

Abstract
Mitochondrial dysfunction plays a primary role in the pathogenesis of Parkinson's disease (PD), particularly in autosomal recessive forms of the disease caused by mutations encoding PINK1. Although mitochondrial pathology can be demonstrated in many cell types, it is neurons that bear the brunt of cell death in PD. We studied the mitochondrial physiology of neurons and muscle cells with loss of function of the nuclear encoded mitochondrial protein PINK1. PINK1 is widely expressed in many types of tissues, but deficiency selectively induces death in neurons. We report here that the same genetic defect results in opposing phenotypes in different cell types, depending on the metabolic properties of the cell. Thus, PINK1-deficient myocytes exhibit high basal mitochondrial membrane potential (Δψm), whereas PINK1-deficient neurons have been shown to exhibit a low Δψm. PINK1 deficiency induces impaired respiration in both cell types, with a concomitant increase in glycolytic activity. We demonstrate that the high glycolytic capacity in myocytes compared with neurons enables them to produce more ATP and, therefore, compensates for the metabolic defects induced by PINK1 deficiency. Furthermore, the high Δψm generated in PINK1 knockout (KO) muscle mitochondria enables them to buffer cytosolic Ca2+ fluxes, rendering them resistant to Ca2+ stress effectively. Conversely, PINK1 KO neurons were previously shown to develop mitochondrial Ca2+ overload and Ca2+-induced mitochondrial depolarisation. Prevention of Ca2+ dysregulation in myocytes might therefore account for the sparing of these cells in PD.
Key words: ATP, PINK1, Mitochondria, Myocyte, Neuron
Introduction
Parkinson's disease (PD) is one of the most common neurodegenerative diseases and is characterised pathologically by loss of dopaminergic neurons in the substantia nigra and the formation of Lewy bodies. Mitochondrial dysfunction is associated with a range of neurodegenerative diseases, and in particular PD (Bueler, 2009). The identification of mutations in genes that cause familial recessive PD (such as DJ-1, PINK1 and parkin) has improved our understanding of the mitochondrial pathophysiology that might occur in both mendelian and sporadic forms of the disease.
One of the major conundrums of mendelian PD remains that the genetic defect is found in all cell types, and the affected proteins are often expressed in all tissues, but it is neurons that are uniquely susceptible to cell death in the human disease. Similarly, in sporadic PD, mitochondrial dysfunction is strongly implicated in the pathogenesis, and mitochondrial abnormalities have been demonstrated in cells other than neurons – for example, reduced complex 1 activity in muscle cells and platelets of patients with PD. However, tissue types that are heavily reliant on mitochondrial function, such as muscle, are typically spared in neurodegenerative disease, but are frequently affected in primary mitochondrial disease, where myopathy is common. In order to investigate the selective vulnerability of neurons compared with myocytes, we used a mendelian PD model, in which mutations in the PINK1 gene result in loss of function of a nuclear-encoded mitochondrial protein. We compared the effects of PINK1 deficiency on cell metabolism and mitochondrial function in two different types of primary PINK1 knockout (KO) mouse cells: neurons and skeletal myocytes.
Mutations in the PINK1 gene have been shown to cause autosomal recessive PD (Valente et al., 2004). PINK1 is a putative serine/threonine-protein kinase, homologous to the kinases of the Ca2+/calmodulin family. The PINK1 protein is predicted to contain a mitochondrial targeting motif and has been shown to be localised to mitochondria (Gandhi et al., 2006; Muqit et al., 2006), although a cytosolic pool of PINK1 has also been described (Beilina et al., 2005; Haque et al., 2008; Lin and Kang, 2008; Weihofen et al., 2008). PINK1 is cleaved by the mitochondrial protease PARL, and this cleavage is important for the function of both PINK1 and mitochondria (Deas et al., 2011; Jin et al., 2010). There are many putative roles for PINK1 in mitochondrial function. A number of studies have demonstrated that PINK1 deficiency results in decreased Δψm, and increased reactive oxygen production and mitochondrial Ca2+ mishandling in mammalian cells (Exner et al., 2007; Piccoli et al., 2008; Wood-Kaczmar et al., 2008; Gegg et al., 2009; Yuan et al., 2010; Gandhi et al., 2009). It has also been suggested that PINK1 affects mitochondrial function by regulating mitochondrial fission and fusion in both mammalian cells and Drosophila (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008; Dagda et al., 2009; Lutz et al., 2009; Cui et al., 2010; Kamp et al., 2010). More recently, several studies have demonstrated that PINK1 can play an important role in recruiting parkin to damaged mitochondria, thus regulating the degradation of mitochondria through autophagy (Geisler et al., 2010; Kawajiri et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010; Ziviani et al., 2010).
In this study, we demonstrate that loss of PINK1 function increased basal Δψm in skeletal myocytes, as opposed to previously reported decreased basal Δψm in neurons. We show that this opposite response in Δψm is due to the difference in ATP metabolism between neurons and myoctyes. Furthermore, the difference in basal Δψm results in differential effects on mitochondrial Ca2+ buffering capacity and on cell survival. Thus, the differential ability of the metabolic environment of muscle cells and neurons to compensate for PINK1 deficiency results in a different phenotype and different susceptibility to cell death in these cell types.
Results
Mitochondrial membrane potential is increased in PINK1 KO myocytes and decreased in PINK1 KO neurons and fibroblasts
PINK1 KO skeletal myocytes exhibited a significant increase in basal Δψm by 98.7±40.5% compared with that of wild-type (WT) myocytes (Fig. 1A). By contrast, the basal Δψm is reduced in primary PINK1 KO midbrain neurons – tetramethyl rhodamine methyl ester (TMRM) fluorescence for PINK1 KO neurons is 63.7±4.2% of WT neurons (Fig. 1A). Similarly in fibroblasts, TMRM fluorescence was reduced for PINK1 KO fibroblasts to 63.4±7.3% compared with that in WT. These data indicate that PINK1 deficiency is associated with the alteration of the basal Δψm, but the direction of this effect varies in different cell types. To further understand the effects of PINK1 KO on the mechanism of maintenance of the Δψm, we assessed the response of Δψm to a range of mitochondrial inhibitors in primary neuronal and myocyte cultures.
