


Abstract
R-loop is the structure co-transcriptionally formed between nascent RNA transcript and DNA template, leaving the non-transcribed DNA strand unpaired. This structure can be involved in the hyper-mutation and dsDNA breaks in mammalian immunoglobulin (Ig) genes, oncogenes and neurodegenerative disease related genes. R-loops have not been studied at the genome scale yet. To identify the R-loops, we developed a computational algorithm and mapped R-loop forming sequences (RLFS) onto 66 803 sequences defined by UCSC as ‘known’ genes. We found that ∼59% of these transcribed sequences contain at least one RLFS. We created R-loopDB (http://rloop.bii.a-star.edu.sg/), the database that collects all RLFS identified within over half of the human genes and links to the UCSC Genome Browser for information integration and visualisation across a variety of bioinformatics sources. We found that many oncogenes and tumour suppressors (e.g. Tp53, BRCA1, BRCA2, Kras and Ptprd) and neurodegenerative diseases related genes (e.g. ATM, Park2, Ptprd and GLDC) could be prone to significant R-loop formation. Our findings suggest that R-loops provide a novel level of RNA–DNA interactome complexity, playing key roles in gene expression controls, mutagenesis, recombination process, chromosomal rearrangement, alternative splicing, DNA-editing and epigenetic modifications. RLFSs could be used as a novel source of prospective therapeutic targets.
INTRODUCTION
R-loop is a stable RNA–DNA hybrid structure in which the RNA strand is base-paired with one DNA strand of a DNA duplex, leaving the opposite DNA strand single-stranded. The R-loop structure has been first characterized over 35 years ago (1). Initial study of R-loop focused on the development of ‘R-loop hybridization technique’ for visualization of the genetic organization of ribosomal RNA genes in yeast via electron microscopy (1–3). The application of this technique also led to the discovery of intron by the observation of splicing of adenovirus 2 late mRNA under electron microscope (4). Since then many subsequent applications of R-loop hybridization have been developed, which are now widely used for the study of gene structure.
In 1995, Drolet and colleagues first demonstrated that R-loop existed in vivo in the bacterial cell (5). In this study, the R-loop formation was shown to be a consequence of transcription process that resulted in hybridization between nascent RNA transcript and DNA template, therefore such process was called ‘co-transcriptional R-loop’ formation. R-loops occur in vivo within sequences that generate G-rich transcripts at the prokaryotic origins of replication, mitochondria and mammalian immunoglobulin (Ig) class switch sequences [see for references (6)]. R-loop forming structure has been documented in mutant yeast that was impaired in RNAP II transcription elongation (7). These and other findings generate interest to study R-loop forming structures and initiate more studies of R-loops in different cells and species. In addition, the in vitro techniques of R-loops detection have been improved and the mechanistic aspects of R-loop formation have been studied. In this article, we focus on the analysis of co-transcriptional R-loops in vivo rather than R-loop hybridization technique.
The two possible mechanisms of R-loop formation proposed by Lieber and Roy are ‘thread back’ and ‘extended hybrid’ mechanisms (6,8,9). According to the thread back mechanism a nascent RNA is single-stranded for a short period of time and then anneals with the template DNA strand. In the extended hybrid mechanism, the nascent RNA that forms upon transcription fails to denature from the template in the transcription bubble, due to the high thermodynamic stability between RNA–DNA hybrids. The R-loop formation also requires some specific pattern of the nucleotide sequence in the DNA template and presence of Li+, Na+, K+ and Cs+ ion to form stable R-loop structure. The R-loop formation in vivo is a dynamic process involving protein–DNA–RNA interactions. Top1 (topoisomerase 1) may prevent an accumulation of negative supercoiling downstream of transcription block and can prevent R-loop formation (10). It was shown that NPH-II helicase can efficiently unwind a RNA–DNA hybrid containing a purine-rich DNA track derived from the 3′-UTR of an early vaccinia gene (11). The negative correlation between R-loop formation and activity of splicing factor ASF/SF2 in chicken cell line has been demonstrated by Li and Manley (12).
In vitro studies showed that R-loop sequences vary in length from 150 to 650 bp in Ig switch region (13), from 110 to 1280 bp in Bcl6 and from 120 to 770 bp in RhoH (14). R-loops are sensitive to over-expression of RNase H, the endonuclease which specifically hydrolyzes RNA–DNA hybrid. Lieber and Roy proposed a R-loop model which depends on the sequence features and its position. It includes three distinct parts: R-loop initiation zone (RIZ), linker and R-loop elongation zone (REZ). They demonstrated that G clusters in RIZ are extremely important for the initiation of R-loop formation (8) but not in other parts while the linker between RIZ and REZ can be of any nucleotide composition. The final part of R-loop, REZ sequence, is required to be of high G density but does not necessarily have to be a G-cluster. This model can be applied for in vivo R-loop detection and facilitate the search of potential R-loop forming sequences (RLFS) in the genome.
Until recently, the studies of R-loops have provided various examples of significance of RNA–DNA interactions in a cell. The formation of R-loops during replication process in both prokaryotes and eukaryotes may lead to replication blockage that is lethal if left unresolved (15). In yeast, inactivation of THO-complex, a conserved eukaryotic nuclear complex containing Tho2, Hpr1, Mft1 and Thp2 proteins, induces R loop formation that results in reduction of transcription elongation efficiency and increases incidence of hyper-recombination (7). R-loop formation can also be associated with occurrence of transcription-associated recombination (TAR) in yeast and mammalian cells (16,17). R-loop formation can initiate various repair systems, such as homologous recombination (HR) that occurs mainly during late S phase of the cell cycle (18,19) and non-homologous end joining (NHEJ) involved in antibody maturation (20). In activated B-lymphocytes of mammals, R-loops contribute to immunoglobulin class switch recombination (Ig-CSR) that generates antibody isotypes (21).
