

Abstract
The JIL-1 kinase localizes specifically to euchromatin interband regions of polytene chromosomes and is the kinase responsible for histone H3S10 phosphorylation at interphase. Genetic interaction assays with strong JIL-1 hypomorphic loss-of-function alleles have demonstrated that the JIL-1 protein can counterbalance the effect of the major heterochromatin components on position-effect variegation (PEV) and gene silencing. However, it is unclear whether this was a causative effect of the epigenetic H3S10 phosphorylation mark, or whether the effect of the JIL-1 protein on PEV was in fact caused by other functions or structural features of the protein. By transgenically expressing various truncated versions of JIL-1, with or without kinase activity, and assessing their effect on PEV and heterochromatic spreading, we show that the gross perturbation of polytene chromosome morphology observed in JIL-1 null mutants is unrelated to gene silencing in PEV and is likely to occur as a result of faulty polytene chromosome alignment and/or organization, separate from epigenetic regulation of chromatin structure. Furthermore, the findings provide evidence that the epigenetic H3S10 phosphorylation mark itself is necessary for preventing the observed heterochromatic spreading independently of any structural contributions from the JIL-1 protein.
Key words: JIL-1 kinase, PEV, Heterochromatin, Gene silencing, Drosophila
Introduction
The JIL-1 kinase is a multidomain protein that localizes specifically to euchromatin interband regions of polytene chromosomes and is the kinase responsible for histone H3S10 phosphorylation at interphase (Jin et al., 1999; Wang et al., 2001). Mutational analyses have shown that the JIL-1 gene is essential for viability (Wang et al., 2001; Zhang et al., 2003) and that a reduction in JIL-1 kinase activity leads to a global disruption of polytene chromosome morphology (Wang et al., 2001; Deng et al., 2005). Furthermore, genetic interaction assays with JIL-1 hypomorphic and null allelic combinations demonstrated that the JIL-1 protein can counterbalance the effect of the three major heterochromatin components Su(var)3–9, Su(var)3–7 and Su(var)2–5 (HP1a) on position-effect variegation (PEV) (Deng et al., 2010; Wang et al., 2011). Based on these observations, it has been proposed that the epigenetic H3S10 phosphorylation mark functions to counteract heterochromatic spreading and gene silencing in Drosophila melanogaster (Ebert et al., 2004; Zhang et al., 2006; Deng et al., 2007; Deng et al., 2010). However, the previous experiments could not exclude the possibility that the effect of the JIL-1 protein on PEV was instead caused by the gross alterations of polytene chromosome morphology observed in the absence of JIL-1, or arose from structural contributions of the JIL-1 protein, independent of its H3S10 phosphorylation activity. In order to distinguish between these scenarios, we have cloned various full length and truncated versions of the JIL-1 protein into the pYES vector that contains a yellow selection marker (Patton et al., 1992), generated transgenic animals and assessed the effect of these lines on PEV of two different reporters – the chromosomal inversion (wm4) and the pericentric insertion line (118E-10). Specifically, we have taken advantage of the finding that the C-terminal domain (CTD) alone, which has no kinase activity, can restore partial viability and fully rescue the gross alteration in polytene chromosome morphology of JIL-1 null mutants (Bao et al., 2008). We show that expression of the CTD in a wild-type background displaces native JIL-1, reduces H3S10 phosphorylation dramatically, and phenocopies the effect of strong JIL-1 hypomorphic mutations on PEV. In addition, we provide evidence that expression of the CTD domain in a JIL-1 null mutant background enhanced the PEV of the 118E-10 allele, even when the overall polytene chromosome morphology was restored back to normal. By contrast, expression of a construct that lacks the CTD domain (ΔCTD), but that retains its ability to phosphorylate H3S10, strongly suppresses PEV of the 118E-10 allele. Taken together, these findings strongly support the hypothesis that the epigenetic H3S10 phosphorylation mark is necessary to counteract heterochromatic spreading and gene silencing.
Results
JIL-1 transgene expression
JIL-1 can be divided into four main domains, including an N-terminal domain (NTD), the first kinase domain (KDI), the second kinase domain (KDII) and a CTD (Jin et al., 1999) (Fig. 1A). To explore further the relative contributions of the different JIL-1 domains and the H3S10 phosphorylation mark to regulation of PEV, we expressed three CFP-tagged JIL-1 upstream activation sequence (UAS) P-element insertion constructs transgenically in wild-type and JIL-1 null mutant animals using the pYES vector (Patton et al., 1992). This vector contains a yellow selection marker to avoid any influence on eye pigmentation levels. A full-length (FL) construct, a construct without the C-terminal domain (ΔCTD) and a construct containing only the CTD were designed (Fig. 1A). All three constructs had properties identical to those previously reported for similar GFP- or CFP-tagged JIL-1 constructs (Wang et al., 2001; Bao et al., 2008). In addition, a transgenic line was selected for each construct that was expressed at levels comparable to those of endogenous JIL-1 by using a da-GAL4 driver line, as illustrated in Fig. 1B. The FL construct rescued all aspects of the JIL-1 null mutant phenotype, including polytene chromosome morphology and viability, and, like endogenous JIL-1, FL was upregulated on the male X chromosome (data not shown). The ΔCTD lacks the C-terminal sequences required for proper chromatin localization, leading to mislocalization of the protein (Bao et al., 2008). However, it does retain its kinase activity, resulting in ectopic histone H3S10 phosphorylation (Bao et al., 2008). Interestingly, the JIL-1Su(var)3-1 allele series generates truncated proteins with C-terminal deletions (Fig. 1A) that also mislocalize to ectopic chromatin sites (Zhang et al., 2006), giving rise to some of the strongest suppressor-of-variegation phenotypes to be described (Ebert et al., 2004; Lerach et al., 2006). The ΔCTD construct rescues autosome polytene chromosome morphology, but only partially rescues that of the male X chromosome in JIL-1 null mutants (Bao et al., 2008). By contrast, the CTD fully restores JIL-1 null mutant chromosome morphology, including that of the male X chromosome (Bao et al., 2008). Furthermore, when the CTD is expressed in a wild-type background, it has a dominant-negative effect and displaces endogenous JIL-1 (Bao et al., 2008), leading to a striking decrease in the levels of histone H3S10 phosphorylation, as shown in Fig. 1C.