Fig. 1.

Δψm in PINK1 KO skeletal myocytes, fibroblasts and neurons. (A) Δψm was measured using TMRM in primary neuronal cultures and skeletal muscle and fibroblast cultures. Δψm was reduced in PINK1 KO neurons and fibroblasts, but increased in PINK1 KO myocytes compared with the WT. A summary of mean values normalised to WT (±s.e.m.) is shown: myocytes, 198.7±40.5%; neurons, 63.7±4.2%; fibroblasts, 63.4±7.3%, *P<0.05. (B) Representative traces showing dynamic response of Δψm in response to oligomycin, rotenone and carbonyl cyanide-p-(trifluoromethoxy) phenylhydrazone (FCCP). In WT myocytes, oligomycin did not affect Δψm; rotenone induced partial depolarization; FCCP induced complete depolarization. In PINK1 KO myocytes, oligomycin induced mitochondrial depolarization. (C) Application of 5 mM pyruvate or 5 mM methyl succinate to PINK1 KO myocytes increased basal Δψm and prevented oligomycin-induced mitochondrial depolarization in PINK KO cells. Error bars represent mean + s.e.m.
Oxidative respiration is impaired in PINK1 KO myocytes
Application of the F1Fo-ATPase inhibitor oligomycin (2 μg/ml) caused marked mitochondrial depolarization in PINK1 KO myocytes (Δψm dropped by 57.9±5.0%, Fig. 1B), indicating that the Δψm in PINK1 KO myocytes must be partially maintained by the hydrolysis of ATP by the F1Fo-ATPase, rather than solely by respiration. By contrast, application of oligomycin in WT myocytes did not alter the Δψm, indicating that the Δψm is usually maintained by respiration. We then exposed cells to respiratory chain substrates. Interestingly, even in conditions of high basal Δψm in PINK1 KO myocytes, application of 5 mM pyruvate or 5 mM methyl succinate induced a further increase in Δψm (Fig. 1C) and prevented the oligomycin-induced depolarisation described above. Therefore, the provision of pyruvate switched the mechanism of maintenance of Δψm to be dependent on the respiratory chain rather than on the hydrolysis of ATP. These results demonstrate that PINK1 KO myocytes, as well as PINK1 KO neurons (Gandhi et al., 2009) maintain their Δψm using the same mechanism of hydrolyisis of ATP when there is an insufficient substrate supply to the respiratory chain.
ATP levels are highest in muscles and lowest in midbrain
We investigated whether the difference in basal Δψm between myocytes and neurons might be attributable to a difference in ATP supply. We therefore measured the ATP concentration in midbrain and skeletal muscle tissues from PINK1 KO mice and WT littermates. As expected, the ATP concentration in skeletal muscle samples was substantially higher than in midbrain samples of the same mouse – WT midbrain: 41.8±11.4 nmoles/μg protein, WT muscle: 465.5±132.0 nmoles/μg protein, KO midbrain: 44.0±9.7 nmoles/μg protein, KO muscle: 835.5±216.2 nmoles/μg protein. The sample size was n=3 animal for each genotype (Fig. 2A).
Fig. 2.
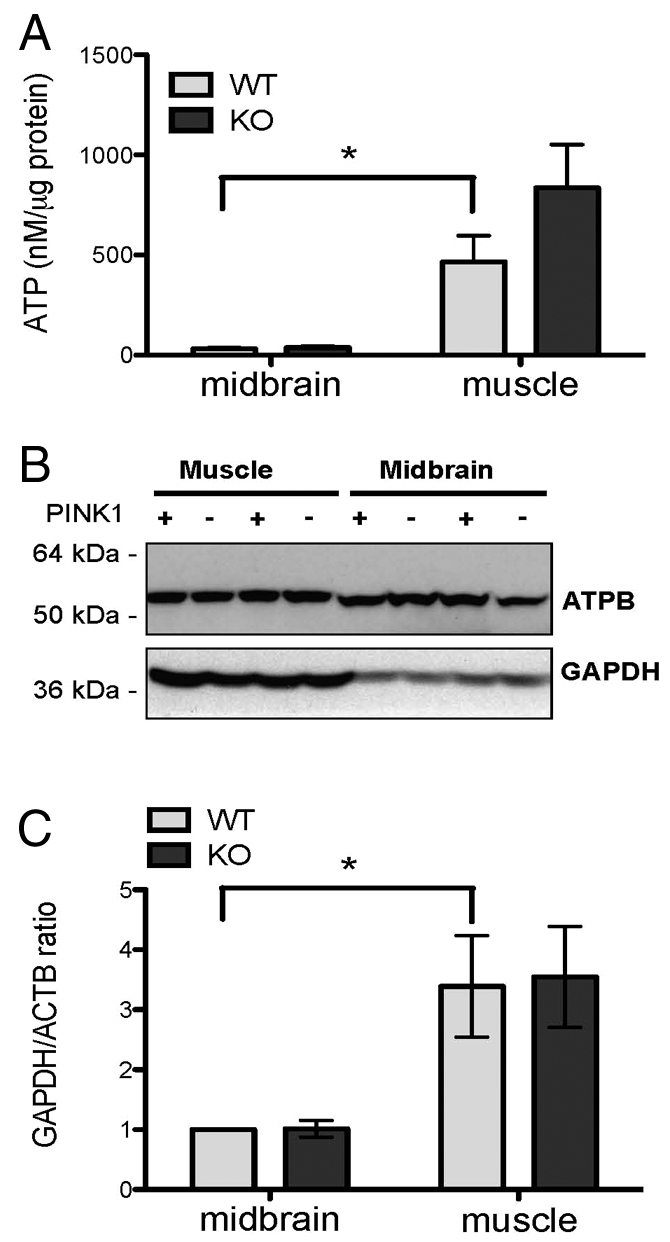
ATP concentration and glycolysis/oxidative phosphorylation ratio. (A) The level of ATP is higher in both PINK1 KO and WT skeletal muscle tissues compared with midbrain tissues from the same animal, *P<0.05. (B) Representative western blot demonstrating the expression of GAPDH and the β-F1-ATPase in midbrain and skeletal muscle tissues of PINK1 KO and WT mice. (C) Densitometry analysis showing the ratio between GAPDH and the β-ATPase. The ratio was higher in skeletal muscle tissues compared with midbrain, and there was no substantial difference between WT and KO tissues. KO midbrain, 1.0±0.1 fold of WT midbrain; WT cortex, 3.4±0.8 fold of WT midbrain; KO cortex, 3.5±0.8 fold of WT midbrain, n=six animals for each genotype, *P<0.05. Error bars represent mean ± s.e.m.