A number of studies proposed and revealed that R-loop formation structure is involved in transcription-associated mutation (TAM) (14,22–25). Recent studies demonstrated a correlation between R-loop formation and activation-induced deaminase (AID) activity, the enzyme which (i) is involved in generation of mutations and recombination events in oncogenes, such as Bcl6 and Myc (14,26), and (ii) may affect genome instability.
Interestingly, R-loops are often associated with neurodegenerative diseases, including spinocerebellar ataxia type 1 (SCA1), myotonic dystrophy (DM1) and fragile X type A (FRAXA) (22,23,25). R-loop forming structures can be found in the Fmr1 and Fxn genes that are responsible for neurodegenerative disease (23,25). It was demonstrated that R-loops could co-localize with some classes of trinucleotide repeat tracks that occur in these genes (23). R-loop structures are found when Fmr1 and Fxn genes are transcribed. The RNA–DNA hybridization via R-loop mechanism can generate genetic instability that may be associated with the expansion of the trinucleotide repeats within the disease related genes (25).
While previous studies outlined several examples of the functional importance of R-loops, there was no systematic analysis done at the genome scale. This analysis can facilitate discovery of new R-loops and their genome localization, which is helpful for better understanding of R-loop structures and their functions, RNA–DNA interactome complexity and diseases. We hypothesize that R-loops can be formed in many genes and may play important roles in a variety of biological processes, including gene expression regulation, development and cell communication.
In this work, we first developed a quantitative model of RLFS, confirmed known RLFS within the genes of the human genome. We focus on the RLFS in the human genes, because genome mapping, data basing and the visualisation of RLFS integrated with other human DNA and RNA data could provide a useful tool for elucidating the role of R-loop formation phenomena in the complexity of function of the genomes and its association with diseases.
Furthermore, we developed a bioinformatics tool for RLFS search and visualization. Our pipeline identified RLFS that have previously been discovered in experimental studies. Based on our computational analysis, we demonstrate for the first time that RLFS are widespread throughout the human genome in genes of diverse functions. We organized our results in R-loopDB database, which collects the information about R-loops in each annotated human gene. The R-loopDB facilitates the interactive and versatile display of R-loops and is integrated into the UCSC Genome Browser for information integration from various sources. We further demonstrate the potential use of our database in the final part of this work.
MATERIALS AND METHODS
Data sources
DNA sequences of UCSC known genes dataset (the human genome; hg18 or NCBI Build 36.1) in FASTA format were downloaded on 23 February 2010. It included 66 803 UCSC known gene IDs that were constructed by automated pipeline from UCSC (27). This dataset contains RefSeq genes and alternative splicing variants of each gene.
R-loop forming DNA sequence model
Based on the experimental study of the characteristic of R-loop formation by Roy and Michael Lieber (8), we propose the following computational model of RLFS. The features of RLFS can be partitioned into three segments, (i) RIZ; (ii) linker and (iii) REZ or
RIZ
The DNA regions of initiation of R-loops are considered as clusters of a few Gs (3–4 nt) in the region. Segment sequence initiates and terminates with G-cluster that contains at least three contiguous Gs, e.g. GGGNGGGNGGG. G-cluster is important for efficient R-loop initiation and this feature is included in our model.
Linker
The DNA sequence region between RIZ and REZ regions is called linker. The nucleotides in this region are not specified in our model. We allow from 0 to 50 nt in the linker region.
REZ
Downstream of RIZ and Linker, REZ can support the extension of R-loop with a high G density (8). REZ has to be G-rich but does not require G-cluster like RIZ. At least 40% of G is required for R-loop formation. In our model, nucleotide number of REZ can vary from 100 to 2000 nt.
The above model of RLFS is used in our algorithm to identify the location of RLFS in the human genes.
Database construction
The results of RLFS identification are collected and included into our R-loopDB. Presently, R-loopDB is accessible via http://rloop.bii.a-star.edu.sg/. The database is managed by a MySQL relational database at the back-end to support user queries. All HTML pages are generated by PHP scripts hosted on an Apache server. The graphical view of gene structure and R-loop is generated by Perl Bio-Graphics Module. The Java script provides interactive interfaces that facilitate site navigation.
Kolmogorov–Waring statistics and parameterization
The Kolmogorov–Waring (K–W) probability function allows description and understanding of evolution patterns in the stochastic birth–death process in complex evolved systems. At near steady-state of the linear birth–death stochastic process, the K–W function can be calculated via the following simple recursive formula (28):
| (1) |
where m = 0, 1,2, … M [M = max(m)]. The inequalities 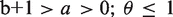 provide the necessary and sufficient conditions for the stable steady state behaviour of the random process (28,29). The parameters a, b and θ, we estimated by a method reported in (28).
provide the necessary and sufficient conditions for the stable steady state behaviour of the random process (28,29). The parameters a, b and θ, we estimated by a method reported in (28).
Querying the database
R-loopDB provides user-friendly accessibility with multiple search options (Figure 1B) that allows user to input official gene symbol, gene family keyword, Ref-Seq ID, gene description keyword, known gene ID and chromosome band as the query term. We recommend user to input known gene ID as the input for users who are interested in specific alternative splicing sequence. Besides searching the genes of interest, R-loopDB provides additional feature of filtering out genes that contain RLFS in the first exon or the first intron. This might be important because R-loop could be formed when the RLFS is located within 5′-end gene region and efficiency of R-loop formation is reduced in the distant downstream regions of the gene (9). The optional search is located in gene search box. User who is interested in finding RLFS located near 5′-end region are recommended to use this option.
Figure 1.

R-loop forming structure and representative screenshots of R-loopDB. (A) Transcription with and without R-loop forming structure. R-loop initiation zone (RIZ) and R-loop elongation zone (REZ) are highlighted in yellow blue, respectively. (B) The search bar. (C) The search result of Bcl6 gene.
The ‘search result’ page (Figure 1C) is designed in the table format including three fields: gene symbol, gene description and chromosome band. The user can click on a gene symbol link to view the detail page for that particular gene.