Fig. 1.

Expression of JIL-1 constructs transgenically in a wild-type background. (A) Diagrams of the JIL-1 CFP-tagged constructs analyzed. The region in the CTD where JIL-1Su(var)3-1 alleles resulting in C-terminally truncated proteins have been mapped (Ebert et al., 2004) is indicated by a bracket. (B) Immunoblot labeled with JIL-1 antibody of protein extracts from wild-type (WT) and flies expressing the FL, the CTD and the ΔCTD constructs, respectively. Labeling with antibody against tubulin was used as a loading control. The relative migration of molecular size markers in kDa is indicated to the left of the immunoblot. (C) Immunoblot, labeled with antibody against phosphorylated H3S10 (H3S10ph) of protein extracts from salivary glands from WT third-instar larvae, from larvae expressing the FL, the CTD and the ΔCTD, respectively, and from JIL-1 null larvae (Z2). Labeling with antibody against histone H3 was used as a loading control.
The effect of CTD expression on regulation of PEV in a wild-type JIL-1 background
PEV in Drosophila occurs when euchromatic genes are transcriptionally silenced as a result of their placement in or near heterochromatin (reviewed in Girton and Johansen, 2008). Silencing typically occurs in only a subset of cells and can be heritable, leading to mosaic patterns of gene expression (Schotta et al., 2003; Delattre et al., 2004). PEV in Drosophila has served as a major paradigm for the identification and genetic analysis of evolutionarily conserved determinants of epigenetic regulation of chromatin structure (reviewed in Girton and Johansen, 2008; Schotta et al., 2003). In previous experiments, we have shown that combinations of strong JIL-1 hypomorphic loss-of-function mutations act as enhancers of PEV of transgenes inserted directly into pericentric heterochromatin (Bao et al., 2007). Furthermore, in the absence of JIL-1 kinase, the major heterochromatin marker H3K9me2 spreads to ectopic locations on the chromosome arms, with the most pronounced increase on the X chromosome (Zhang et al., 2006; Deng et al., 2007). These findings suggest a model for a dynamic balance between euchromatin and heterochromatin, where the boundary between these two chromatin types is regulated by the state of histone H3S10 phosphorylation (Ebert et al., 2004; Zhang et al., 2006; Deng et al., 2007; Deng et al., 2010). To determine whether the H3S10 phosphorylation mark itself was required to control this balance, we explored the effect of CTD expression on the regulation of PEV caused by both the P-element insertion of a reporter gene (118E-10) and a chromosome rearrangement (wm4).
Insertion of the P element [P(hsp26-pt, hsp70-w)] into euchromatic sites results in a uniform red-eye phenotype, whereas its insertion into a known heterochromatic region of the fourth chromosome (as is the case for line 118E-10) results in a variegating eye phenotype (Fig. 2A) (Wallrath and Elgin, 1995; Wallrath et al., 1996; Cryderman et al., 1998; Bao et al., 2007). We compared the eye pigment levels of flies homozygous for the transgenic reporter line 118E-10 in transgenic lines expressing the CTD or the ΔCTD, respectively. Pigment assays were essentially performed as in Kavi and Birchler (Kavi and Birchler, 2009) using three sets of ten pooled fly heads from each genotype. Both male and female flies were scored – however, owing to differences between the sexes, only the results from male flies are shown – nevertheless, the trend observed in female flies was identical to that of male flies. The expression of the CTD enhances PEV, as indicated by the increased proportion of white ommatidia and a 55% decrease in the optical density (OD) of the eye pigment levels (0.0168±0.0031, n=3) when compared with that of control flies (0.0370±0.0035, n=3) (Fig. 2A,B) – this reduction was statistically significant (P<0.002). By contrast, expression of the ΔCTD suppresses PEV, as indicated by an increase in the proportion of red ommatidia and a statistically significant (P<0.0001) 305% increase in the OD of the eye pigment levels (0.1130±0.0074, n=3). These opposing effects of the CTD and ΔCTD on PEV correlate with the finding that expression of the CTD depressed histone H3S10 phosphorylation, whereas levels of H3S10 phosphorylation were increased upon ΔCTD expression (Fig. 1C). Furthermore, the polytene squash preparations from larvae expressing the CTD (Fig. 3) showed that the heterochromatic H3K9me2 mark spread to the chromosome arms. Spreading on the X chromosome was especially pronounced in both males and females, as would be predicted by the model in the absence of H3S10 phosphorylation (Deng et al., 2007; Deng et al., 2010).
Fig. 2.

The effect on PEV of the 118E-10 allele by expression of the CTD or the ΔCTD. (A) Examples of the degree of PEV in the eyes of wild-type JIL-1 flies (cont), wild-type JIL-1 flies expressing the CTD and wild-type JIL-1 flies expressing the ΔCTD in a 118E-10/118E10 background. All images are from male flies. (B) Histograms showing the levels of eye pigment of wild-type JIL-1 flies (cont), wild-type JIL-1 flies expressing the CTD and wild-type JIL-1 flies expressing the ΔCTD in a male 118E-10/118E10 background. The average pigment level when the CTD or the ΔCTD was expressed was compared with the control level using a two-tailed Student's t-test.
Fig. 3.

The effect on H3K9me2 localization in polytene chromosomes expressing the CTD. The polytene squash preparations were labeled with antibody against H3K9me2 (in red) and with Hoechst (DNA, in blue or grey). The X chromosome is indicated by an X. Preparations from wild-type (control) and male and female larvae expressing the CTD are shown. In wild-type preparations, H3K9me2 labeling was mainly localized to and abundant at the chromocenter – however, when the CTD was expressed, the H3K9me2 labeling spread to the autosomes and particularly to the X chromosome in both males and females.