PINK1 KO myocytes exhibit a higher ratio of glycolysis:oxidative phosphorylation than PINK1 KO neurons
To investigate the major source of ATP production – that is, oxidative phosphorylation or glycolysis we assessed the expression of the β-subunit of the F1-ATPase (β-F1-ATPase) and the glycolytic enzyme GAPDH (glycolytic glyceraldehyde-3-phosphate dehydrogenase) in midbrain and skeletal muscle from 9–11-month-old PINK1 KO mice and WT littermates (Fig. 2B,C). The ratio between β-F1-ATPase and GAPDH demonstrates the ratio between oxidative phosphorylation and glycolysis and has been previously reported as a marker for alteration in bioenergetics in tumours (Cuezva et al., 2002). In WT and KO mice, the GAPDH:β-F1-ATPase ratio is 3.4±0.8 and 3.5±0.8 fold higher in the skeletal muscle compared with midbrain (n=6 animals for each genotype, Fig. 2C), confirming the existence of higher levels of glycolysis in muscle compared with midbrain. For each type of tissue, the GAPDH:β-F1-ATPase ratio is similar between WT and KO mice.
PINK1 KO triggers an increase in glycolysis in both myocytes and neurons
We assessed the metabolic rate of the cells by measuring the ATP depletion rate, in the presence of inhibitors of glycolysis and/or oxidative phosphorylation. The Mag-Fura-2 AM (hereafter referred to as Mag-Fura) fluorescent probe was used as an indicator of ATP consumption. The energy capacity of the cell is defined as the time between application of inhibitors of glycolysis and/or ATP-synthase (i.e. cessation of ATP production) to the time of cell lysis (i.e. energetic collapse due to total ATP depletion and inability to maintain Ca2+ homeostasis).
Inhibition of glycolysis by 1 μM iodoacetic acid (IAA), resulted in a progressive increase in cellular Mg2+ ([Mg2+]c) owing to ATP depletion, and eventually led to cell lysis (Fig. 3A).
Fig. 3.

ATP metabolism in PINK1 KO and WT neurons and myocytes. (A) Measurements of [Mg2+] indicate the rate of ATP depletion following inhibition of glycolysis. Both WT and PINK1 KO Mag-Fura loaded neurons were exposed to iodoacetic acid (IAA), inducing a slow progressive increase in [Mg2+] that culminated in cell lysis (abrupt increase of signal owing to cell ATP collapse). (B) Time to cell lysis and the rate of ATP depletion (measured by [Mg2+]) in neurons and myocytes in response to the inhibitor of glycolysis IAA. In PINK1 KO neurons compared with WT, there was a significant increase in ATP depletion rate and earlier cell lysis. There was no difference in either measurement in PINK1 KO and WT myocytes. (C) Time to cell lysis and ATP depletion rate in response to NaN3 and oligomycin. In PINK1 KO compared with WT, there was a decrease in ATP depletion rate and prolongation of the time to cell lysis. The effect was the same in both neurons and myocytes. (D) Time to cell lysis and ATP depletion rate in response to inhibition of ATP synthesis (both glycolysis and oxidative phosphorylation). In PINK1 KO neurons, there was an increase in ATP depletion rate and a decrease in time to cell lysis compared with that of the WT. There was no significant difference between PINK1 KO and WT myocytes. Error bars represent mean ± s.e.m; *P<0.05.
Inhibition of glycolysis in myocytes did not result in a significant difference in the ATP depletion rate or the energy capacity of the cell when comparing WT with PINK1 KO myocytes – the Mag-Fura 340 nm and 380 nm emission ratio (hereafter referred to as Mag-Fura 340:380 ratio) change per minute was measured as: WT myocytes 0.0053±0.00039, KO myocytes 0.0057±0.00046 (Fig. 3B). In contrast to myocytes, the rate of ATP depletion was substantially higher in PINK1 KO neurons – the Mag-Fura 340:380 ratio change per minute was: WT neurons 0.00060±0.00025, KO neurons 0.0023±0.00007 (Fig. 3B). Furthermore, the time for the cells to consume all their ATP (cell lysis time) was notably shorter in the PINK1 KO neurons compared with the WT: WT neurons 98.67±5.52 minutes, KO neurons 69.27±2.25 minutes (Fig. 3B). It is possible that the higher rate of ATP depletion and the reduced energy capacity observed in PINK1 KO neurons are attributable to the hydrolysis of ATP by F1Fo-ATPase in order to maintain the Δψm in neurons.
Next, we applied an inhibitor of oxidative phosphorylation, oligomycin (2 μg/ml), and an inhibitor of the respiratory chain, NaN3, and measured the ATP depletion rate and the energy capacity of the cell once again. Inhibition of oxidative phosphorylation in PINK1 KO neurons led to a substantial decrease in the ATP depletion rate. The Mag-Fura 340:380 ratio change per minute was: WT neurons 0.011±0.0025, KO neurons 0.0036±0.0002 (Fig. 3C). Furthermore, this was associated with an increase in the time to cell lysis of the KO compared with WT neurons: WT neurons 18.31±0.94 minutes, KO neurons 23.15±0.79 minutes (Fig. 3C). Similar effects were observed in myocytes. Inhibition of oxidative phosphorylation led to a decrease in the ATP depletion rate in PINK1 KO myocytes – Mag-Fura 340:380 ratio change per minute: WT myocytes 0.0031±0.00016, KO myocytes 0.0024±0.00013 (Fig. 3C), and an increase in cell lysis time compared with the WT: WT myocytes 207.5±2.54 minutes, KO myocytes 246.9±7.28 (Fig. 3C).