Output interface
R-loopDB allows visualization of RLFS in the selected gene (Figure 2) on (i) a gene map (Figure 2A); (ii) details of the RLFS sequence structure (Figure 2B); (iii) RLFS mapped on the UCSC browser known gene (Figure 2C) and (iv) annotation of the gene by NCBI search (Figure 2D). The user can navigate to any RLFS (see green box in Figure 2A) which is located in a region of the gene of interest and see details of the RLFS sequence as shown in Figure 2B. This figure provides high-lighted sub-sequences of RLFS including RIZ, linker, REZ and G (guanine)-cluster (see ‘Materials and Methods’ section). To ensure that users interested in R-loop can conveniently find a wide range of information for genes of interest, we provide linkage to external databases including UCSC Genome Browser and NCBI Entrez Gene. This enables integration of other information of genomic context, expression data and updated information for the gene of interest.
Figure 2.

Snapshot of a representative R-loopDB results pages for Bcl6 gene. (A) Overview figure that shows all known transcripts of the gene and RLFS mapping results. (B) Detailed summary of the RLFS (in green box of A), including sequence structure, location, length and G-cluster. (C) Link from RLFS mapping result to UCSC database tracks (URL: http://genome.ucsc.edu/cgi-bin/) (in red box of A), (D) Link from RLFS mapping result to NCBI Entrez gene database (URL: http://www.ncbi.nlm.nih.gov/gene/) (in blue box of A).
RESULTS AND DISCUSSION
Data validation
To validate our findings, we compared predictions from our model with previously reported data describing R-loop-positive and R-loop-negative genes. Previously, R-loop structures have been detected only in a few mammalian genes: Ig switch region, Bcl6, Myc, Rhoh, Fmr1 and Fxn (14,21,23,25,26,30). In two other genes, Ig variable heavy chain and a-Myb, no R-loop structure have been reported in gene regions (14). We compared our prediction results with experimental data for these genes and the results were completely consistent with the observation. This suggests that our RLFS identification method produces reliable results.
Figure 2 shows an example of analysis of RLFSs within Bcl6 gene region. Panel A shows that five RLFSs can be found in this gene region and all of these five RLFSs are located in the first intron. Panel B provides detailed visualization of RLFS, demonstrating explicit location of the RIZ in the 5′-end of the sequence and the REZ in the 3′-end of the sequence. In this figure, G-clusters are highlighted. Panel C shows results of our application integrated in the UCSC browser viewer. This integration allows user to connect information about RLFS localization with many annotation tracks available in UCSC browser, which provides more information, such as intron or exon localization of RLFS, co-localization of RLFS with important regulatory signals [histone methylation, CpG islands, repeat elements, transcription factor-binding sites (TFBSs), etc.] Panel D provides characteristics of a gene of interest (Bcl6) via link to NCBI Entrez gene annotation list.
Prevalence of R-loops in the human genes
In total 66 803 sequences of UCSC known genes and splice variants were downloaded and studied. We found that 59% (39 720/66 803) of UCSC known genes and their splice variants contain at least one RLFS. We then counted the number of RLFS in each UCSC known gene sequence. Overall, 245 181 RLFSs from 39 720 UCSC known gene sequences were found and stored in the R-loopDB.
To prevent over-counting of RLFS location events on our further statistical analysis, we merged overlapping RLFSs sharing at least 1 nt into single longest DNA segment. After overlapped RLFS merging the number of RLFSs is 140 106. Figure 3A demonstrates that the frequency distribution of the number of such RLFSs follows the skewed power-law like frequency distribution and it can be described well with the K–W birth–death evolution model (28). This function is used for statistical characterisation of the frequency distribution of occurrence of diverse structurally and functionally important signals, for instance TFBSs in a gene promoter region of a given eukaryotic genome (29), domains or structure motifs in a protein of a given proteome (28). Such type frequency distributions are sample size-dependent (not scale-free) and are naturally occurred in complex organisms in the course of evolution as the result of positive selection ‘useful’ structure/functional elements (28). Figure 3A suggests that evolution of the RLFSs follows a similar statistical rule.
Figure 3.

Statistic of RLFS in a gene of the human genome. (A) Numerical characteristics of RLFS distribution. (B) Observed frequency distribution of RLFS in a gene of the human genome and its fitting by K–W probability function (see ‘Materials and Methods’ section). This model fits empirical frequency distribution at θ = 0.9905; a = 1.90, b = 3.83506.
We also analysed the frequency of RLFS in each UCSC known gene and their splice variants. The distribution of RLFS per gene is shown in the Figure 3A. We found that ∼60% of UCSC known gene and splice variant sequences contained only one or two RLFS. However, many genes and their isoforms carry very large number (>100) of RLFS (Figure 3A and B). Eleven of UCSC known gene sequences containing more than 100 RLFSs are represented by four gene IDs: IgH (14q32), Ptprn2 (7q36), Mad1l1 (7p22) and Sorcs2 (4p16). IgH, Ptprn2, Mad1l1 and Sorcs2 have 105, 140, 104 and 115 RLFSs respectively.
RLFSs occur multiple times in 35% of known genes and their splice variants
Interestingly, RLFSs occur in 16 362 known genes and their splice variants only once, whereas 35% (23 358/66 803) of the 66 803 genes and their splice variants contain multiple RLFS (Figure 3). This finding implies that multiple occurrences of RLFS may play important roles in gene expression regulation.
Immunoglobulin class switch recombination (Ig-CSR) is the process in which IgM changes to IgG, IgA, or IgE by DNA rearrangement of the Ig heavy chain from IgHµ to IgHγ, IgHα, or IgHε (31). It occurs at class switch sequences located upstream of the corresponding constant domain exons. It was demonstrated that R-loops form at Ig-CSR regions in activated B lymphocytes. According to R-loop model, inversions of switch regions reduce their efficiency (32). It was suggested that R-loop structures are necessary for enhancing the CSR process. In particular IgH is one of the activated B lymphocyte genes in which R-loop formation was reported (21). Our analysis reveals 105 RLFSs in IgH. We suggest that abundance of R-looping regions may play an important role in Ig-CSR.