The In(1)wm4 X chromosome contains an inversion that juxtaposes the euchromatic white (w) gene and centric heterochromatic sequences distal to the nucleolus organizer (Muller, 1930; Pirrotta et al., 1983). The resulting somatic variegation of wm4 expression occurs in clonal patches in the eye, reflecting heterochromatic spreading from the inversion breakpoint that silences wm4 expression in the white patches and euchromatic packaging of the w gene in the red patches (reviewed in Grewal and Elgin, 2002). Studies of this effect suggest that the degree of spreading depends on the level of heterochromatic factors at the breakpoint (reviewed by Weiler and Wakimoto, 1995; Girton and Johansen, 2008). Interestingly, strong hypomorphic combinations of JIL-1 alleles, in which heterochromatic factors spread to ectopic locations (Zhang et al., 2006; Deng et al., 2007), act as suppressors not enhancers of PEV of the wm4 allele (Lerach et al., 2006). Based on these findings, Lerach and colleagues (Lerach et al., 2006) proposed a model whereby the suppression of PEV of wm4 in strong JIL-1 hypomorphic backgrounds occurs because of a reduction in the level of heterochromatic factors at the pericentromeric heterochromatin near the inversion breakpoint site, decreasing its potential for heterochromatic spreading and silencing. Thus, a prediction of this model is that expression of the CTD and the ΔCTD should both lead to suppression of PEV of wm4. To test this hypothesis, we expressed the CTD and the ΔCTD in wm4/Y flies. Fig. 4A,B illustrates that expression of the CTD suppressed PEV, as indicated by the increased proportion of red ommatidia and a 343% increase in the OD of the eye pigment levels (0.1223±0.0120, n=3) when compared with control flies (0.0357±0.0038, n=3) – this increase was statistically significant (P<0.0005). Expression of the ΔCTD also suppressed PEV, as indicated by an increase in the proportion of red ommatidia and a statistically significant (P<0.005) 348% (0.1243±0.0214, n=3) increase in the OD of the eye pigment levels, strongly supporting the hypothesis of Lerach and colleagues (Lerach et al., 2006).
Fig. 4.

The effect on PEV of the wm4 allele by expression of the CTD or the ΔCTD. (A) Examples of the degree of PEV in the eyes of wild-type JIL-1 flies (cont), wild-type JIL-1 flies expressing the CTD and wild-type JIL-1 flies expressing the ΔCTD in a wm4/Y background. (B) Histograms showing the levels of eye pigment of wild-type JIL-1 flies (cont), wild-type JIL-1 flies expressing the CTD and wild-type JIL-1 flies expressing the ΔCTD in a wm4/Y background. The average pigment level when the CTD or the ΔCTD was expressed was compared with the control level using a two-tailed Student's t-test.
The effect of CTD expression on regulation of PEV in a JIL-1 null background
The finding that CTD or ΔCTD expression can partially rescue the viability of JIL-1 null mutants allowed us to examine further the effect that expression of these constructs would have on PEV of the 118E-10 allele in the absence of endogenous JIL-1. First, we recombined the da-GAL4 driver onto the JIL-1z2 chromosome in order to generate ‘JIL-1 transgene’/+; JIL-1z2/ JIL-1z2 da-GAL4; 118E-10/+ flies. The JIL-1z2 allele is a true null allele, generated by P-element mobilization (Wang et al., 2001; Zhang et al., 2003). As illustrated in Fig. 5, when the FL was expressed within this genetic background, it led to a variegated eye phenotype with an eye pigment level OD of 0.0163±0.0007 (n=3). However, when the CTD was expressed, PEV was enhanced, as indicated by a decrease in the proportion of red ommatidia and a statistically significant (P<0.0001) 54% decrease in the OD (0.0075±0.0002, n=3) of eye pigment levels (Fig. 5A,B). By contrast, expression of the ΔCTD led to suppression of PEV, as indicated by an increase in the proportion of red ommatidia and a statistically significant (P<0.0001) 151% increase in the OD (0.0246±0.0018, n=3) of eye pigment levels when compared with the FL (Fig. 5A,B). Immunoblot analysis demonstrated that the expression levels of FL, CTD, and ΔCTD were comparable (Fig. 6A). Furthermore, while the FL JIL-1 transgenic protein could phosphorylate histone H3S10 to levels close to those of the wild-type, the ΔCTD transgenic protein phosphorylated H3S10 at enhanced levels. Conversely, there to levels no detectable levels of H3S10 phosphorylation when the CTD was expressed (Fig. 6B). Thus, the observed effects of expression of these constructs on PEV correlated with the degree to which the constructs phosphorylated histone H3S10.
Fig. 5.

The effect on PEV of the 118E-10 allele in JIL-1 null flies expressing the FL, the CTD or the ΔCTD. (A) Examples of the degree of PEV in the eyes of JIL-1z2/JIL-1z2 null flies expressing the FL, the CTD or the ΔCTD in a 118E-10/+ background. All images are from male flies. (B) Histograms showing the levels of eye pigment of male JIL-1z2/JIL-1z2 null flies expressing the FL, the CTD or the ΔCTD in a 118E-10/+ background. The average pigment level when the CTD or the ΔCTD was expressed was compared with the level when the FL was expressed using a two-tailed Student's t-test.
Fig. 6.
