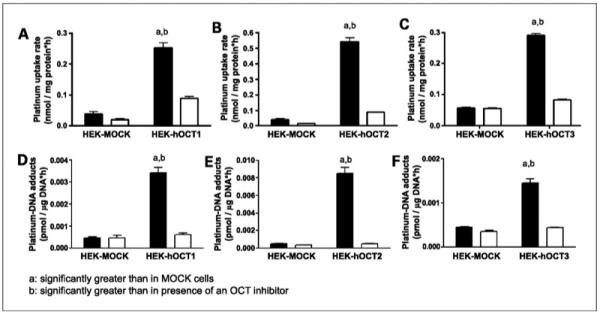
Effects of OCTs on uptake (A–C) and DNA adduct formation (D-F) of picoplatin. Studies were done in OCT1-transfected (A and D), OCT2-transfected (B and E), and OCT3-transfected (C and F) cells and the corresponding EV-transfected HEK cells after 2-h exposure to picoplatin in the presence (white columns) and absence (black columns) of an OCT inhibitor (150 μmol/L disopyramide for OCT1, 1.5 mmol/L cimetidine for OCT2, and 10 μmol/L corticosterone for OCT3). Briefly, HEK293 cells were incubated in an antibiotic-free medium containing 10 μmol/L picoplatin for 2 h, after which platinum concentration inside the cells and bound to DNA was determined as described in Materials and Methods. Columns, mean of three independent experiments; bars, SEM.
Experiments were done to identify the chemical species that may be recognized and transported by OCTs. Picoplatin undergoes aquation in water with a rate constant of 1.47 ± 0.32 × 10–5 per second (14). Clearly, a monocationic complex, [Pt(NH3)(2-Pic)(H2O)(Cl)]+, exists outside the cell, in addition to a possible diaquated, dicationic complex, [Pt(NH3)(2-Pic)(H2O)2]2+. OCTs typically transport small positively charged cations, thereby making the monoaqua and diaqua complexes candidates for transport. We determined the cellular uptake of platinum from three distinct solutions: (a) a solution of picoplatin (10 μmol/L) in a chloride-containing buffer (PB-Cl), in which picoplatin would be expected to equilibrate with the monoaqua complex [but not the diaqua complex due to short (30 minutes) incubation time]; (b) a solution of a synthesized sample of the monoaqua complex of picoplatin in a chloride-free buffer (PB-SO4), in which formation of picoplatin is impossible; and (c) a solution of a synthesized diaqua complex in a chloride-free buffer (PB-SO4; Supplementary Fig. S1). It was observed that platinum gets uptaken from the solutions 1 and 2 with an equal additional preference in OCT1-expressing cells over the corresponding EV cells, whereas no such preference is seen in case of solution 3. This clearly suggests that the monoaqua complex of picoplatin (but not the diaqua complex) is a substrate for OCT1 and may be chemical species that is transported.
A similar trend was observed after comparison of concentrations of platinated DNA in OCT-expressing HEK cells and those in HEK-EV cells. The platinum-DNA adduct concentration after 2-hour exposure to picoplatin in HEK-hOCT1 cells was 7.39-fold higher than that in HEK-EV cells [0.463 ± 0.057 pmol/(mg DNA-hour); P < 0.001; Fig. 2D]. This platinum-DNA adduct formation was inhibitable by an OCT1 inhibitor, disopyramide, in HEK-hOCT1 cells (P < 0.001) but not in HEK-EV cells (P > 0.05; Fig. 2D). Similar results were observed in HEK-OCT2 cells, wherein □ 18.4-fold excess formation of picoplatin-DNA adduct was observed over that in HEK-EV cells [8.509 ± 0.650 pmol/(mg DNA-hour) versus 0.463 ± 0.057 pmol/(mg DNA-hour); P < 0.001; Fig. 2E]. Coincubation with cimetidine caused a 16.4-fold reduction (P < 0.001) in picoplatin-DNA adduct concentration in HEK-hOCT2 cells, whereas only 1.37-fold reduction was observed in HEK-EV cells (P > 0.05; Fig. 2E). Similarly, the increase in picoplatin-DNA adduct level in HEK-hOCT3 [1.448 ± 0.101 pmol/(mg DNA-hour)] cells was 3.21-fold over HEK-EV cells (P < 0.001; Fig. 2F), and it was completely inhibited by corticosterone (10 μmol/L) in HEK-hOCT3 cells (P < 0.001; Fig. 2F) but not in HEK-EV cells (P > 0.05; Fig. 2F).