Taken together, these data suggest two features: first, PINK1-deficient cells have increased glycolysis to drive ATP production, and thus, inhibition of glycolysis results in faster ATP depletion and reduced energy capacity. Second, the major source of ATP consumption in PINK1 KO cells is the hydrolysis of ATP by the F1Fo-ATPase, and thus, inhibition of the ATPase results in a slowing of the ATP depletion rate and an increase in the energy capacity of the cell.
In order to estimate the overall ATP consumption in WT and PINK1 KO myocytes and neurons, we applied both inhibitors of glycolysis [iodoacetic acid (IAA), 20 μM] and F1Fo-ATP synthase (oligomycin, 2 μg/ml). As shown in Fig. 3C, there is no statistically significant difference between the rate of ATP consumption and cell lysis time in response to both inhibitors between PINK1 KO and WT myocytes. However, neurons with PINK1 KO demonstrated a higher rate of ATP consumption, suggesting a higher metabolic rate in PINK1 KO neurons compared with WT neurons (WT neurons: 0.0023±0.00021, KO neurons: 0.0029±0.00012, Fig. 3D).
Increased glycolysis induced by PINK1 KO can protect cells against ischaemia
In order to assess whether the increased glycolysis in PINK1 KO cells has an effect on cell survival, neurons and skeletal myocytes were exposed to 50 minutes ischaemia followed by 30 minutes reoxygenation (the time taken for the loading of the dyes). This ischaemic injury results in significant levels of cell death in WT neurons (40.3±3.0%, Fig. 4). Interestingly, the same ischaemic injury in PINK1 KO neuronal cultures led to only 23.8±3.3% (Fig. 4) cell death. In myocytes, ischaemia treatment also had a greater effect on WT compared with PINK1 KO cells (3.1±0.5% vs 5.1±0.8%) (Fig. 4), although this difference is not statistically significant.
Fig. 4.
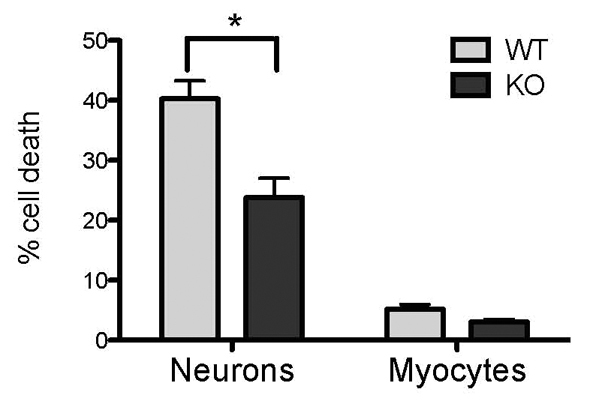
Cell death in response to ischaemic injury. PINK1 KO and WT neuron and myocyte cultures were exposed to 50 minutes of ischaemia with a normal concentration of glucose (10 mM) in the media. The percentage of cell death is reduced in PINK1 KO cells. WT neurons, 40.3±3.0%; KO neurons, 23.8±3.3%; WT myocytes, 5.1±0.8%; KO myocytes, 3.3±0.5%, *P<0.05. Error bars represent mean ± s.e.m.
It is well established that neurons are dependent on oxidative phosphorylation, and therefore, more susceptible to ischaemic injury. Here we demonstrate that the increase in glycolysis triggered by the PINK1 deficient state results in protection from hypoxic-ischaemic injury. Myocytes are less susceptible to such injury owing to their high glycolytic capacity.
The basal increase in Δψm protects PINK1 KO myocytes from Ca2+ stress
We investigated the effect of PINK1 deficiency on Ca2+ homeostasis in myocytes. We used Fura-2 AM (hereafter referred to as Fura-2) as an indicator of the concentration of cellular free Ca2+ ([Ca2+]c), and applied caffeine to initiate the ryanodine-receptor-operated Ca2+ signal (as a physiological calcium stimulus). Application of 10 mM caffeine induced a transient increase in Ca2+ in both WT and PINK1 KO myocytes (Fig. 5A,B). The Ca2+ signal in response to caffeine was in fact lower in the PINK1 KO myocytes compared with WT (75.7±6.1%, Fig. 5C). As Ca2+ uptake into mitochondria is determined partly by the electrochemical gradient of Ca2+, we hypothesise that the higher basal Δψm in PINK1 KO myocytes results in a higher mitochondrial buffering capacity for Ca2+ compared with neurons. Of note, the high [Ca2+]c signal induced by 10 mM caffeine did not lead to depolarization of the mitochondria owing to the premature opening of the permeability transition pore (PTP) (Fig. 5A,B). This is in marked contrast to the effect of physiological Ca2+ stimuli in PINK1 KO neurons, which induces mitochondrial Ca2+ overload, premature opening of the PTP and profound mitochondrial depolarisation (Gandhi et al., 2009).
Fig. 5.

Skeletal muscle cell response to calcium stimuli. (A,B) PINK1 KO and WT myocytes were loaded with 5 μM Fura-2 and 500 nM TMRM. 10 mM caffeine produced a rise in [Ca2+]c in both KO and WT cells, and this rise in [Ca2+]c is associated with a slight decrease in TMRM fluorescence, indicating hyperpolarisation of the Δψm. (C) The caffeine-induced rise in [Ca2+]c signal was reduced in PINK1 KO myocytes compared with the WT, suggesting higher mitochondrial Ca2+ uptake. Summary of mean values normalised to WT are shown. KO, 75.6±6.1%, *P<0.05. Error bars represent mean ± s.e.m.