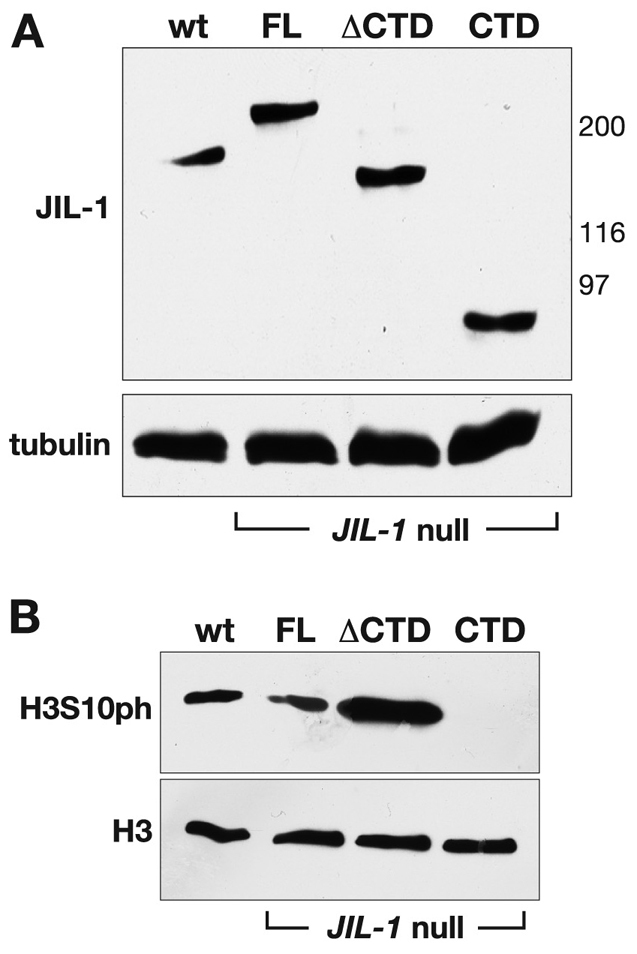
Expression of transgenic JIL-1 constructs in JIL-1 null flies. (A) Immunoblot of protein extracts from wild-type (wt) and from JIL-1z2/JIL-1z2 null flies expressing the FL, the CTD and the ΔCTD, respectively (labeled with JIL-1 antibody). Labeling with tubulin antibody was used as a loading control. The relative migration of molecular size markers in kDa is indicated to the right of the immunoblot. (B) Immunoblot of protein extracts from salivary glands from wild-type third-instar larvae (wt) and from JIL-1z2/JIL-1z2 null larvae expressing the FL, the CTD and the ΔCTD, respectively [labeled with phosphorylated (H3S10ph) antibody]. Labeling with antibody against histone H3 was used as a loading control.
In order to demonstrate directly that phosphorylated H3S10 and H3K9me2 levels at the hsp70-white gene reporter in the P-element insertion line 118E-10 were affected in the experiments, we performed chromatin immunoprecipitation (ChIP) assays as described by Legube and colleagues (Legube et al., 2006). Chromatin was immunoprecipitated from the salivary glands of ‘JIL-1 transgene’/+; JIL-1z2/JIL-1z2 da-GAL4; 118E-10/+ larvae using one of the following antibodies: a rabbit antibody against phosphorylated H3S10; a purified rabbit IgG antibody (negative control); or monoclonal antibodies against H3K9me2 or glutathione S-transferase (GST) as a negative control. Primers corresponding to the hsp70-white gene were used to amplify the precipitated material (Hines et al., 2009). Experiments were performed in duplicate, and relative enrichment of hsp70-white DNA from the phosphorylated H3S10 and H3K9me2 immunoprecipitates was normalized to the corresponding control antibody immunoprecipitates, which were performed in tandem for each experimental sample. In FL-expressing salivary glands, there was an approximately fivefold relative enrichment of phosphorylated-H3S10-immunoprecipitated hsp70-white DNA compared with the control immunoprecipitate (Fig. 7A). This enrichment increased to about 100-fold when the ΔCTD was expressed. By contrast, in CTD-expressing salivary glands, the relative enrichment was close to control levels (Fig. 7A). Fig. 7B shows that, when the FL or ΔCTD were expressed, the relative enrichment of H3K9me2-immunoprecipitated hsp70-white DNA was very low. However, when the CTD was expressed, there was an approximately three to four fold increase in the relative enrichment level compared with when FL or ΔCTD was expressed. These experiments indicate that phosphorylated-H3S10 and H3K9me2 levels at the hsp70-white reporter gene in 118E-10 correlated directly with the different H3S10 phosphorylation capabilities of FL, CTD and ΔCTD. Furthermore, the results show that, in the absence of H3S10 phosphorylation (such as in CTD-expressing salivary glands), there was an accompanying increase in H3K9me2 levels at the hsp70-white reporter gene.
Fig. 7.

Chip analysis of the reporter gene hsp70-white in the 118E-10 P-element insertion. (A) Histograms of the relative enrichment of chromatin immunoprecipitated by antibody against phosphorylated H3S10 (H3S10ph) from the salivary glands of JIL-1z2/JIL-1z2 null third-instar larvae expressing the FL, the CTD or the ΔCTD in a 118E-10/+ background. For each experimental condition, the relative enrichment was normalized to the corresponding control immunoprecipitation with purified rabbit IgG antibody (cont). The graph shows the results from two independent experiments. (B) Histograms of the relative enrichment of chromatin immunoprecipitated by anti-H3K9me2 mAb from salivary glands of JIL-1z2/JIL-1z2 null third-instar larvae expressing the FL, the CTD or the ΔCTD in a 118E-10/+ background. For each experimental condition, the relative enrichment was normalized to the corresponding control immunoprecipitation with GST mAb 8C7 (cont). The graph shows the results from two independent experiments.