OCT1 was expressed in lung cancer cell lines and tumor samples
Consideration of picoplatin for treatment of NSCLC prompted us to study the expression level of OCTs in lung cancer cell lines (A549, A460, H838, and H1703) and in lung tumor samples. The expression of OCT1 mRNA was detected in all four lung cancer cell lines tested, but there was no substantial difference in the expression level in these cell lines (Fig. 3A; Supplementary Table S1), whereas the expression of OCT2 was undetectable in these samples. Similarly, the Western blot analysis of these cell lines could detect OCT1 protein (Fig. 3B) but not OCT2 (data not shown). The mRNA level of OCT1 was detected in seven of eight normal lung samples, whereas two of eight lung tumor samples displayed OCT1 at a level comparable with that in normal tissues. Although one normal lung sample and three of eight lung tumor samples expressed OCT2, the levels were dramatically lower than that of OCT1. The expression of OCT3 was high and variable in the normal and tumor samples (data not shown).
Figure 3.
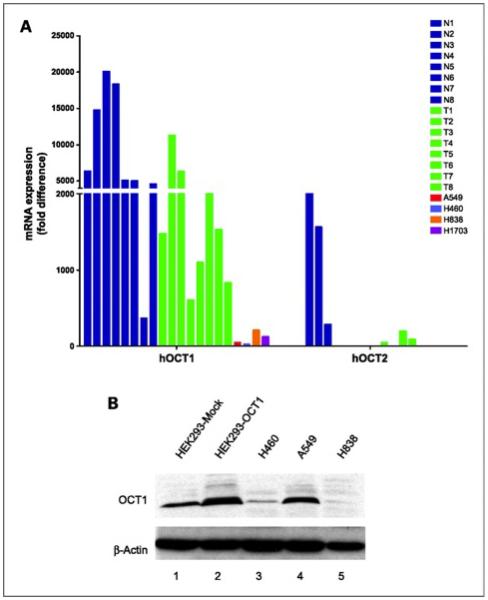
A. expression levels of mRNA transcripts for OCT1 and OCT2 in normal lung tissue samples (N), lung tumor tissue samples (T), and lung cancer cell lines. Total RNA was isolated from four lung cancer cell lines and eight samples from normal and tumor lung tissues. The mRNA expression was quantitated by quantitative reverse transcription-PCR as described in Materials and Methods. B. Western blot expression analysis of OCT1 in lung cancer cell lines. Cell extracts from HEK293 cells transfected with EV or OCT1 (lanes 1 and 2) and lung cancer cell lines (lanes 3-5) were probed with anti-OCT1 antibody. β-Actin was used as a loading standard. The presence of OCT1 in these samples was detected as a band at □ 90 kDa.
An OCT inhibitor, cimetidine, reduced the cytotoxicity of picoplatin in lung cancer cell lines
To delineate the potential role played by OCTs in the cytotoxicity of picoplatin, we determined the sensitivity of the four lung cancer cell lines to picoplatin. The IC50 values were determined after 4-hour exposure to picoplatin in the presence or absence of an OCT inhibitor, cimetidine (3 mmol/L). Here, the modification factor is expressed as the ratio of IC50 value in the presence of cimetidine to that in its absence. As shown in Table 2, there was a significant increase in IC50 values of picoplatin in all four lung cancer cell lines in the presence of cimetidine (P < 0.05). Under similar conditions, there was no significant change in the IC50 values of cisplatin and carboplatin, which are poorly recognized by OCTs, specifically OCT1 and OCT3, and the modification factor ranged from 1.1 to 1.5. Further, although cisplatin is a good substrate of OCT2, its negligible expression in these lung cancer cell lines resulted in minimal inhibition of its cytotoxicity by an OCT inhibitor.
Table 2.
Cytotoxicity of picoplatin in lung cancer cell lines in the presence and absence of an OCT inhibitor, cimetidine
| Platinum drug |
IC50 in lung cancer cell lines (μmol/L) |
|||||||
|---|---|---|---|---|---|---|---|---|
| H460 |
A549 |
H838 |
H1703 |
|||||
| Control | Cimetidine treated |
Control | Cimetidine treated |
Control | Cimetidine treated |
Control | Cimetidine treated |
|
| Picoplatin | 135 ±40.5 | 297 ± 73.0 | 114 ±25.5 | 408 ± 54.7 | 159 ± 66.7 | 359 ± 70.0 | 179 ±20.6 | 492 ± 96.1 |
| Cisplatin | 13.9 ±9.32 | 15.6 ±9.15 | 12.8 ±4.94 | 19.4 ±8.23 | 52.9 ±31.7 | 81.3 ±11.5 | 56.8 ±14.1 | 70.0 ± 23.1 |
| Carboplatin | 128 ±41,3 | 107 ± 8.43 | 190 ±18.6 | 164 ± 16.5 | 224 ±12.6 | 265 ± 16.7 | 630 ±10.8 | 640 ± 26.9 |
NOTE: In a typical MTT assay, cells were exposed to platinum drug for 4 h with and without 3 mmol/L cimetidine as described in Materials and Methods. Trie IC50 values were determined by fitting the observed data by nonlinear regression analysis in GraphPad Prism software. Data are expressed as the mean ± SEM of three independent experiments.