We also found that Mad1l1, Ptprn2, Sorcs2 as well as IgH are also highly abundant in RLFSs (Figure 3B). It has been reported that copy number gains and losses in Mad1l1, Ptprn2 and Sorcs2 can be associated with various diseases (33–39). Previous studies also suggested an association between R-loop formation and mutations in non-Ig genes (14,26). We used COSMIC database (URL: http://www.sanger.ac.uk/genetics/CGP/cosmic/) to determine mutations in Mad1l1, Ptprn2 and Sorcs2 genes across cancer tissue samples. We found mutations in Mad1l1 and Sorcs2 in the glioma patient samples, and mutations in Ptprn2 in ovarian cancers patient samples. We analysed the distances of mutated sites in these genes and the location of RLFS. Interestingly, mutated sites and RLFS locations overlap in Mad1l1 and are in close proximity in Sorcs2 (0.98 kb) and Ptprn2 (0.29 kb). These findings suggest that R-loop may contribute to mutagenesis in these genes and abundance of R-looping regions might raise the risk of mutagenesis. The R-loop mediated mutagenesis and its link with single nucleotide polymorphisms (SNPs) and recombination events remains an interesting field for further investigation.
RLFSs can be co-localized with mutation and recombination regions
Single-stranded DNA associated with persisting R-loop is less protected from mutagens and thus contributes to occurrence of TAMs, including single-base substitutions, insertions and deletions. To find evidence for the mutations caused by R-loop formation, we integrated SNP data from dbSNP database (40) and RLFSs. We found that SNPs could be localized in RLFS regions. In particular, Figure 4A shows that SNPs in the first exon of Krt14 are strongly enriched within RLFS and thus this RLFS could be associated with TAM. Interestingly, among the SNPs, there are four non-synonymous (i.e. resulting in amino acid changes) SNPs: [rs28928893 (41), rs60171927 (42), rs60399023 (43) and rs58330629 (43)]. Each of these SNPs is known to cause epidermolysis bullosa simplex disease (43). This finding may give insight in association of R-loop formation with disease caused mutations.
Figure 4.

R-loops co-localization with mutations and recombination regions. (A) R-loop association with transcription-associated mutation (TAM). The first annotation track illustrates SNPs location retrieved from dbSNP build 130 (40). SNPs are enriched in RLFS (pink colour) of Krt14 gene. The second annotation track shows non-synonymous SNP of Krt14 gene overlap with RLFS. These SNPs are associated to epidermolysis bullosa simplex disease (41–43). (B) R-loop associated to replication-induced recombination (RIR). The first annotation track (black colour) illustrates the RIR breakpoint that occurs in S phase of Top1-deficient cells (47). The second annotation track (brown colour) shows deleted regions found in the human lung adenocarcinoma cell samples. The third annotation track (green colour) demonstrates the region of CpG island that may play a role in R-loop-mediated recombination. (C) R-loop associated to AID-dependent translocation. The first annotation track shows the region of R-loop forms in vitro (red track) with the regions of Myc that undergoes AID-dependent translocation in B-cell lymphoma (26). The second annotation track demonstrates the positions of translocation breakpoints (brown track) between Myc and Ig switch regions in Burkitt's lymphoma patients (49,50). The third annotation track (green colour) demonstrates the region of CpG island that may play a role in R-loop-mediated recombination.
Besides mutations, R-loops could also be linked to TAR (16,17). When DNA replication and RNA synthesis are co-directional, R-loop can produce a replication fork stalling and collapse, thus inducing DNA strand breaks. To reduce the impact of DNA breaks, DNA repair system, such as template switching via homologous recombination process can be activated (44,45). In mammalian B lymphocytes, R-loop and AID can trigger class switching in Ig gene to form DSBs, which in turn cause chromosomal translocations via NHEJ (16).
Besides Ig gene, R-loop can also be detected in oncogenes (e.g. Bcl6 and Myc), providing a link to such hallmarks of cancer as hypermutation and genome rearrangement (14). Defects in the repair of DNA strand breaks underpin many hereditary diseases such as neurodegeneration and immune dysfunction (46). In addition, recombination is not a risk-free event; for example there is a chance of loss of heterozygozity (LOH), which may eventually lead to development of cancer and other genetic diseases. We suggest that our DB could be useful for finding important associations between RLFS and such types of genome abnormalities. We assume that R-loops can initiate recombination during late S phase of the cell cycle and contribute to AID-dependent translocation of many oncogenes.
To elucidate the association between RLFS and TAR phenomena, we integrated R-loop data with recombination breakpoint data from (i) replication-induced recombination (47) and (ii) AID-dependent translocation data set (26). The data set (47) contains the chromosome locations of breakpoints found in Top1-deficient human colorectal carcinoma cells. Top1 is a key enzyme that plays an important role in the removal of DNA supercoiling associated with replication and transcription, leading to suppression of genomic instability by preventing interference between replication and transcription. The authors found that Top1-deficient cells accumulated replication forks stalling and recombination breakpoints in the S phase. In absence of Top1 protein, defective RNA processing leads to the formation of R-loops. That could block fork progression and finally generate DNA breaks. By over-expressing exogenous RNAaseH1 in the Top1-deficient cells, the authors produced evidence that degradation of RNA–DNA hybrids prevents R-loop formation during gene transcription.