In order to determine how this effect on PEV correlated with polytene chromosome morphology and H3K9me2 localization, we performed immunolabeling of polytene squash preparations. In a JIL-1z2 null background without transgene expression, polytene morphology is greatly perturbed, with ectopic spreading of the H3K9me2 mark, and this is especially prominent on the X chromosome (Fig. 8). Expression of the FL or ΔCTD constructs restored the chromosome morphology and prevented the H3K9me2 from spreading (Fig. 8). Interestingly, however, expression of the CTD restored chromosome morphology without counteracting the heterochromatic spreading of the H3K9me2 mark in both males and females. Taken together, these findings suggest that the H3S10 phosphorylation mark is required to counteract heterochromatic spreading and that this effect is independent of any potential structural contributions from the JIL-1 protein. However, these experiments could not exclude the possibility that, in the absence of kinase activity, the presence of the NTD and/or the kinase domains in the full-length JIL-1 protein prevent the spreading of the H3K9me2 mark. Thus, in order to address this issue further we expressed a ‘kinase dead’ version of full-length JIL-1, in which the lysine crucial for catalytic activity in each of the two kinase domains (K293 and K652) was changed to alanine – this has previously been shown to lack kinase activity (Deng et al., 2008) in a JIL-1 null mutant background. As illustrated in Fig. 9, expression of this construct, which only differs from wild-type JIL-1 at two amino acid positions, did not prevent the spreading of the heterochromatic mark H3K9me2. By contrast, preparations expressing the ΔCTD, which retains its phosphorylated H3S10 kinase activity (Bao et al., 2008) showed no evidence of heterochromatic spreading (Fig. 8) – this strongly suggests that the H3S10 phosphorylation mark is required to counteract this activity.
Fig. 8.

The effect on H3K9me2 localization in polytene chromosomes from JIL-1 null larvae expressing the FL, the CTD or the ΔCTD. The polytene squash preparations were labeled with antibody against H3K9me2 (in red) and with Hoechst (DNA, in blue or grey). The X chromosome is indicated by an X. Preparations from JIL-1z2/JIL-1z2 null larvae expressing either the FL, the ΔCTD or the CTD are shown. For comparison, the top panel shows a preparation from a JIL-1z2/JIL-1z2 null larvae without transgene expression.
Fig. 9.

The effect on H3K9me2 localization in polytene chromosomes from JIL-1 null larvae expressing a ‘kinase dead’ JIL-1 construct. The polytene squash preparation is from a male JIL-1 null (JIL-1z2/JIL-1z2) third-instar larvae triple labeled with Hoechst (DNA, in blue or grey), H3K9me2 antibody (in red) and JIL-1 antibody (in green). Note that although expression of the ‘kinase dead’ construct is near wild-type levels, and is localized on the chromosome arms and upregulated on the male X chromosome (X), the chromosome morphology as well as the spreading and upregulation of histone H3K9 dimethylation on the X chromosome are indistinguishable from those observed in JIL-1 null third-instar larvae (Fig. 7).
Discussion
We have explored the hypothesis that the epigenetic H3S10 phosphorylation mark is required to counteract heterochromatic spreading and gene silencing in Drosophila. We show that, when the CTD-domain, which displaces endogenous JIL-1, was expressed in a wild-type background, it had a dominant-negative effect and essentially phenocopied the effect of hypomorphic JIL-1 alleles on PEV. These effects on PEV correlated with the spreading of the heterochromatic mark H3K9me2 to the chromosome arms and a decrease in H3S10 phosphorylation levels. Furthermore, we demonstrate that expression of the CTD-domain in a JIL-1 null mutant background enhanced PEV of the 118E-10 allele compared with when the wild-type JIL-1 construct was expressed. Interestingly, although spreading of the heterochromatic H3K9me2 mark was not counteracted by expression of the CTD in the absence of H3S10 phosphorylation, the grossly perturbed polytene chromosomes of the JIL-1 null mutant salivary glands were restored to essentially wild-type morphology. Moreover, a ‘kinase dead’ version of JIL-1 that only differed from wild-type JIL-1 at two amino acid positions did not prevent the heterochromatic spreading. Taken together, these findings suggest that: (1) the gross perturbation of polytene chromosome morphology observed in JIL-1 null mutants is unrelated to gene silencing in PEV and is likely to occur as a result of faulty polytene chromosome alignment and/or organization separate from the epigenetic regulation of chromatin structure; (2) structural contributions from the JIL-1 protein are unlikely to play a role in counteracting heterochromatic spreading and gene silencing in PEV; and (3) the epigenetic H3S10 phosphorylation mark is required for preventing the observed heterochromatic spreading as well as gene silencing in PEV assays.
It has recently been demonstrated that JIL-1 can interact directly with Su(var)3–9, and can potentially regulate the function of that protein by phosphorylating it at residue S191 (Boeke et al., 2010). However, phosphorylation of Su(var)3–9 by JIL-1 did not affect the enzymatic activity of Su(var)3–9 or its ability to repress transcription (Boeke et al., 2010) – furthermore, the direct protein–protein interaction is mediated by the C-terminus of JIL-1 (Boeke et al., 2010). As expression of the ΔCTD, which lacks this interaction domain, prevented heterochromatic spreading in a JIL-1 mutant background, it is unlikely that phosphorylation of Su(var)3–9 by JIL-1 is involved in regulating the role of Su(var)3–9 in PEV. However, an interesting possibility is that direct interactions between JIL-1 and Su(var)3–9 can contribute to other aspects of the JIL-1 null phenotype. For example, in genetic interaction assays monitoring the lethality as well as the polytene chromosome morphology defects associated with the JIL-1 null phenotype, among the three major heterochromatin components only a reduction in the dose of the Su(var)3–9 gene rescued both phenotypes (Zhang et al., 2006; Deng et al., 2007; Deng et al., 2010). A reduction of Su(var)3–7 rescued the lethality, but not the chromosome defects (Deng et al., 2010), and no genetic interactions were detectable between JIL-1 and Su(var)2–5 in these assays (Deng et al., 2007). These observations indicate that while Su(var)3–9 activity might be a contributing factor in the lethality and polytene chromatin structural perturbations associated with loss of the JIL-1 histone H3S10 kinase, these effects are likely to be uncoupled from HP1a and to a lesser degree from Su(var)3–7. Therefore, these findings provide additional evidence that such parameters are probably independent of, and separate from, the mechanisms of classical heterochromatin assembly and gene silencing. This hypothesis is supported by experiments probing for dynamic interactions between loss-of-function alleles of JIL-1 and Su(var)3–9, Su(var)3–7 or Su(var)2–5 using PEV assays (Deng et al., 2010; Wang et al., 2011), where a direct antagonistic and counterbalancing effect on gene expression between JIL-1 and all three heterochromatic factors has been demonstrated.