OCT1 and OCT2 polymorphisms modulated picoplatin uptake and DNA adduct formation
In a large-scale screening study in DNA samples from ethnically diverse U.S. populations (13), we discovered several nonsynonymous variants of OCT1 and OCT2. In previous studies, we determined that several of these variant transporters exhibit reduced uptake of the model organic cation MPP+ and, more recently, with metformin and paraquat (13, 15). In the current study, we observed that 9 of 12 variants of OCT1 showed statistically significant reduction in picoplatin uptake (P < 0.01; Fig. 4A) with respect to its uptake in the OCT1 wild-type transporter-expressing cells. Further, only three of the nine variants showed decreased picoplatin-DNA adduct levels compared with the reference OCT1 (P < 0.05; Fig. 4B). The common variant of OCT2, A270S, showed increased picoplatin uptake over the reference OCT2 (P < 0.001; Fig. 5A), but DNA adducts were actually a little lower in the A270S cell lines (Fig. 5B). This lack of agreement could be explained by enhanced picoplatin binding to A270S, resulting in greater apparent “uptake” but not in greater binding to DNA.
Figure 4.
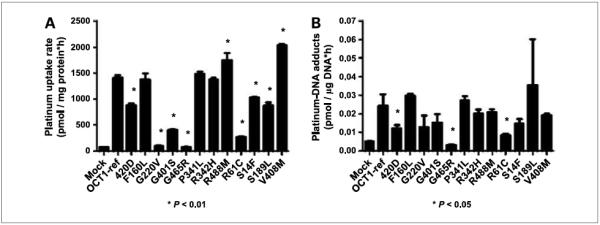
Effect of missense variants of OCT1 on the uptake (A) and DNA adduct formation of picoplatin (B). Cells were exposed for 2 h to picoplatin. Platinum concentration inside the cells and bound to DNA was determined as described in Materials and Methods. Picoplatin uptake and DNA adducts in cells expressing the variant OCT1 transporter were compared with the wild-type OCT1-expressing cells using Student’s t test. Three independent experiments were conducted and similar results were obtained.
Figure 5.
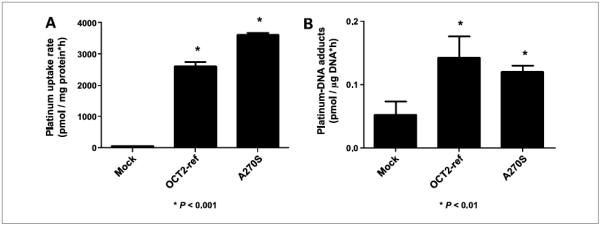
Effect of a common variant of OCT2, OCT2-A270S, on the uptake (A) and DNA adduct formation (B) of picoplatin. Platinum concentration inside the cells and bound to DNA was determined as described in Materials and Methods. Picoplatin uptake and DNA adducts in cells expressing the variant OCT2 transporter were compared with the wild-type OCT2-expressing cells using Student’s t test. Three independent experiments were conducted and similar results were obtained.
Picoplatin distribution and pharmacokinetics were not changed substantially in Oct1−/− and Oct1/2−/− mice
We examined the role of OCT1 on picoplatin disposition in vivo. The total plasma platinum levels following i.v. administration of picoplatin (20 mg/kg) in wild-type and knockout (Oct1 and Oct1/2) mice fitted a two-compartment model, and at all time points tested, the mean plasma concentration of picoplatin in Oct1−/− and Oct1/2−/− mice was higher than that in the wild-type mice (Supplementary Table S2; Supplementary Fig. S2). The t1/2 of picoplatin elimination from blood (Supplementary Table S2) was slightly longer (1.24-fold) in Oct1−/− mice than in Oct1+/+ mice (P = 0.05). This resulted in a trend toward increase in the area under the curve and Cmax for Oct1−/− mice compared with that for Oct1+/+ mice. There was no difference in pharmacokinetic profiles of picoplatin in wild-type, Oct1−/−, and Oct1/2−/− mice. Furthermore, there was a trend toward reduced picoplatin accumulation in the kidney and to some extent in the liver of Oct1−/− and Oct1/2−/− mice compared with those of Oct1+/+ mice, but the difference did not reach statistical significance (Supplementary Fig. S3). The tissue distribution in wild-type mice showed preferentially higher platinum uptake in OCT-expressing tissues (liver, kidney, and intestine). These data, together with the tissue accumulation data, suggest that OCT1 has little effect on the disposition of picoplatin.