We compared the regions of breakpoints (47) in transcribed genes with predicted RLFSs. We found overlaps of breakpoint and RLFS regions in several cancer-associated genes. For instance, Figure 4B shows chromosome map of Foxo3, as an example of co-localization of predicted RLFSs and experimentally induced replication-induced recombination breakpoints (48). Foxo3 belongs to the O-subclass of the fork head family of transcription factors that protect cells against a wide range of physiological stresses and is known as a tumour suppressor. Foxo3 has been recently reported to be a novel target of deletion in human lung adenocarcinoma (48). The Foxo3 deletion regions co-localize with the lung adenocarcinoma replication-induced recombination breakpoint region and RLFSs defined by our model. These findings suggest a causal role of R-loop formation in generation of replication-induced recombination breakpoints. One more compelling example is the co-localization of R-loop with the deletion regions of glycine dehydrogenase (GLDC) gene. GLDC is a component of the multiple-enzyme glycine cleavage system involved in the major pathway for degradation of glycine. The deletion in this gene is a major cause of non-ketotic hyperglycinaemia, an inborn error of glycine metabolism characterized by accumulation of glycine in body fluids leading to various neurological symptoms (49). However, the precise mechanism of deletions in GLDC has not been elucidated. Recently the sequence boundaries of the deletion regions in GLDC were identified (49). It was found that the most 5′end deletion breakpoints were located within 5′end gene region. 72% (18 out of 25) 5′end deletion breakpoints include exon1 - exon4 of 25 GLDC exons (49). We found 10 RLFSs; all the RLFSs were clustered within exon1, intron1, intron2 and intron4 (see “GLDC” in R-loopDB). Our database search result suggests that R-loop-mediated recombination in GLDC could be related to mechanisms caused non-ketotic hyperglycinaemia.
Another piece of evidence supporting direct association of our RLFS models with translocation break-points is the study of AID-dependent translocation breakpoints of Myc gene reported by Duquette et al. (26). Translocations of Myc to the Igh switch regions are typical for sporadic Burkitt's lymphomas (50,51). However, the detection of Igh-Myc translocations was found only in the wild-type, but not AID-deficient Il6-transgenic mice, implying involvement of AID in Igh-Myc translocation (52). Importantly, Duquette et al. reported the in vitro formation of R-loop in Myc gene. AID requires ssDNA substrate that can be generated by R-loop. To validate and show the association of R-loop with AID-dependent translocation breakpoints, we compared breakpoints of Myc gene to computationally predicted RLFSs. Figure 4C demonstrates that our model predicted RLFSs in the region overlapping AID-dependent translocation breakpoints and located near the translocations identified from Burkitt's lymphomas tissues and cell lines. These data support a causal role of R-loop formation in generation of AID-dependent translocation breakpoints.
RLFSs can be involved in alternative splicing
The connection between R-loop formation and activity of splicing factor ASF/SF2 in chicken cell line has been demonstrated by Li and Manley (12). The authors reported the unexpected finding that genetic inactivation of ASF/SF2 protein splicing factor, which is essential for alternative splicing process, resulted in the R-loop formation. The observation that ASF/SF2 protein prevents R-loop formation suggests function of ASF/SF2 protein in pre-mRNA processing and the location of R-loop formation next to the splice sites (53). However, the association between R-loop formation and splicing factors activity in the human genome is not clear. Linking the R-loopDB and the UCSC genome browser allows users to study associations between RLFS and various signals important for gene expression and genome alterations. Besides the alterations on the DNA sequence level, it may be interesting to study the connection of RLFS and alternative splicing process. As an example of such kind of analysis, we studied the localization of the RLFSs and the splice sites in Sorcs2 via UCSC genome browser integration. We explored the location of RLFS in this gene and found that RLFS overlapped with two start sites immediately after first exon (Figure 5A). We also found additional 15 regions where RLFSs co-localize with splice sites of Sorcs2 gene. Output from our analysis with co-localization information is presented in Table 1. Association of R-loop formation with exon skipping mechanism could be considered to support our findings. Figure 5B shows an example of such association. This is the first evidence of R-loop-mediated mRNA splicing in the human genes.
Figure 5.

RLFS associated with splice variants and exon skipping sites. (A) RLFS located near spliced sites of Sorcs2 gene. Blue line represents RLFS. Sorcs2 encodes sortilin-related vacuolar protein sorting 10 (VPS10) domain containing receptor 2, one family member of VPS10 domain-containing receptor proteins. The roles of VPS10P-domain receptors are regulation of neuronal viability and regulation of protein transport and signal transduction (58). This gene is strongly expressed in the central nervous system. The variation of Sorcs2 allele has been implicated in bipolar disorders (59). Additional 15 regions where RLFSs are co-localized with splice sites of Sorcs2 gene were defined in 1-kb region of splice sites. (B) RLFS are upstream located of exon skipping sites in Mad1l1 and Ptprn2 genes.
Table 1.
Regions where RLFSs co-localize with splice sites of Sorcs2 gene
| no. | RLFS ID | Chromosome and Coordinate of splice variant | Distance between RLFS and splice variant (bp) |
|---|---|---|---|
| 1 | RL041260 | chr4:7433013–7795463 | 864 |
| 2 | RL041261 | chr4:7434526–7793293 | 230 |
| 3 | RL041276 | chr4:7482922–7487600 | 710 |
| 4 | RL041295 | chr4:7518228–7526195 | 719 |
| 5 | RL041300 | chr4:7522978–7526195 | overlap |
| 6 | RL041305 | chr4:7534409–7535529 | overlap |
| 7 | RL041322 | chr4:7612776–7616372 | 569 |
| 8 | RL041336 | chr4:7691063–7795454 | 183 |
| 9 | RL041347 | chr4:7720979–7721178 | 931 |
| 10 | RL041349 | chr4:7742156–7767357 | 620 |
| 11 | RL041364 | chr4:7719787–7749888 | 437 |
| 12 | RL041368 | chr4:7735368–7755992 | 252 |
| 13 | RL041378 | chr4:7767811–7776361 | overlap |
| 14 | RL041380 | chr4:7780978–7789800 | 813 |
| 15 | RL041382 | chr4:7786907–7793133 | overlap |
RLFSs in cancer and neurodegenerative diseases related genes
Besides previously reported genes, we also identified novel RLFS in more than 200 important genes associated with cancer e.g. Tp53, BRCA1/BRCA2 and Kras (Figure 6A), genes common for central nervous system and neurodegenerative diseases e.g. ATM, Park2 and Ptprd (Figure 6B). According to our study R-loop forming mechanism can be associated with other cell types and diseases (data not presented). Information about RLFS abundance in the above mentioned genes is presented in the Figure 3B. This figure shows that genes related to cancer and neurodegenerative disease have low abundance of RLFS. Figure 5A and B confirms our discussion of RLFS co-localization with alternative splicing sites. Interestingly, several genes linked to cancer are also the targets of the mutator enzyme called AID.