Almost all known histone modifications correlate with activating or repressive functions, depending on which histone variant or amino acid residue is modified (Allis et al., 2007). However, these histone modifications do not occur in isolation but rather in a combinatorial manner, leading to both synergistic and antagonistic pathways (Allis et al., 2007) in which the same mark can participate (Berger, 2007). This has made it difficult to establish a defined causative biological effect of the addition or removal of a single mark in vivo. We have provided evidence that the histone H3S10 phosphorylation mark at euchromatic regions is required to counteract the spreading of heterochromatic factors and gene silencing. This repression of gene activity is likely to be independent of a direct effect on the transcriptional machinery, as it has been demonstrated that RNA polymerase-II-mediated transcription occurs at robust levels in the absence of H3S10 phosphorylation in Drosophila (Cai et al., 2008). Furthermore, Deng and colleagues used a LacI-tethering system to provide direct evidence that phosphorylation of the histone H3S10 residue by JIL-1 can play a causative role in establishing euchromatic chromatin regions (Deng et al., 2008). These findings, together with those of the present study, strongly support the hypothesis that a function of the epigenetic histone H3S10 phosphorylation mark is to antagonize heterochromatization by participating in a dynamic balance between factors promoting repression and activation of gene expression.
Materials and Methods
JIL-1 CFP-tagged fusion constructs
A full length JIL-1 (1–1207) construct (FL), a ΔCTD construct containing residues 1–926 and a CTD construct containing sequences from amino acids 927 to1207 with an in-frame CFP-tag were cloned into the pYES vector (Patton et al., 1992) using standard methods (Sambrook and Russell, 2001). For the CTD construct that did not contain the endogenous JIL-1 nuclear localization sequence (NLS), situated in the NTD (Jin et al., 1999), the NLS-pECFP vector from Clontech was added to the N-terminus. The fidelity of all constructs was verified by sequencing at the Iowa State University Sequencing facility.
Drosophila melanogaster stocks
Fly stocks were maintained at 25°C according to standard protocols (Roberts, 1998). The JIL-1z2 null allele has been described previously (Wang et al., 2001; Zhang et al., 2003). JIL-1 construct pYES lines were generated by standard P-element transformation (BestGene) and expression of the transgenes was driven using a da-GAL4 driver introduced by standard genetic crosses. Recombinant JIL-1z2 da-GAL4 chromosomes were generated as described previously (Ji et al., 2005) and the presence of JIL-1z2 was confirmed by PCR (Zhang et al., 2003). Expression levels of each of the JIL-1 constructs were monitored by immunoblot analysis as described below. The ‘kinase dead’ LacI–JIL-1 construct has been described previously (Deng et al., 2008) and driven using the Sgs3-GAL4 driver. All driver lines and the In(1)wm4 allele were obtained from the Bloomington Stock Center (Bloomington, IN). The P-element insertion line 118E-10 was the generous gift of Lori Wallrath. Balancer chromosomes and markers have been described previously (Lindsley and Zimm, 1992).
PEV assays were performed as previously described (Lerach et al., 2006; Bao et al., 2007; Deng et al., 2010; Wang et al., 2011). In short, we generated flies expressing the various JIL-1 constructs in a background of the two PEV arrangements (118E-10 or wm4) by standard crossing. To quantify the variegated phenotype, adult flies were collected from the respective crosses at eclosion, aged 6 days at 25°C, frozen in liquid nitrogen, and stored at −80°C until they were assayed. The pigment assays were essentially performed as previously described (Kavi and Birchler, 2009) using three sets of ten fly heads for each genotype collected from males and females, respectively. For each sample, the heads from the ten flies were homogenized in 200 μl of methanol with 0.1% hydrochloric acid, centrifuged, and the OD of the supernatant was spectrophotometrically measured at a wavelength of 480 nm. Statistical comparisons were performed using a two-tailed Student's t-test. The eyes of representative individuals from these crosses were photographed using an Olympus Stereo Microscope and a SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI).
Immunohistochemistry
Standard polytene chromosome squash preparations were performed as described previously (Cai et al., 2010) using either 1 or 5 minute fixation protocols, and labeled with antibody as previously described (Jin et al., 1999; Wang et al., 2001). In some preparations, the male X chromosome was identified by double labeling with MSL antibody as described previously (Jin et al., 2000). Primary antibodies used in this study include: rabbit antibody against phosphorylated H3S10 (Cell Signaling); rabbit anti-histone H3 (Cell Signaling); rabbit anti-MSL-2 (generous gift of Mitzi Kuroda, Harvard University, Boston, MA); rabbit anti-H3K9me2 (Upstate Biotechnology); mouse anti-tubulin (Sigma); rabbit anti-JIL-1 (Jin et al., 1999); chicken anti-JIL-1 (Jin et al., 2000); and anti-JIL-1 mAb 5C9 (Jin et al., 2000). DNA was visualized by staining with Hoechst 33258 (Molecular Probes) in PBS. The appropriate species- and isotype-specific Texas Red-, TRITC- and FITC-conjugated secondary antibodies (Cappel/ICN, Southern Biotech) were used (1:200 dilution) to visualize primary antibody labeling. The final preparations were mounted in 90% glycerol containing 0.5% n-propyl gallate. The preparations were examined using epifluorescence optics on a Zeiss Axioskop microscope, and images were captured and digitized using a cooled SPOT CCD camera. Images were imported into Photoshop, where they were pseudocolored, image processed and merged. In some images, non-linear adjustments were made to the channel with Hoechst labeling for optimal visualization of chromosomes.