OCT1 enhanced the antitumor effects of picoplatin in mice with xenografts of HEK293 cells
In our preliminary experiments in nude mice, we determined the maximum tolerated dose for picoplatin using five dosing regimens (5, 10, 20, 30, and 40 mg/kg) and found that the once a week i.v. 20 mg/kg regimen was well tolerated with no change in body weight over 4 weeks (data not shown). Next, to assess the role of OCT1 in vivo in tumor responsiveness to picoplatin, we designed a proof-of-concept in vivo study using xenografts of stably transfected OCT1 cell line. HEK293 cells were chosen for this study because of their modest tumor growth rates (doubling time, 14-17 days) and their well-documented ability to readily express transfected mammalian proteins (16). Two weeks after tumor induction in nude mice through s.c. injection of HEK-EV and HEK-hOCT1 cells, mice were randomized into picoplatin and saline treatment groups. The treatment consisted of once a week i.v. tail vein injection of picoplatin (20 mg/kg) and saline for 3 weeks in both tumor types (OCT1 and EV). After an initial single tail vein dose of picoplatin, tumor growth slowed down substantially, with the relative change in picoplatin-treated OCT1 tumors being significantly lower than that of the corresponding saline-treated tumors on days 5 and 8 after injection of the first dose of picoplatin (P < 0.05; Fig. 6). The same trend was followed until day 18 from the first dose of picoplatin treatment, after which a dramatic decrease in OCT1 expression was observed in OCT1-expressing tumors along with a corresponding reduction in the efficacy of picoplatin in these tumors. Picoplatin also effected a trend toward reducing the volume of HEK-EV xenografts; however, this trend was not statistically significant (data not shown). Similar statistically significant reductions in OCT1 tumor volumes were observed with oxaliplatin, a well-established OCT1 substrate, until day 18 from the beginning of oxaliplatin treatment.
Figure 6.
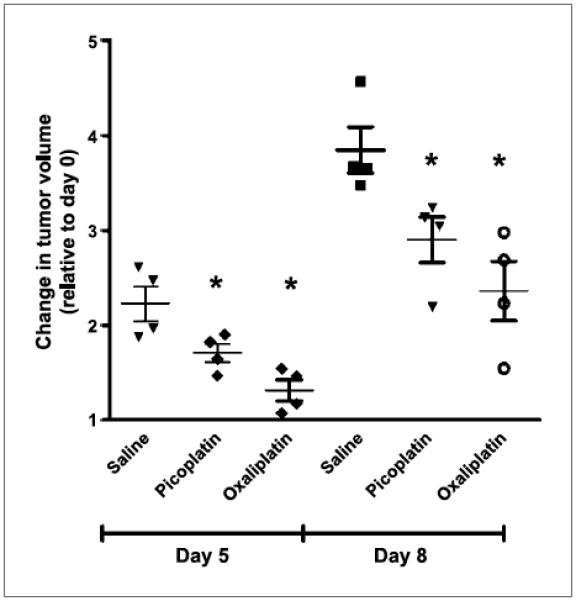
Change in volume of xenografts of OCT1-transfected HEK293 cells in nude mice. Tumor volume measurements were carried out in nude mice (eight per group) that received once weekly dose of picoplatin (20 mg/kg) or saline. The relative change in tumor volume in each group is plotted against time, which refers to the change in tumor size at each time point with respect to the tumor size before treatment initiation (i.e., at day 0). Data are expressed as an aligned dot plot, with the horizontal line showing the mean and error bar representing the SD. *, P < 0.05.
Discussion
Platinum anticancer therapy remains among the most effective in the treatment of various solid tumors. The distinct spectrum of activity of earlier-generation platinums, cisplatin and oxaliplatin, was attributed to their difference in DNA adduct formation accompanied by differences in the ability of nucleotide excision repair machinery to repair the platinum DNA adducts. However, in recent studies, we and others showed that oxaliplatin is an excellent substrate of OCTs, whereas cisplatin and carboplatin are not (10, 11). The studies also showed that OCTs were highly expressed in colorectal cancer cell lines and tumor samples, suggesting that interaction with OCTs may be a contributory factor for the efficacy of oxaliplatin against colon cancer.