Figure 6.


RLFSs associated with essential genes. (A) RLFSs on cancer-related genes (B) RLFSs on central nervous system and neurodegenerative diseases.
RLFSs as possible targets of epigenetic reprogramming
RLFS can result in extension of transcription bubble of non-template DNA strand and may play important role in gene modification and epigenetic reprogramming. Activation-induced cytidine deaminase/apolipoprotein B RNA-editing catalytic component (AID/APOBEC) is a group of enzymes capable of editing nucleic acid through deamination of cytosines to uracils. The recent discoveries indicated that AID is critical for epigenetic reprogramming in mammals (54,55). AID needs ssDNA substrate, and thus R-loop forming mechanism could provide a substrate for AID. This enzyme is active in primordial germ cells (PGCs) and in early embryos where demethylation occurs. The rate of methylation was found to be up to three fold higher in wild-type PGCs comparing to AID-deficient PGCs (54,55). AID-mediated demethylation occurred throughout the genome at specific target regions rather than globally and a mechanism regulating this demethylation is unknown. We hypothesize that R-loop structure may be a potential target of AID-mediated epigenetic reprogramming.
To support this hypothesis, we identified co-localization of RLFSs in Dazl and Foxo1 genes. These genes are known to become demethylated during PGC development and more highly methylated in AID-deficient PGCs (55). In the recent study, it has been shown that incorrect DNA methylation of Dazl gene is associated with defective human sperm (56). Figure 7 demonstrates that the predicted RLFSs of Dazl and Foxo1 genes are located in the demethylated area processed by AID. Interestingly, RLFSs are co-localized in the first intron and CpG islands of both genes. These findings and our other observations revealed by using R-loopDB search tool imply an association of RLFS with epigenetic modification and transcription initiation and elongation. Thus our preliminary study using R-loopDB suggests (i) an association of RLFS with AID activity which may be functional not only in case of Ig genes but also other genes related to epigenetic reprogramming and (ii) the RLFS model should be used in future study of a role of R-loop forming mechanism in AID-mediated epigenetic reprogramming. Other interesting directions of the implementation of predicted RLFSs (and R-loop formation) may be relevant to the mechanisms that underlie the RNA-directed transcription gene silencing (57) and Dnmt1- mediated DNA methylation in non-CpG context in DNA bubbles leading to silencing of DNA replication and transcriptionally active loci (60).
Figure 7.

Dazl and Foxo1 are demethylated by AID during PGC development and contain RLFS. Pink lines represent predicted RLFS.
Future experimental and technological approaches to analysis of RLFS and R-loops
The formation of R-loops using short RNA probes having RIZ and REZ sequences, predicted and collected in our R-loop DB can have several technological applications. Using computationally predicted RNA sequences, a method for directing the enzymatic double-stranded scission of RLFS DNA could be developed. A protocol of such ‘R-loop-extraction assay’ should consist of the following steps (i) sequence-specific R-loop formation; (ii) chemical modification of the displaced single strand of DNA with base-specific modification reagents to stabilize the R-loop such as neomycin (61–64) and block renaturation of DNA; (iii) hydrolysis of RNA used for R-loop formation to render both DNA strands sensitive for scission/cleavage at either end of single-stranded bubble formed by R-loop formation; (v) amplification of the specific RLFS DNA and (vi) computational analysis of the reaction products. Finally using the next generation sequencing (NGS) technique such method could be implemented in the highly specific assay to study the structural and functional roles of naturally occurring and artificially generated R-loop formation sequences in the individual human genes, different gene groups and genome regions.
CONCLUSION
In this work, we described a quantitative model of RLFS and created the R-loopDB, the first database of RLFS intended for detailed investigation of their sequences, location and RLFS-containing genes. Our web implementation supports various types of query that allows user to find not only genes of interest, but also their splice variants and the regions of epigenetic modifications associated with RLFSs. These regulatory signals can provide novel understanding of the gene expression regulation and complexity of RNA–DNA interactions in the genome and transcriptome functions.
The prediction of RLFSs in over half of the human genes reveals a novel level of RNA–DNA interactome complexity that perhaps will lead to a better understanding of the role of R-loop forming structure in gene expression controls and epigenetic modifications. The specific conformation of RNA–DNA hybrid formation also provides a unique target for controlling the transfer of genetic information through binding by small molecules. The knowledge of R-loop studies show that RNA can interact with DNA and generates a few beneficial effects and a lot of harmful effects in cells. In our study, we provide biological insights into the R-loop structure in several molecular machineries. In particular, our findings suggested that (i) over half of transcripts contain at least one R-loop indicating that RLFSs present a common regulatory element essential for gene expression controls and epigenetic modifications; (ii) multiple occurrences of the RLFS in essential genes suggest specific role of RLFS in these genes; (iii) R-loops may be directly involved in alternative splicing process; (iv) mutation and genome variations may be associated with R-loop formation and (v) RLFS may help AID in epigenetic reprogramming in development. Finally, our database provides comprehensive analysis of R-loops in essential genes related to cancer, neurodegenerative diseases and many genetic diseases.
We provide a workflow of R-loop extraction assay, which could be used for implementation of our R-loopDB predictions. Identification of RLFS in personal human genomes, mammalian and non-mammalian species and analysis of conservation and evolution of RLFS will be studied in the further study.
We found that R-loops are widely encountered in a vast majority of genes of the human genome. R-loopDB provides the first comprehensive catalogue of RLFS, which could be used in the systematic studies of the structures and functions of R-loops in normal and abnormal cells, as well as in the drug industry and clinical research applications. We expect that R-loopDB will help researchers in the R-loop analysis and design of the experiments aimed to discover mutated sites and epigenetic modifications in RLFS- identified genes. We also believe that R-loopDB will be useful for drug discovery and identification of new classes of therapeutic targets.