Immunoblot analysis
Protein extracts were prepared from adult flies or from dissected third-instar larval salivary glands homogenized in a buffer containing: 20 mM Tris-HCl pH 8.0; 150 mM NaCl; 10 mM EDTA; 1 mM EGTA; 0.2% Triton X-100; 0.2% NP-40; 2 mM Na3VO4; 1 mM PMSF; and 1.5 μg/ml aprotinin. Proteins were separated by SDS-PAGE according to standard procedures (Sambrook and Russell, 2001). Electroblot transfer was performed as described previously (Towbin et al., 1979) with transfer buffer containing 20% methanol and in most cases including 0.04% SDS. For these experiments, we used the Bio-Rad Mini PROTEAN III system, electroblotting to 0.2 μm nitrocellulose and HRP-conjugated secondary antibodies against mouse or rabbit (Bio-Rad) (1:3000) for visualization of the primary antibody. Antibody labeling was visualized using chemiluminescent detection methods (SuperSignal West Pico Chemiluminescent Substrate, Pierce). The immunoblots were digitized using a flatbed scanner (Epson Expression 1680).
Chromatin immunoprecipitation
For ChIP experiments, 50 pairs of salivary glands per sample were dissected from third instar larvae and fixed for 15 minutes at room temperature in 1 ml of fixative (50 mM HEPES at pH 7.6, 100 mM NaCl, 0.1 mM EDTA at pH 8, 0.5 mM EGTA at pH 8, 2% formaldehyde). Preparation of chromatin for immunoprecipitation was performed as previously described (Legube et al., 2006). Rabbit antibody against phosphorylated H3S10 (Cell Signaling), purified rabbit IgG antibody (Sigma), anti-H3K9me2 mAb (Abcam), or anti-GST mAb 8C7 (Rath et al., 2004) were used for immunoprecipitation. For each sample, the chromatin lysate was divided into equal amounts and immunoprecipitated with experimental and control antibody, respectively. DNA from the immunoprecipitated chromatin fragments (average length, 500 bp) was purified using a Wizard SV DNA purification kit (Promega). The isolated DNA was used as a template for quantitative real-time (qRT) PCR performed with the Stratagene Mx4000 real-time cycler. The PCR mixture contained Brilliant II SYBR Green QPCR Master Mix (Stratagene) as well as the corresponding primers: hsp70-white-forward 5′-GCAACCAAGTAAATCAACTGC-3′, hsp70-white-reverse 5′-GTTTTGGCACAGCACTTTGTG-3′, which amplify region +149 to +250 (Hines et al., 2009). Cycling parameters were 10 minutes at 95°C, followed by 40 cycles of 30 seconds at 95°C, 30 seconds at 55°C and 30 seconds at 72°C. Fluorescence intensities were plotted against the number of cycles using an algorithm provided by Stratagene. DNA levels were quantified by using a calibration curve based on the dilution of concentrated DNA. For each experimental condition, the relative enrichment was normalized to the corresponding control immunoprecipitation from the same chromatin lysate.
Acknowledgements
We thank members of the laboratory for discussion, advice and critical reading of the manuscript. We also acknowledge Kevin Bieniek for technical assistance. We especially thank Lori Wallrath, Pamela Geyer and Mitzi Kuroda for providing fly stocks and reagents.
Footnotes
Funding
This work was supported by National Institutes of Health [grant number GM062916 to K.M.J. and J.J.]. Deposited in PMC for release after 12 months.
References
- Allis C. D., Jenuwein T., Reinberg D. (2007). Epigenetics. Cold Spring HarborNew York: Cold Sping Harbor Laboratory Press; [Google Scholar]
- Bao X., Deng H., Johansen J., Girton J., Johansen K. M. (2007). Loss-of-function alleles of the JIL-1 histone H3S10 kinase enhance position-effect-variegation at pericentric sites in Drosophila heterochromatin. Genetics 176, 1355-1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X., Cai W., Deng H., Zhang W., Krencik R., Girton J., Johansen K. M. (2008). The COOH-terminal domain of the JIL-1 histone H3S10 kinase interacts with histone H3 and is required for correct targeting to chromatin. J. Biol. Chem. 283, 32741-32750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S. L. (2007). The complex language of chromatin regulation during transcription. Nature 447, 407-412 [DOI] [PubMed] [Google Scholar]
- Boeke J., Regnard C., Cai W., Johansen J., Johansen K. M., Becker P. B., Imhof A. (2010). Phosphorylation of SU(VAR)3-9 by the chromosomal kinase JIL-1. PLoS ONE. 5, e10042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W., Bao X., Deng H., Jin Y., Girton J., Johansen J., Johansen K. M. (2008). RNA polymerase II-mediated transcription at active loci does not require histone H3S10 phosphorylation in Drosophila. Development 135, 2917-2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W., Jin Y., Girton J., Johansen J., Johansen K. M. (2010). Preparation of polytene chromosome squashes for antibody labeling. J. Vis. Exp. 9, pii 1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryderman D. E., Cuaycong M. H., Elgin S. C. R., Wallrath L. L. (1998). Characterization of sequences associated with position-effect-variegation at pericentric sites in Drosophila heterochromatin. Chromosoma 107, 277-285 [DOI] [PubMed] [Google Scholar]
- Delattre M., Spierer A., Jaquet Y., Spierer P. (2004). Increased expression of Drosophila Su(var)3-7 triggers Su(var)3-9-dependent heterochromatin formation. J. Cell. Sci. 117, 6239-6247 [DOI] [PubMed] [Google Scholar]
- Deng H., Zhang W., Bao X., Martin J. N., Girton J., Johansen J., Johansen K. M. (2005). The JIL-1 kinase regulates the structure of Drosophila polytene chromosomes. Chromosoma 114, 173-182 [DOI] [PubMed] [Google Scholar]
- Deng H., Bao X., Zhang W., Girton J., Johansen J., Johansen K. M. (2007). Reduced levels of Su(var)3-9 but not Su(var)2-5 (HP1) counteract the effects on chromatin structure and viability in loss-of-function mutants of the JIL-1 histone H3S10 kinase. Genetics 177, 79-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Bao X., Cai W., Blacketer M. J., Belmont A. S., Girton J., Johansen J., Johansen K. M. (2008). Ectopic histone H3S10 phosphorylation causes chromatin structure remodeling in Drosophila. Development 135, 699-705 [DOI] [PubMed] [Google Scholar]