Due to the structural similarity of oxaliplatin and picoplatin, we hypothesized that OCTs may also play a role in enhancing the antitumor effects of picoplatin. Our initial efforts sought to understand the interaction between picoplatin and OCTs. The reduction in the IC50 of picoplatin in HEK293 cells transfected with OCT1 (6.74-fold) and OCT2 (10.7-fold) compared with empty vector-transfected cells showed the potential role of these transporters in picoplatin cytotoxicity. The maximum degree of selectivity for the transporter of interest was achieved by drug exposure for 7 hours compared with 24 or 48 hours at which a slight decrease in the modification factor (selectivity) was observed. Furthermore, a dramatically higher uptake of picoplatin in these cell lines established it as an excellent substrate of OCT1 and OCT2. Results of DNA adduct formation experiments agreed well with our cytotoxicity experiments and proved that the intracellular picoplatin is available for reaction with its cellular target—DNA. These effects were blocked by known OCT inhibitors. Further mechanistic investigation of picoplatin uptake in OCT1-transfected cells suggests that picoplatin itself, along with the positively charged monoaqua complex of picoplatin and not the diaqua complex, is likely to be transported by OCT1 (Supplementary Fig. S1). OCT3 had little effect on picoplatin cytotoxicity, consistent with its small effect in enhancing picoplatin-DNA adduct formation (Fig. 2F). Although the exact mechanism for the low OCT3-mediated picoplatin binding to DNA is not known, it may be due to binding of picoplatin to intracellular histamine, the endogenous substrate of OCT3. Histamine-platinum complexes have been described (17) and would reduce the availability of intracellular picoplatin for interaction with DNA. Such effects would not be observed for OCT1 and OCT2 because histamine is a poorer substrate for these transporters.
We also determined the interaction of picoplatin with Ctr1, MATE1, and MATE2K. Ctr1 has been previously shown to be responsible for cisplatin accumulation and cytotoxicity. Furthermore, cisplatin and oxaliplatin have been indicated to be good substrates for MATE1 and MATE2K, respectively (18, 19). All of these transporters, in our in vitro experiments, failed to show any significant contribution to picoplatin cytotoxicity (Supplementary Table S3). Our mechanistic studies involving DNA unwinding and transcription inhibition studies have shown a slightly reduced DNA binding potency of picoplatin over cisplatin but with significantly reduced glutathione reactivity (data not shown). Cisplatin reacts with cellular thiols glutathione and metallothionine, which limit the amount of drug available to bind to its biological target, DNA. This reactivity is considered to be one of the mechanisms of resistance to cisplatin therapy. Improved safety profile of picoplatin over cisplatin, along with its reduced reactivity toward glutathione, although compromised DNA reactivity, makes it a promising clinical candidate.
Picoplatin has shown activity in a variety of solid tumors, including lung, ovarian, colorectal, and hormone-refractory prostrate cancers. Clinical data show its activity in platinum-sensitive, platinum-resistant, and platinum-refractory disease. Results of phase II clinical trials of picoplatin have shown survival benefit in ovarian, NSCLC, and SCLC patients (20). The chemotherapeutic treatment of NSCLC by platinum agents has been hampered by rapid development of resistance (21). Consideration of picoplatin as a potential treatment for NSCLC led us to determine the role of OCT-mediated uptake in its lung cancer efficacy. The expression of OCT1 and OCT2 was detectable in normal and tumor lung tissue samples and lung cancer cell lines, with the expression of OCT2 being the lowest (Fig. 3; Supplementary Table S1). Our Western blot studies showed that OCT1 was also present in lung cancer cell lines. Reduction in cytotoxic potency of picoplatin but not that of cisplatin and carboplatin, in the presence of an OCT inhibitor in the lung cancer cell lines, corroborates the involvement of OCTs in picoplatin lung cancer efficacy. Cisplatin is effectively transported by OCT2, but its negligible expression in the lung cancer cell lines tested resulted in only minimal inhibition of its cytotoxicity.
OCT1 and OCT2 genes are highly polymorphic, and the reduced/nonfunctional polymorphic variants of OCT1 and OCT2 identified in our previous studies (13, 15) had significant effect on picoplatin uptake and DNA adduct formation. In particular, the common OCT1 variants 420D, G465R, and R61C showed significant reduction in cellular accumulation and DNA adduct formation of picoplatin when compared with the reference OCT1 transporter. The common OCT2 variant, A270S, exhibited increased uptake of picoplatin compared with the reference OCT2 transporter. Similar results were obtained previously in our laboratory with metformin (13, 22) for interaction with the same variants of OCT1 and OCT2, respectively, when compared with the reference transporter. All the variants of OCTs tested had similar levels of mRNA expression (13, 22). However, the levels of the proteins on the plasma membranes from some of the variants of OCT1, including R61C, are reduced presumably due to poor trafficking or altered stability of the transporter, thus leading to functional differences. Higher capacity of OCT2 variant A270S to transport metformin was alluded to be due to higher membrane protein expression compared with the reference OCT2 transporter, which might be the case with picoplatin (22). However, data from our laboratory are not consistent with recent data (23) and may be related to differences in the OCT2 cell lines being used between laboratories. The presence of OCT1 in lung tumor samples at high levels and the highly polymorphic nature of this gene led us to look further into its effect on picoplatin pharmacokinetics and in vivo tumor efficacy.