FUNDING
Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore. Funding for open access charge: Bioinformatics Institute, A*Star, Singapore.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors are grateful to Dr Micheal R. Lieber for initiation of our interest to R-loops and the discussion of the parameters of the R-loop model and Dr Aliaksandr Yarmishyn for useful comments and suggestion to improve the manuscript.
REFERENCES
- 1.Thomas M, White RL, Davis RW. Hybridization of RNA to double-stranded DNA: formation of R-loops. Proc. Natl Acad. Sci. USA. 1976;73:2294–2298. doi: 10.1073/pnas.73.7.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosbash M, Blank D, Fahrner K, Hereford L, Ricciardi R, Roberts B, Ruby S, Woolford J. R-looping and structural gene indentification of recombinant DNA. Methods Enzymol. 1979;68:454–469. doi: 10.1016/0076-6879(79)68035-7. [DOI] [PubMed] [Google Scholar]
- 3.Woolford JL, Jr, Rosbash M. The use of R-looping for structural gene identification and mRNA purification. Nucleic Acids Res. 1979;6:2483–2497. doi: 10.1093/nar/6.7.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell. 1977;12:1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- 5.Drolet M, Phoenix P, Menzel R, Masse E, Liu LF, Crouch RJ. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl Acad. Sci. USA. 1995;92:3526–3530. doi: 10.1073/pnas.92.8.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy D, Yu K, Lieber MR. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 2008;28:50–60. doi: 10.1128/MCB.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell. 2003;12:711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Roy D, Lieber MR. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol. Cell. Biol. 2009;29:3124–3133. doi: 10.1128/MCB.00139-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol. Cell Biol. 2010;30:146–159. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 11.Taylor SD, Solem A, Kawaoka J, Pyle AM. The NPH-II helicase displays efficient DNA x RNA helicase activity and a pronounced purine sequence bias. J. Biol. Chem. 2010;285:11692–11703. doi: 10.1074/jbc.M109.088559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duquette ML, Huber MD, Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res. 2007;67:2586–2594. doi: 10.1158/0008-5472.CAN-06-2419. [DOI] [PubMed] [Google Scholar]
- 15.Camps M, Loeb LA. Critical role of R-loops in processing replication blocks. Front Biosci. 2005;10:689–698. doi: 10.2741/1564. [DOI] [PubMed] [Google Scholar]
- 16.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 17.Gottipati P, Cassel TN, Savolainen L, Helleday T. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol. Cell. Biol. 2008;28:154–164. doi: 10.1128/MCB.00816-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair. 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Soulas-Sprauel P, Rivera-Munoz P, Malivert L, Le Guyader G, Abramowski V, Revy P, de Villartay JP. V(D)J and immunoglobulin class switch recombinations: a paradigm to study the regulation of DNA end-joining. Oncogene. 2007;26:7780–7791. doi: 10.1038/sj.onc.1210875. [DOI] [PubMed] [Google Scholar]
- 21.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 22.Lin Y, Dent SY, Wilson JH, Wells RD, Napierala M. R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl Acad. Sci. USA. 2010;107:692–697. doi: 10.1073/pnas.0909740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIvor EI, Polak U, Napierala M. New insights into repeat instability: Role of RNA.DNA hybrids. RNA Biol. 2010;7:551–558. doi: 10.4161/rna.7.5.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naik AK, Lieber MR, Raghavan SC. Cytosines, but not purines, determine recombination activating gene (RAG)-induced breaks on heteroduplex DNA structures: implications for genomic instability. J. Biol. Chem. 2010;285:7587–7597. doi: 10.1074/jbc.M109.089631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2010;39:1749–1762. doi: 10.1093/nar/gkq935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duquette ML, Pham P, Goodman MF, Maizels N. AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation. Oncogene. 2005;24:5791–5798. doi: 10.1038/sj.onc.1208746. [DOI] [PubMed] [Google Scholar]
- 27.Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 28.Kuznetsov VA. Family of skewed distributions associated with the gene expression and proteome evolution. Sign. Process. 2003;83:889–910. [Google Scholar]
- 29.Kuznetsov VA, Singh O, Jenjaroenpun P. Statistics of protein-DNA binding and the total number of binding sites for a transcription factor in the mammalian genome. BMC Genomics. 2010;11(Suppl. 1):S12. doi: 10.1186/1471-2164-11-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu K, Roy D, Bayramyan M, Haworth IS, Lieber MR. Fine-structure analysis of activation-induced deaminase accessibility to class switch region R-loops. Mol. Cell. Biol. 2005;25:1730–1736. doi: 10.1128/MCB.25.5.1730-1736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunnick W, Hertz GZ, Scappino L, Gritzmacher C. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 1993;21:365–372. doi: 10.1093/nar/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nat. Immunol. 2003; 4:435–441. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- 33.Richards EG, Zaveri HP, Wolf VL, Kang SH, Scott DA. Delineation of a less than 200 kb minimal deleted region for cardiac malformations on chromosome 7p22. Am. J. Med. Genet. A. 2011;155:1729–1734. doi: 10.1002/ajmg.a.34041. [DOI] [PubMed] [Google Scholar]
- 34.Xu B, Woodroffe A, Rodriguez-Murillo L, Roos JL, van Rensburg EJ, Abecasis GR, Gogos JA, Karayiorgou M. Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc. Natl Acad. Sci. USA. 2009;106:16746–16751. doi: 10.1073/pnas.0908584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coe BP, Lee EH, Chi B, Girard L, Minna JD, Gazdar AF, Lam S, MacAulay C, Lam WL. Gain of a region on 7p22.3, containing MAD1L1, is the most frequent event in small-cell lung cancer cell lines. Genes Chromosomes Cancer. 2006;45:11–19. doi: 10.1002/gcc.20260. [DOI] [PubMed] [Google Scholar]