These data strongly suggest that the metabolic environment created by PINK1 deficiency in myocytes, that is, high levels of glycolysis with increased ATP levels and higher basal Δψ results in a notable effect on the ability of muscle mitochondria to buffer Ca2+ compared with that of neuronal mitochondria. This, in turn, would be predicted to protect muscle cells from the Ca2+-induced PTP opening that leads to neuronal cell death.
Discussion
We have previously demonstrated that PINK1 deficiency in mouse and human neurons results in impaired Ca2+ efflux from the mitochondria via the mitochondrial Na+/Ca2+ exchanger, which causes Ca2+ accumulation inside the mitochondria (Gandhi et al., 2009). Mitochondrial Ca2+ overload stimulates reactive oxygen species (ROS) production, leading to inhibition of the glucose transporter and subsequent reduction of substrate supply. Reduction of substrate delivery results in decreased respiration and decreased basal Δψm, which can be restored by provision of substrates of mitochondrial respiratory chain complexes I and II. Both the decreased basal Δψm and altered Ca2+ homeostasis induced by PINK1 deficiency results in an increased vulnerability of neurons to open the permeability transition pore, followed by cell death in response to physiological Ca2+ signals.
PINK1 is present in many brain regions. Furthermore, PINK1 mRNA is expressed in all adult (human) tissues, with the most abundant levels of expression observed in the heart, skeletal muscle and testis, and intermediate expression observed in the liver, kidney, pancreas and brain (Unoki and Nakamura, 2001; Gandhi et al., 2006; Taymans et al., 2006). Thus, PINK1 deficiency can occur in all human tissues, but only neurons are dysfunctional, and only selective subgroups of neurons die in PINK1-associated PD. The reason for this selectivity of cell type is unclear, as the genetic and molecular defect occurs in all cells expressing PINK1. Interestingly, other cells that are rich in mitochondria, such as myocytes, do not undergo cell death in mammalian models (e.g. human disease and transgenic mice). We note that the PINK1-deficient Drosophila models do exhibit significant muscle mitochondrial pathology, with much-less-dramatic neuronal loss (Yang et al., 2006). This can be attributed to differences in the rate of energetic metabolism in Drosophila cells compared with mammalian cells (Noma, 2005).
It is well established that PINK1 deficiency leads to decreased Δψm in mammalian neurons as well as some non-neuronal cells, suggesting a possible widespread mitochondrial phenotype induced by PINK1 deficiency. For example, PINK1 deficiency resulted in an ~16% decrease of Δψm in human neurons and a >40% decrease of Δψm in human neuroblastoma cell lines (Gandhi et al., 2006; Sandebring et al., 2009). In fibroblasts taken from PD patients harbouring the V170G mutation, the Δψm is also ~20% lower compared with that of control cells (Grunewald et al., 2009). In this study, we have demonstrated a cell-specific effect of PINK1 deficiency on the basal Δψm. PINK1 deficiency led to a decreased Δψm in neurons and fibroblasts, which is consistent with previous reports. In skeletal myocytes, however, the basal Δψm is substantially higher when PINK1 is absent. An increased basal Δψm has not been demonstrated previously in PINK1 loss of function, and also not in any PD models, and has major implications on the function of mitochondria in muscle cells compared with neurons.
When cellular respiration is compromised and is insufficient to maintain the Δψm, the F1Fo-ATP synthase can switch from producing ATP to consuming ATP to maintain the Δψm (Campanella et al., 2008). This reverse action of F1Fo-ATP synthase as an ATPase has been observed previously in models of cellular stress, such as ischaemia. In our previous study we showed that, in PINK1 KO neurons, the Δψm is partially maintained by the F1Fo-ATP synthase working in reverse as a proton-motive ATPase. The decrease in basal Δψm in PINK1 KO neurons is due to reduced substrate supply, and can be restored by the provision of mitochondrial complex I and II substrates (Gandhi et al., 2009). In this study, we found that PINK1 KO skeletal myocytes utilise the same mechanism for maintaining their mitochondrial membrane potential as PINK1 KO neurons – that is, the Δψm is partially maintained by hydrolysis of ATP in the F1Fo-ATPase, rather than only by respiration. Furthermore, this mechanism can be reversed by the provision of complex I substrates. As the mechanism of maintaining the Δψm is the same in both neurons and myocytes, this raises the question of why PINK1 deficiency leads to such a different outcome in the basal mitochondrial potential of these cell types.
We propose that the difference in the Δψm is due to the difference in ATP levels in these different cell types, and we have confirmed here that the ATP level is substantially higher in skeletal muscle compared with brain. We have also confirmed that the source of ATP production arises from increased glycolysis in myocytes compared with neurons. We have shown that PINK1 deficiency in neurons and myocytes promotes an increase in glycolysis, presumably in order to compensate for the impairment in mitochondrial respiration. In fact, this increase in glycolytic activity in neurons is sufficient to protect neurons from ischaemia-induced cell death. However, there is an inherent difference in the glycolytic capacity between neurons and myocytes, and it is well established that the skeletal myocytes have a much higher glycolytic capacity compared with that of neurons (Bolanos et al., 2010). Thus, their compensatory mechanism for the impairment in respiration induced by PINK1 deficiency is highly effective, resulting in higher levels of ATP. This leads to increased activity of the ATPase in order to maintain a higher basal Δψm than that of neurons.
It has been suggested in the literature that, under stress conditions, in which oxidative phosphorylation is impaired, neurons are unable to invoke glycolysis to maintain ATP production (Almeida et al., 2001; Herrero-Mendez et al., 2009), which is why they are more sensitive to stress. However, those experiments inhibited oxidative phosphorylation by the application of inhibitors. This acute treatment might not allow the neurons enough time to compensate for impaired respiration through increased glycolysis. In the case of PINK1 KO, neurons are exposed to prolonged impairment of oxidative phosphorylation and therefore, have sufficient time to increase their glycolytic activity. Of note, however, although the elevated glycolysis might provide more energy to the neurons, it might also lead to increased oxidative stress. A recent study has shown that the activation of glycolysis in neurons by overexpression of Pfkfb3 (a key regulator of glyolysis) is accompanied by increased oxidative stress and apoptosis. This was because of a marked decrease in the oxidation of glucose through the pentose phosphate pathway – a metabolic route involved in the regeneration of reduced glutathione (Herrero-Mendez et al., 2009). Therefore, the PINK1 KO-induced increase in glycolysis might also contribute to the increased oxidative stress observed in neuronal models.