In vivo pharmacokinetic experiments did not reveal any statistically significant differences in the pharmacokinetic profiles of picoplatin in Oct1−/− and Oct1/2−/− mice; however, the knockout mice exhibited a trend toward higher mean plasma concentration when compared with the wild-type mice (Supplementary Table S2; Supplementary Fig. S2). The tissue distribution studies suggest a role of OCTs in platinum uptake in the liver, kidney, and heart (Supplementary Fig. S3). These three tissues express substantial levels of OCT1 (liver), OCT2 (kidney), and OCT3 (heart). Our findings that picoplatin accumulation is highest in liver, kidney, and heart are consistent with the high expression levels of OCTs in the three organs. However, it is possible that species differences in the interaction of picoplatin between mouse and human orthologs of OCT1 may exist and that the effect of OCT1 on picoplatin pharmacokinetics may be more pronounced in humans. Further, we observed compensatory upregulation of OCT2 and OCT3 in the liver and kidney of Oct1−/− mice (Supplementary Fig. S4), which may have obscured the effects of OCT1 on the tissue distribution and pharmacokinetics of picoplatin. Due to similar compensatory upregulation mechanisms pertaining to OCT2 (2- to 2.5-fold) and OCT3 (1.4- to 1.7-fold), our in vitro experiments in HCT-116 cell line using small interfering RNA (siRNA) against OCT1 failed to yield a conclusive result. However, the combination of OCT1-siRNA with an OCT inhibitor, cimetidine, and/or OCT2-siRNA indeed resulted in □ 40% reduction in picoplatin uptake (data not shown).
To establish the link between OCT1 expression and antitumor efficacy of picoplatin, we prepared xenografts of HEK293 cells stably transfected with hOCT1 and the corresponding empty vector. At initial time points of 5 and 8 days, picoplatin treatment was associated with a statistically significant reduction in the volume of OCT1 xenografts compared with control saline-treated xenografts (Fig. 6), which was not observed in the case of the empty vector xenografts. Similar results were obtained with oxaliplatin, a well-established substrate of OCT1. Although this trend was evident also on days 14 and 18 of picoplatin treatment initiation, it did not reach statistical significance at those time points, probably due in part to a reduction in OCT1 expression in the HEK-hOCT1 xenografts (data not shown). However, there was a trend toward reduction in tumor volume by picoplatin in the HEK-EV xenografts, suggesting that OCT1 contributes significantly to, but is not essential for, the antitumor efficacy of picoplatin.
The results of our in vitro studies and preliminary in vivo experiments highlight the significance of OCTs in picoplatin antitumor efficacy. A phase III clinical trial of picoplatin is currently under way for lung cancer. The variable expression of OCTs in tumors along with their genetic variants could contribute to any observed variation in response to picoplatin treatment. Our study has provided a basis for consideration of OCTs as possible markers for picoplatin efficacy. Development of new platinum-based anticancer agents with modification in picoplatin structure would be an interesting avenue to explore to obtain a clinical candidate with improved safety and efficacy profiles. Our data suggest that consideration of picoplatin in the treatment of colorectal cancer may be justified, as well as consideration for other cancers that express OCTs. Our data suggest that OCTs could serve as biomarkers in tumors for susceptibility to picoplatin, thus justifying thorough experimentation pertaining to the evaluation of picoplatin against OCT-expressing tumors.
Supplementary Material
Figure S1. Cellular platinum accumulation after incubation with picoplatin in PB-Cl buffer or [Pt(2-Pic))(H2O)(Cl)]1+ and [Pt(2-Pic)(H2O)2]2+ in PB-SO4 buffer. HEK293 cells were incubated with picoplatin and its mono- and diaqua-complexes (10 μmol/L) in the corresponding phosphate buffer at 37 °C and 5% CO2 for 30 minutes. Data are represented as the mean ± SEM of three independent experiments.