- Deng H., Cai W., Wang C., Lerach S., Delattre M., Girton J., Johansen J., Johansen K. M. (2010). JIL-1 and Su(var)3-7 interact genetically and counter-balance each others' effect on position effect variegation in Drosophila. Genetics 185, 1183-1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A., Schotta G., Lein S., Kubicek S., Krauss V., Jenuwein T., Reuter G. (2004). Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev. 18, 2973-2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girton J., Johansen K. M. (2008). Chromatin structure and regulation of gene expression: the lessons of PEV in Drosophila. Adv. Genet. 61, 1-43 [DOI] [PubMed] [Google Scholar]
- Hines K. A., Cryderman D. E., Flannery K. M., Yang H., Vitalini M. W., Hazelrigg T., Mizzen C. A., Wallrath L. L. (2009). Domains of Heterochromatin Protein 1 required for Drosophila melanogaster heterochromatin spreading. Genetics 182, 967-977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y., Rath U., Girton J., Johansen K. M., Johansen J. (2005). D-Hillarin, a novel W180-domain protein, affects cytokinesis through interaction with the septin family member Pnut. J. Neurobiol. 64, 157-169 [DOI] [PubMed] [Google Scholar]
- Jin Y., Wang Y., Walker D. L., Dong H., Conley C., Johansen J., Johansen K. M. (1999). JIL-1: a novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol. Cell 4, 129-135 [DOI] [PubMed] [Google Scholar]
- Jin Y., Wang Y., Johansen J., Johansen K. M. (2000). JIL-1, a chromosomal kinase implicated in regulation of chromatin structure, associates with the MSL dosage compensation complex. J. Cell Biol. 149, 1005-1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavi H. H., Birchler J. A. (2009). Interaction of RNA polymerase II and the small RNA machinery affects heterochromatic silencing in Drosophila. Epigenetics Chromatin 2, 15-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legube G., McWeeney S. K., Lercher M. J., Akhtar A. (2006). X-chromosome-wide profiling of MSL-1 distribution and dosage compensation in Drosophila. Genes Dev. 20, 871-883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerach S., Zhang W., Bao X., Deng H., Girton J., Johansen J., Johansen K. M. (2006). Loss-of-function alleles of the JIL-1 kinase are strong suppressors of position effect variegation of the wm4 allele in Drosophila. Genetics 173, 2403-2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley D. L., Zimm G. G. (1992). The genome of Drosophila melanogaster. New York: Academic Press; [Google Scholar]
- Muller H. J. (1930). Types of visible variegations induced by X-rays in Drosophila. J. Genetics 22, 299-335 [Google Scholar]
- Patton J. S., Gomes X. V., Geyer P. K. (1992). Position-independent germline transformation in Drosophila using a cuticle pigmentation gene as a selectable marker. Nucleic Acids Res. 20, 5859-5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V., Hadfield C., Pretorius G. H. J. (1983). Microdissection and cloning of the white locus and the 3B1 - 3C2 region of the Drosophila X chromosome. EMBO J. 2, 927-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath U., Wang D., Ding Y., Xu Y.-Z., Qi H., Blacketer M. J., Girton J., Johansen J., Johansen K. M. (2004). Chromator, a novel and essential chromodomain protein interacts directly with the putative spindle matrix protein Skeletor. J. Cell. Biochem. 93, 1033-1047 [DOI] [PubMed] [Google Scholar]
- Roberts D. B. (1998). In Drosophila: a Practical Approach. OxfordUK: IRL Press; [Google Scholar]
- Sambrook J., Russell D. W. (2001). Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Schotta G., Ebert A., Dorn R., Reuter G. (2003). Position-effect variegation and the genetic dissection of chromatin regulation in Drosophila. Semin. Cell Dev. Biol. 14, 67-75 [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350-4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrath L. L., Elgin S. C. R. (1995). Position effect variegation in Drosophila is associated with altered chromatin structure. Genes Dev. 9, 1263-1277 [DOI] [PubMed] [Google Scholar]
- Wallrath L. L., Guntur V. P., Rosman L. E., Elgin S. C. R. (1996). DNA representation of variegating heterochromatic P-element inserts in diploid and polytene tissues of Drosophila melanogaster. Chromosoma 104, 519-527 [DOI] [PubMed] [Google Scholar]
- Wang C., Girton J., Johansen J., Johansen K. M. (2011). A balance between euchromatic (JIL-1) and heterochromatic (SU(VAR)2-5 and SU(VAR)3-9) factors regulates position-effect variegation in Drosophila. Genetics 188, 745-748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhang W., Jin Y., Johansen J., Johansen K. M. (2001). The JIL-1 tandem kinase mediates histone H3 phosphorylation and is required for maintenance of chromatin structure in Drosophila. Cell 105, 433-443 [DOI] [PubMed] [Google Scholar]
- Weiler K. S., Wakimoto B. T. (1995). Heterochromatin and gene expression in Drosophila. Annu. Rev. Genet. 29, 577-605 [DOI] [PubMed] [Google Scholar]
- Zhang W., Jin Y., Ji Y., Girton J., Johansen J., Johansen K. M. (2003). Genetic and phenotypic analysis of alleles of the Drosophila chromosomal JIL-1 kinase reveals a functional requirement at multiple developmental stages. Genetics 165, 1341-1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Deng H., Bao X., Lerach S., Girton J., Johansen J., Johansen K. M. (2006). The JIL-1 histone H3S10 kinase regulates dimethyl H3K9 modifications and heterochromatic spreading in Drosophila. Development 133, 229-235 [DOI] [PubMed] [Google Scholar]