Aside from generating energy through ATP production, one of the main functions of mitochondria is maintaining Ca2+ homeostasis. Dysfunction of Ca2+ homeostasis has been implicated in a number of neurodegenerative diseases, including Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis and PD (Abramov et al., 2004; Mattson, 2007). Rapid uptake of cytosolic Ca2+ by mitochondria is crucial for the survival of neurons and muscle cells, as both of them are exposed to frequent and large influxes of cytosolic Ca2+, which must be buffered by the mitochondria (Chan et al., 2007). Recent studies have shown that neurons lacking PINK1 have a reduced mitochondrial Ca2+ buffering capacity, owing to increased basal mitochondrial free Ca2+ concentration ([Ca2+]m) and decreased Δψm (Gandhi et al., 2009). Here we demonstrated that, in contrast to neurons, the absence of PINK1 in skeletal myocytes in fact resulted in an increased mitochondrial Ca2+ buffering capacity. This can, in part, be explained by the high Δψm in PINK1 KO myocytes, enabling Ca2+ to be taken up into the mitochondria. Furthermore, in PINK1 KO neurons, stimuli that induce a [Ca2+]c signal can lead to extensive mitochondrial depolarization and ultimately, cell death (Gandhi et al., 2009). This is particularly problematic in dopaminergic neurons, which utilise a Ca2+ pacing mechanism and are, therefore, exposed to high cytosolic Ca2+ fluxes. However, in skeletal myocytes, we have shown that stimulation of a physiological [Ca2+]c signal did not result in any mitochondrial depolarization. These results indicate that myocytes are able to overcome the Ca2+ deregulation induced by PINK1 deficiency. We propose that this is because myocytes are able to increase their glycolytic activity in order to generate ATP, thereby maintaining a higher basal Δψm. Thus, in the face of PINK1 deficiency, myocytes generate a metabolic environment that does not sensitise them to the effects of Ca2+ toxicity or stress.
In summary, we have demonstrated that PINK1 KO leads to impaired mitochondrial respiration, not only in neurons, but also in other cells, such as skeletal myocytes. However, the response to mitochondrial impairment varies in different cell types, which might be because of a difference in metabolic capacity in the different cell types. Our data demonstrated that skeletal myocytes were able to maintain mitochondrial health, even when their oxidative phosphorylation was impaired owing to PINK1 deficiency, which could be a result of their capacity to generate more ATP through glycolysis. In the case of neurons, which have a relatively low glycolytic capacity compared with that of myocytes, the cell and mitochondrial function are heavily impaired, although glycolysis is upregulated in response to PINK1 KO (for a summary, see Fig. 6). These inherent differences between neurons and myocytes might help to explain the selective vulnerability of neurons in neurodegenerative disease, such as PINK1-induced PD. Further work in this area could help to explain why other mitochondria-rich cells, such as myocytes, are not affected by the genetic defect.
Fig. 6.

Scheme summarising differential effects of PINK1 deficiency in neurons and myocytes. See text for further details.
Materials and Methods
Animals
PINK1 KO mice were generated by Lexicon Genetics (The Woodlands, TX), and their details have been published elsewhere (Wood-Kaczmar et al., 2008). Animal husbandry and experimental procedures were performed in full compliance with the UK Animal (Scientific Procedures) Act of 1986.
Cell culture
Mixed cultures of midbrain neurons and glial cells were prepared as described previously (Vaarmann et al., 2010). Mice pups were obtained by crossing two heterozygote animals and comparing homozygote KO or WT animals within a litter. Cells at days 10–20 in vitro were used.
To prepare mixed cultures of skeletal myocytes and fibroblast cells, hind legs of postnatal day 2–5 pups were taken – skins and fat tissues were carefully removed. The tissues were then digested in 0.2% collagenase solution at 37°C for 30–40 minutes. The reaction was stopped by adding 2–3 ml fetal bovine serum (FBS), and the tissue suspension was centrifuged at 2010 rpm (700 g) for 10 minutes. The cell pellet was resuspended in pre-warmed growth medium [Dulbecco's modified Eagle's medium (DMEM), containing 20% FBS, 100 i.u./ml penicillin and 200 i.u./ml streptomycin], and plated on 22 mm coverslips that were pre-coated with 0.2% gelatin for 1 hour at 37°C. Cells were then cultured in a humidified CO2 incubator (5% CO2 in air) at 37°C. After 2 or 3 days, the medium was changed to a low-serum medium (DMEM containing 10% FBS without antibiotics), and was subsequently changed every 3 or 4 days. All experiments were performed in vitro between days 5 and 11 in culture.
Imaging mitochondrial membrane potential
For measurements of Δψm, we used tetramethyl rhodamine methyl ester (TMRM), [Invitrogen]. A reduction in TMRM fluorescence represents Δψm depolarisation. Neuronal cultures or myocyte cultures were loaded with 40 nM TMRM in a HEPES-buffered salt solution (HBSS) [composed of (mM): 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose and 10 HEPES; pH adjusted to 7.35 with NaOH] for 40 minutes at room temperature – the dye was present during the experiment. Z-stack confocal images were obtained using a Zeiss LSM 510 confocal microscope and a 40× oil-immersion objective. TMRM was excited using the 543 nm laser line and fluorescence measured using a 560 nm long-pass filter. TMRM fluorescence intensity was quantified by removing all background signals and measuring the mean TMRM fluorescence intensity in the pixels containing mitochondria – therefore, the signal is independent of mitochondrial mass.