- 36.Bullinger L, Kronke J, Schon C, Radtke I, Urlbauer K, Botzenhardt U, Gaidzik V, Cario A, Senger C, Schlenk RF, et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2010;24:438–449. doi: 10.1038/leu.2009.263. [DOI] [PubMed] [Google Scholar]
- 37.Roversi G, Pfundt R, Moroni RF, Magnani I, van Reijmersdal S, Pollo B, Straatman H, Larizza L, Schoenmakers EF. Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006;25:1571–1583. doi: 10.1038/sj.onc.1209177. [DOI] [PubMed] [Google Scholar]
- 38.Olejniczak ET, Van Sant C, Anderson MG, Wang G, Tahir SK, Sauter G, Lesniewski R, Semizarov D. Integrative genomic analysis of small-cell lung carcinoma reveals correlates of sensitivity to bcl-2 antagonists and uncovers novel chromosomal gains. Mol. Cancer Res. 2007;5:331–339. doi: 10.1158/1541-7786.MCR-06-0367. [DOI] [PubMed] [Google Scholar]
- 39.Prakash SK, LeMaire SA, Guo DC, Russell L, Regalado ES, Golabbakhsh H, Johnson RJ, Safi HJ, Estrera AL, Coselli JS, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 2010;87:743–756. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shemanko CS, Mellerio JE, Tidman MJ, Lane EB, Eady RA. Severe palmo-plantar hyperkeratosis in Dowling-Meara epidermolysis bullosa simplex caused by a mutation in the keratin 14 gene (KRT14) J. Invest. Dermatol. 1998;111:893–895. doi: 10.1046/j.1523-1747.1998.00388.x. [DOI] [PubMed] [Google Scholar]
- 42.Pfendner EG, Sadowski SG, Uitto J. Epidermolysis bullosa simplex: recurrent and de novo mutations in the KRT5 and KRT14 genes, phenotype/genotype correlations, and implications for genetic counseling and prenatal diagnosis. J. Invest. Dermatol. 2005;125:239–243. doi: 10.1111/j.0022-202X.2005.23818.x. [DOI] [PubMed] [Google Scholar]
- 43.Coulombe PA, Hutton ME, Letai A, Hebert A, Paller AS, Fuchs E. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: genetic and functional analyses. Cell. 1991;66:1301–1311. doi: 10.1016/0092-8674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- 44.Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011;25:2041–2056. doi: 10.1101/gad.17010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Gonzalez B, Felipe-Abrio I, Aguilera A. The S-phase checkpoint is required to respond to R-loops accumulated in THO mutants. Mol. Cell. Biol. 2009;29:5203–5213. doi: 10.1128/MCB.00402-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu. Rev. Genomics Hum. Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 47.Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009;11:1315–1324. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mikse OR, Blake DC, Jr, Jones NR, Sun YW, Amin S, Gallagher CJ, Lazarus P, Weisz J, Herzog CR. FOXO3 encodes a carcinogen-activated transcription factor frequently deleted in early-stage lung adenocarcinoma. Cancer Res. 2010;70:6205–6215. doi: 10.1158/0008-5472.CAN-09-4008. [DOI] [PubMed] [Google Scholar]
- 49.Kanno J, Hutchin T, Kamada F, Narisawa A, Aoki Y, Matsubara Y, Kure S. Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J Med Genet. 2007;44:e69. doi: 10.1136/jmg.2006.043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muller JR, Janz S, Potter M. Differences between Burkitt's lymphomas and mouse plasmacytomas in the immunoglobulin heavy chain/c-myc recombinations that occur in their chromosomal translocations. Cancer Res. 1995;55:5012–5018. [PubMed] [Google Scholar]
- 51.Neri A, Barriga F, Knowles DM, Magrath IT, Dalla-Favera R. Different regions of the immunoglobulin heavy-chain locus are involved in chromosomal translocations in distinct pathogenetic forms of Burkitt lymphoma. Proc. Natl Acad. Sci. USA. 1988;85:2748–2752. doi: 10.1073/pnas.85.8.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Aguilera A. mRNA processing and genomic instability. Nat. Struct. Mol. Biol. 2005;12:737–738. doi: 10.1038/nsmb0905-737. [DOI] [PubMed] [Google Scholar]
- 54.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Navarro-Costa P, Nogueira P, Carvalho M, Leal F, Cordeiro I, Calhaz-Jorge C, Goncalves J, Plancha CE. Incorrect DNA methylation of the DAZL promoter CpG island associates with defective human sperm. Hum. Reprod. 2010;25:2647–2654. doi: 10.1093/humrep/deq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc. Natl Acad. Sci. USA. 2007;104:12422–12427. doi: 10.1073/pnas.0701635104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Willnow TE, Petersen CM, Nykjaer A. VPS10P-domain receptors - regulators of neuronal viability and function. Nat. Rev. Neurosci. 2008;9:899–909. doi: 10.1038/nrn2516. [DOI] [PubMed] [Google Scholar]
- 59.Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, Schulze TG, Cichon S, Rietschel M, Nothen MM, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry. 2008;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arya DP, Coffee RL, Jr, Willis B, Abramovitch AI. Aminoglycoside-nucleic acid interactions: remarkable stabilization of DNA and RNA triple helices by neomycin. J. Am. Chem. Soc. 2001;123:5385–5395. doi: 10.1021/ja003052x. [DOI] [PubMed] [Google Scholar]
- 62.Charles I, Xi H, Arya DP. Sequence-specific targeting of RNA with an oligonucleotide-neomycin conjugate. Bioconjug. Chem. 2007;18:160–169. doi: 10.1021/bc060249r. [DOI] [PubMed] [Google Scholar]
- 63.Shaw NN, Arya DP. Recognition of the unique structure of DNA:RNA hybrids. Biochimie. 2008;90:1026–1039. doi: 10.1016/j.biochi.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 64.Shaw NN, Xi H, Arya DP. Molecular recognition of a DNA:RNA hybrid: sub-nanomolar binding by a neomycin-methidium conjugate. Bioorg. Med. Chem. Lett. 2008;18:4142–4145. doi: 10.1016/j.bmcl.2008.05.090. [DOI] [PubMed] [Google Scholar]