{kind=link}
Figure S2. Time Profiles of Plasma Concentration of Picoplatin in OCT1−/−, OCT1/2-/1 and OCT WT Mice. Total plasma platinum concentration was plotted against time after a single intravenous injection of picoplatin (20 mg/kg) in mice (3 per group).
{kind=link}
Figure S3. Tissue accumulation of picoplatin in Oct1 wild type (black stripes) and knockout mice (white and black bars). The total platinum concentration in various tissues was determined 24 hours after a single intravenous bolus dose of picoplatin (20 mg/kg) in nine weeks old male mice (3 per group) and was normalized to plasma platinum concentration. Data are expressed as mean platinum concentration the SD.
{kind=link}
Figure S4.Expression levels of OCT2 and OCT3 in the liver and kidney of Oct1−/− mice. Total RNA was isolated from liver and kidney of wildtype and knockout OCT1 mice and mRNA expression levels of OCT2 and OCT3 were determined by qRT-PCR as described in Materials and Methods.
{kind=link}
Table S1. Expression levels of mRNA transcripts for OCT1 and OCT2 in 8 normal lung tissue samples (N), 8 lung tumor tissue samples (T) and in 4 lung cancer cell lines (corresponds to figure 3). The mRNA expression was quantitated by qRTPCR as described in Materials and Methods. The expression of OCT1 and OCT2 transcripts relative to the control gene, GAPDH, was expressed as the relative levels to the expression of OCT2 in a lung cancer cell line “H460”. Data are expressed as mean ± SD of three measurements (ND = Not Detected; below limit of quantitation).
{kind=link}
Table S2. Plasma pharmacokinetic analysis of total plasma platinum versus time curves following a single intravenous dose of picoplatin (20 mg/kg) in mice (3 per group). The pharmacokinetic parameters were obtained by two compartmental analysis using WinNonlin 4.0. AUC = area under the curve; CL = clearance; Vss = steady state volume of distribution; T½ = elimination half life; MRT = mean residence time.
{kind=link}
Table S3. Effect of Ctr1 and MATE expression on cytotoxicity of picoplatin.
{kind=link}
Acknowledgments
Grant Support: Tobacco-Related Disease Research Program (TRDRP) grant 17RT-0126, a grant from the NIH, GM61390 and the UCSF Helen Diller Family Comprehensive Cancer Center Preclinical Therapeutics Core.
Footnotes
*Note: Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Zeller JL, Lynm C, Glass RM. Lung cancer. JAMA. 2007;297:1022. doi: 10.1001/jama.297.9.1022. [DOI] [PubMed] [Google Scholar]
- 2.Gkiozos I, Charpidou A, Syrigos K. Developments in the treatment of non-small cell lung cancer. Anticancer Res. 2007;27:2823–7. [PubMed] [Google Scholar]
- 3.Cosaert J, Quoix E. Platinum Drugs In The Treatment Of Non-Small-Cell Lung Cancer. Br J Cancer. 2002;87:825–33. doi: 10.1038/sj.bjc.6600540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 5.Bunn PA., Jr. Platinums in lung cancer: sufficient or necessary? J Clin Oncol. 2005;23:2882–3. doi: 10.1200/JCO.2005.11.930. [DOI] [PubMed] [Google Scholar]
- 6.Shah N, Dizon DS. New-generation platinum agents for solid tumors. Future Oncol. 2009;5:33–42. doi: 10.2217/14796694.5.1.33. [DOI] [PubMed] [Google Scholar]
- 7.Holford J, Sharp SY, Murrer BA, Abrams M, Kelland LR. In vitro circumvention of cisplatin resistance by the novel sterically hindered platinum complex AMD473. Br J Cancer. 1998;77:366–73. doi: 10.1038/bjc.1998.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song IS, Savaraj N, Siddik ZH, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol Cancer Ther. 2004;3:1543–9. [PubMed] [Google Scholar]
- 9.Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci. 2002;99:14298–302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Lovejoy KS, Shima JE, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–57. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoo S, Masuda S, Yonezawa A, Terada T, Katsura T, Inui K. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab Dispos. 2008;36:2299–306. doi: 10.1124/dmd.108.023168. [DOI] [PubMed] [Google Scholar]
- 12.Battle AR, Choi R, Hibbs DE, Hambley TW. Platinum(IV) analogues of AMD473 (cis-[PtCl2(NH3)(2-picoline)]): preparative, structural, and electrochemical studies. Inorg Chem. 2006;45:6317–22. doi: 10.1021/ic060273g. [DOI] [PubMed] [Google Scholar]