For measurements of Ca2+-induced mitochondrial depolarisation, TMRM was again used in ‘redistribution mode’. In these experiments, the TMRM dye was present continuously at 500 nM and allowed to equilibrate. Mitochondrial depolarisation was then measured by the movement of dye from mitochondria into the cytosol – that is, by observing the increase of TMRM signal in the cytosol.
ATP concentration measurements
PINK1 KO mice and WT littermates were culled by cervical dislocation. Midbrain, cortex and skeletal muscle from the hind legs were dissected out and kept on ice. The tissues were homogenised in 0.1 M Tris-acetate buffer containing 2 mM EDTA (pH 7.75), and heated to 100°C for 90 seconds. ATP levels in the midbrain, cortex and skeletal muscle of the PINK1 KO mice and WT littermates were measured using the ATP Biolumiescent Assay Kit (Sigma). Standard curves were generated, and the amount of ATP in the samples was calculated according to the standard curve.
SDS-PAGE and immunoblot analysis
Brain and skeletal muscle tissues were obtained from PINK1 KO and WT littermates aged between 3 and 6 months. Tissues were lysed in 2× Laemmli buffer. SDS-PAGE and immunoblotting were performed as described previously (Wood-Kaczmar et al., 2008). Band densitometry was performed using Quantity One software (BioRad).
Measurements of [Ca2+]c
All fluorescent dyes were obtained from Invitrogen. Mag-Fura AM or Fura-2 AM (referred to as Mag-Fura and Fura-2, respectively) were used at 5 μM and loaded with 0.005% Pluronic P-123 for 30 minutes at room temperature followed by two washes with HBSS. Fluorescence measurements were obtained using an Olympus microscope with a CCD camera. Both Mag-Fura and Fura-2 were excited alternately at 340 nm and 380 nm. Emitted fluorescence was reflected through a 515 nm long-pass filter to a CCD camera. Images were analysed using Lucida software.
Indirect measurements of cellular ATP by MagFura-2
As both primary neuronal and myocyte cultures contain a mixture of cell types, including astrocytes and fibroblasts, kinetic measurements of ATP using luminescence of luciferin–luciferase from whole cultures are unable to provide information about ATP in specific cell types. Mg2+ is released from MgATP upon the hydrolysis of ATP (Leyssens et al., 1996), and therefore measurement of cellular free magnesium ([Mg2+]c) using the Mg2+-sensitive fluorescent probe Mag-Fura can be used as an indication of ATP consumption. Application of inhibitors of glycolysis and/or oxidative phosphorylation blocks ATP production in cells, which eventually leads to ATP depletion and subsequent Mg2+ release. This results in an increase in the Mag-Fura fluorescence. In addition to binding Mg2+, Mag-Fura dye is also used as a low-affinity Ca2+ indicator, enabling the energy capacity of the cell to be estimated. The energy capacity of the cell is defined as the time between application of inhibitors of glycolysis/ATP-synthase (i.e. cessation of ATP production) and the time of cell lysis (i.e. energetic collapse due to total ATP depletion and inability to maintain Ca2+ homeostasis) (Abramov et al., 2007).
Cell death analysis
Cells were incubated with 10 μM propidium iodide (PI) and 1 μg/μl Hoechst 33258 for 15 minutes and analysed using an Olympus microscope. Hoechst 33258 stains all nuclei, while PI stains only cells with a disrupted plasma membrane. Dead cells (PI positive) were counted and expressed as a percentage of total nuclei.
Statistical analysis
All data were obtained from at least three independent experiments. All statistical analyses were performed using a t-test. In all cases where WT cells were set at 100% (i.e. the mitochondrial membrane potential, GAPDH: ATPbeta ratio, and basal Ca2+ level), a one-sample t-test was performed.
Acknowledgements
The PINK1 KO mice were a kind gift from Julian Downward, Cancer Research UK.
Footnotes
Funding
This work was supported in part by the Wellcome Trust and UK Medical Research Council Joint Call in Neurodegeneration award [grant number WT089698] to the UK Parkinson's disease Consortium whose members are from the UCL Institute of Neurology, the University of Sheffield; and the MRC Protein Phosphorylation Unit at the University of Dundee. A.Y.A. is a Parkinson's UK Senior Research Fellow. Z.Y. is a NEUROTRAIN Early Stage Research Training fellow funded through the European Union research program FP6. S.G. was supported by a Wellcome Clinical Research Training Fellowship [grant number WT073531]. N.W.W. is supported by MRC programme grant [grant number G0400000]. Deposited in PMC for release after 6 months.
References
- Abramov A. Y., Canevari L., Duchen M. R. (2004). Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta. 1742, 81-87 [DOI] [PubMed] [Google Scholar]
- Abramov A. Y., Scorziello A., Duchen M. R. (2007). Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci 27, 1129-1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A., Almeida J., Bolanos J. P., Moncada S. (2001). Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc. Natl. Acad. Sci. USA 98, 15294-15299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A., Van Der Brug M., Ahmad R., Kesavapany S., Miller D. W., Petsko G. A., Cookson M. R. (2005). Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 102, 5703-5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos J. P., Almeida A., Moncada S. (2010). Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145-149 [DOI] [PubMed] [Google Scholar]
- Bueler H. (2009). Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease. Exp. Neurol. 218, 235-246 [DOI] [PubMed] [Google Scholar]
- Campanella M., Casswell E., Chong S., Farah Z., Wieckowski M. R., Abramov A. Y., Tinker A., Duchen M. R. (2008). Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 8, 13-25 [DOI] [PubMed] [Google Scholar]
- Chan C. S., Guzman J. N., Ilijic E., Mercer J. N., Rick C., Tkatch T., Meredith G. E., Surmeier D. J. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature 447, 1081-1086 [DOI] [PubMed] [Google Scholar]
- Cuezva J. M., Krajewska M., de Heredia M. L., Krajewski S., Santamaria G., Kim H., Zapata J. M., Marusawa H., Chamorro M., Reed J. C. (2002). The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 62, 6674-6681 [PubMed] [Google Scholar]
- Cui M., Tang X., Christian W. V., Yoon Y., Tieu K. (2010). Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 285, 11740-11752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda R. K., Cherra S. J., 3rd, Kulich S. M., Tandon A., Park D., Chu C. T. (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843-13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E., Plun-Favreau H., Gandhi S., Desmond H., Kjaer S., Loh S. H., Renton A. E., Harvey R. J., Whitworth A. J., Martins L. M., et al. (2011). PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867-879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Dodson M. W., Huang H., Guo M. (2008). The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 105, 14503-14508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner N., Treske B., Paquet D., Holmstrom K., Schiesling C., Gispert S., Carballo-Carbajal I., Berg D., Hoepken H. H., Gasser T., et al. (2007). Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 27, 12413-12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S., Muqit M. M., Stanyer L., Healy D. G., Abou-Sleiman P. M., Hargreaves I., Heales S., Ganguly M., Parsons L., Lees A. J., et al. (2006). PINK1 protein in normal human brain and Parkinson's disease. Brain 129, 1720-1731 [DOI] [PubMed] [Google Scholar]
- Gandhi S., Wood-Kaczmar A., Yao Z., Plun-Favreau H., Deas E., Klupsch K., Downward J., Latchman D. S., Tabrizi S. J., Wood N. W., et al. (2009). PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627-638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg M. E., Cooper J. M., Schapira A. H., Taanman J. W. (2009). Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 4, e4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S., Holmstrom K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119-131 [DOI] [PubMed] [Google Scholar]
- Grunewald A., Gegg M. E., Taanman J. W., King R. H., Kock N., Klein C., Schapira A. H. (2009). Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp. Neurol. 219, 266-273 [DOI] [PubMed] [Google Scholar]
- Haque M. E., Thomas K. J., D'Souza C., Callaghan S., Kitada T., Slack R. S., Fraser P., Cookson M. R., Tandon A., Park D. S. (2008). Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 105, 1716-1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Mendez A., Almeida A., Fernandez E., Maestre C., Moncada S., Bolanos J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747-752 [DOI] [PubMed] [Google Scholar]
- Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., Youle R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933-942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F., Exner N., Lutz A. K., Wender N., Hegermann J., Brunner B., Nuscher B., Bartels T., Giese A., Beyer K., et al. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571-3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawajiri S., Saiki S., Sato S., Sato F., Hatano T., Eguchi H., Hattori N. (2010). PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 584, 1073-1079 [DOI] [PubMed] [Google Scholar]
- Leyssens A., Nowicky A. V., Patterson L., Crompton M., Duchen M. R. (1996). The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J. Physiol. 496, 111-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Kang U. J. (2008). Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 106, 464-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz A. K., Exner N., Fett M. E., Schlehe J. S., Kloos K., Lammermann K., Brunner B., Kurz-Drexler A., Vogel F., Reichert A. S., et al. (2009). Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938-22951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (2007). Calcium and neurodegeneration. Aging Cell 6, 337-350 [DOI] [PubMed] [Google Scholar]
- Muqit M. M., Abou-Sleiman P. M., Saurin A. T., Harvey K., Gandhi S., Deas E., Eaton S., Payne Smith M. D., Venner K., Matilla A., et al. (2006). Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J. Neurochem. 98, 156-169 [DOI] [PubMed] [Google Scholar]
- Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma T. (2005). Dynamics of nucleotide metabolism as a supporter of life phenomena. J. Med. Invest. 52, 127-136 [DOI] [PubMed] [Google Scholar]
- Piccoli C., Sardanelli A., Scrima R., Ripoli M., Quarato G., D'Aprile A., Bellomo F., Scacco S., De Michele G., Filla A., et al. (2008). Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem. Res. 33, 2565-2574 [DOI] [PubMed] [Google Scholar]
- Poole A. C., Thomas R. E., Andrews L. A., McBride H. M., Whitworth A. J., Pallanck L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 105, 1638-1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandebring A., Thomas K. J., Beilina A., van der Brug M., Cleland M. M., Ahmad R., Miller D. W., Zambrano I., Cowburn R. F., Behbahani H., et al. (2009). Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE 4, e5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymans J. M., Van den Haute C., Baekelandt V. (2006). Distribution of PINK1 and LRRK2 in rat and mouse brain. J. Neurochem. 98, 951-961 [DOI] [PubMed] [Google Scholar]
- Unoki M., Nakamura Y. (2001). Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 20, 4457-4465 [DOI] [PubMed] [Google Scholar]
- Vaarmann A., Gandhi S., Gourine A. V., Abramov A. Y. (2010). Novel pathway for an old neurotransmitter: dopamine-induced neuronal calcium signalling via receptor-independent mechanisms. Cell Calcium 48, 176-182 [DOI] [PubMed] [Google Scholar]
- Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., et al. (2004). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158-1160 [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 107, 378-383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A., Ostaszewski B., Minami Y., Selkoe D. J. (2008). Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum. Mol. Genet. 17, 602-616 [DOI] [PubMed] [Google Scholar]
- Wood-Kaczmar A., Gandhi S., Yao Z., Abramov A. Y., Miljan E. A., Keen G., Stanyer L., Hargreaves I., Klupsch K., Deas E., et al. (2008). PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3, e2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J. W., Yang L., Beal M. F., Vogel H., Lu B. (2006). Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA 103, 10793-10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Ouyang Y., Yang L., Beal M. F., McQuibban A., Vogel H., Lu B. (2008). Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 105, 7070-7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X. L., Guo J. F., Shi Z. H., Xiao Z. Q., Yan X. X., Zhao B. L., Tang B. S. (2010). R492X mutation in PTEN-induced putative kinase 1 induced cellular mitochondrial dysfunction and oxidative stress. Brain Res. 1351, 229-237 [DOI] [PubMed] [Google Scholar]
- Ziviani E., Tao R. N., Whitworth A. J. (2010). Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 107, 5018-5023 [DOI] [PMC free article] [PubMed] [Google Scholar]