- 13.Shu Y, Sheardown SA, Brown C, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–31. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holford J, Raynaud F, Murrer BA, Grimaldi K, Hartley JA, Abrams M, Kelland LR. Chemical, biochemical and pharmacological activity of the novel sterically hindered platinum coordination complex, cis-[amminedichloro(2-methylpyridine)]platinum(II) (AMD473) Anticancer Drug Des. 1998;13:1–18. [PubMed] [Google Scholar]
- 15.Chen Y, Zhang S, Sorani M, Giacomini KM. Transport of paraquat by human organic cation transporters and multidrug and toxic compound extrusion family. J Pharmacol Exp Ther. 2007;322:695–700. doi: 10.1124/jpet.107.123554. [DOI] [PubMed] [Google Scholar]
- 16.Cheng JD, Valianou M, Canutescu AA, et al. Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol Cancer Ther. 2005;4:351–60. doi: 10.1158/1535-7163.MCT-04-0269. [DOI] [PubMed] [Google Scholar]
- 17.Tomilets VA, Dontsov VI, Zakharova IA, Klevtsov AV. Histamine releasing and histamine binding action of platinum and palladium compounds. Arch Immunol Ther Exp (Warsz) 1980;28:953–7. [PubMed] [Google Scholar]
- 18.Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family) J Pharmacol Exp Ther. 2006;319:879–86. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- 19.Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74:477–87. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Eckardt JR, Bentsion DL, Lipatov ON, et al. Phase II study of picoplatin as second-line therapy for patients with small-cell lung cancer. J Clin Oncol. 2009;27:2046–51. doi: 10.1200/JCO.2008.19.3235. [DOI] [PubMed] [Google Scholar]
- 21.d’Amato TA, Landreneau RJ, Ricketts W, et al. Chemotherapy resistance and oncogene expression in non-small cell lung cancer. J Thorac Cardiovasc Surg. 2007;133:352–63. doi: 10.1016/j.jtcvs.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Li S, Brown C, et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics. 2009;19:497–504. doi: 10.1097/FPC.0b013e32832cc7e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zolk O, Solbach TF, König J, Fromm MF. Functional characterization of the human organic cation transporter 2 variant p.270Ala>Ser. Drug Metab Dispos. 2009;37:1312–18. doi: 10.1124/dmd.108.023762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cellular platinum accumulation after incubation with picoplatin in PB-Cl buffer or [Pt(2-Pic))(H2O)(Cl)]1+ and [Pt(2-Pic)(H2O)2]2+ in PB-SO4 buffer. HEK293 cells were incubated with picoplatin and its mono- and diaqua-complexes (10 μmol/L) in the corresponding phosphate buffer at 37 °C and 5% CO2 for 30 minutes. Data are represented as the mean ± SEM of three independent experiments.
Figure S2. Time Profiles of Plasma Concentration of Picoplatin in OCT1−/−, OCT1/2-/1 and OCT WT Mice. Total plasma platinum concentration was plotted against time after a single intravenous injection of picoplatin (20 mg/kg) in mice (3 per group).
Figure S3. Tissue accumulation of picoplatin in Oct1 wild type (black stripes) and knockout mice (white and black bars). The total platinum concentration in various tissues was determined 24 hours after a single intravenous bolus dose of picoplatin (20 mg/kg) in nine weeks old male mice (3 per group) and was normalized to plasma platinum concentration. Data are expressed as mean platinum concentration the SD.
Figure S4.Expression levels of OCT2 and OCT3 in the liver and kidney of Oct1−/− mice. Total RNA was isolated from liver and kidney of wildtype and knockout OCT1 mice and mRNA expression levels of OCT2 and OCT3 were determined by qRT-PCR as described in Materials and Methods.
Table S1. Expression levels of mRNA transcripts for OCT1 and OCT2 in 8 normal lung tissue samples (N), 8 lung tumor tissue samples (T) and in 4 lung cancer cell lines (corresponds to figure 3). The mRNA expression was quantitated by qRTPCR as described in Materials and Methods. The expression of OCT1 and OCT2 transcripts relative to the control gene, GAPDH, was expressed as the relative levels to the expression of OCT2 in a lung cancer cell line “H460”. Data are expressed as mean ± SD of three measurements (ND = Not Detected; below limit of quantitation).
Table S2. Plasma pharmacokinetic analysis of total plasma platinum versus time curves following a single intravenous dose of picoplatin (20 mg/kg) in mice (3 per group). The pharmacokinetic parameters were obtained by two compartmental analysis using WinNonlin 4.0. AUC = area under the curve; CL = clearance; Vss = steady state volume of distribution; T½ = elimination half life; MRT = mean residence time.
Table S3. Effect of Ctr1 and MATE expression on cytotoxicity of picoplatin.