


Abstract
RNA junctions are important structural elements that form when three or more helices come together in space in the tertiary structures of RNA molecules. Determining their structural configuration is important for predicting RNA 3D structure. We introduce a computational method to predict, at the secondary structure level, the coaxial helical stacking arrangement in junctions, as well as classify the junction topology. Our approach uses a data mining approach known as random forests, which relies on a set of decision trees trained using length, sequence and other variables specified for any given junction. The resulting protocol predicts coaxial stacking within three- and four-way junctions with an accuracy of 81% and 77%, respectively; the accuracy increases to 83% and 87%, respectively, when knowledge from the junction family type is included. Coaxial stacking predictions for the five to ten-way junctions are less accurate (60%) due to sparse data available for training. Additionally, our application predicts the junction family with an accuracy of 85% for three-way junctions and 74% for four-way junctions. Comparisons with other methods, as well applications to unsolved RNAs, are also presented. The web server Junction-Explorer to predict junction topologies is freely available at: http://bioinformatics.njit.edu/junction.
INTRODUCTION
Our fascination with RNA has grown enormously in recent years due to the many newly discovered structured RNAs with diverse functions (1–3). Indeed, RNA's diversity in size, shape and function is well recognized, from the small (∼23 nt) microRNA elements involved in post-transcriptional regulation of genes within plants and animals (4) to large ribosomal RNAs (∼3200 nt) responsible for protein synthesis (5–7). A thorough understanding of RNA structures and functions requires knowledge of RNA structure and dynamics. Although research over the past 30 years has produced many advances in RNA secondary structure prediction, RNA 3D structure prediction remains elusive for the large part, mainly due to the difficulty in recognizing long-range interactions (8), especially without resorting to manual manipulation and intuition (9,10).
An RNA ‘junction’, also known as multi-branch loop, is the point of connection between different helical (double-stranded) segments (11) (Figure 1a). This secondary structure element is common to many RNA molecules and is involved in a wide range of functional roles, including the self-cleaving catalytic domain of the hammerhead ribozyme (12), the recognition of the binding pocket domain by purine riboswitches (13) and the translation initiation of the hepatitis C virus at the internal ribosome entry site (14). Since junctions serve as major architectural features in RNA, it is essential to understand their structural, energetic and dynamic properties.
Figure 1.

(a) (left) 2D diagram of a three-way junction and (right) its 3D representation element composed of three helices labeled and color-coded by H1 (red), H2 (purple), and H3 (green), and the corresponding single stranded loop regions labeled L1 to L3 with nucleotides color-coded in gray. Helices and loop regions are labeled in a unique way according to the 5′ to 3′ orientation of the entire RNA structure, by labeling H1 as the first helix encountered, while entering the junction region, as one moves along the nucleotide chain in the 5′ to 3′ direction and so forth. (b) Histogram from a total of 216 RNA junctions sorted by branching degree ranging from 3 (3WJ) to 10 (10WJ).
Junctions can be described in terms of the ‘coaxial stacking’ of helices, a stacking of two separate helical elements that form a contiguous helix. Coaxial stacking motifs occur in several large RNA structures, including tRNA (15), pseudoknots (16), group II intron (17) and the large ribosomal subunits (5–7) (see examples in Figure 2). Coaxial stacking provides thermodynamic stability to the molecule as a whole (18,19) and reduces the separation between loop regions within junctions (20). Moreover, coaxial stacking interactions form cooperatively with long-range interactions in many RNAs (21) and are thus essential features that distinguish different junction topologies.
Figure 2.

Examples of three- and four-way junction families presented in 3D representations. RNA three- and four-way junctions can be classified into 3 and 9 families, respectively, according to their coaxial stacking patterns and topology (22,23). Helical elements are color coded in cyan, purple, red and green, respectively. Junctions are labeled according to Supplementary Table S1.
Analyses of solved crystal structures have shown that RNA junctions can be grouped into families according to 3D shape or topology. Lescoute and Westhof (22) categorized topologies of three-way junctions in folded RNAs as families A, B and C (Figure 2); in most of three-way junctions, two helices stack coaxially. Laing and Schlick (23) grouped four-way junctions into 9 families, namely H, cH, cL, cK, π, cW, ψ, cX and X (Figure 2), according to coaxial stacking interactions and helical conformation signatures. They showed that coaxial stacking likely forms when the loop between helices is short, though other factors like sequence content (24), base-stacking interactions of non-canonical base pairs (25) and protein binding (26) also affect coaxial stacking orientations. Laing et al. (27) later showed that higher order junctions (5 or more helices) can be structurally decomposed into sub-junctions resembling local helical configurations found in three- and four-way junctions. These and other studies (28,29) underscore the notion that RNA junctions tend to organize their helical components in parallel and perpendicular helical configurations.
Experimental and computational studies of specific RNAs have also advanced our understanding of the structural and dynamical properties of RNA junctions. Lilley et al. (30–32) studied the helical organization of junctions in DNA and RNA using Fluorescence resonance energy transfer (FRET), and observed transitional changes and flexibility in their helical configuration under Mg2+ and Na+ concentration variations. Analyses of the ribosome crystal structure from the Steitz lab have shown that junctions are more flexible in the absence of binding proteins (33). Tyagi and Mathews (34) predicted coaxial stacking for pairs of consecutive helices with one or none intervening mismatch loops, by free energy minimization. Recent molecular dynamics studies on three-way junctions by the Leontis and Šponer (35) groups have demonstrated the high flexibility of helical elements which can lead to hinge-like motions as well as other small localized fluctuations, to accommodate other RNAs. Aalberts and Nandagopal (20) reported that coaxial stacking interactions formed in adjacent helical elements provide an entropic free-energy benefit that can be used for RNA secondary structure prediction. Of interest is also the database RNAJunction by Bindewald et al. (36), containing information on RNA structural elements including junctions. In the goal of facilitating the initial stage of predicting RNA tertiary structure, we develop here a computational approach based on the established random forests method to predict the arrangement of helices within junctions.
The data mining algorithm called random forests (37) uses a set of input parameters (feature vectors) for training the prediction protocol. We develop here feature vectors composed of structural information from solved RNA structures (see ‘Materials and Methods’ section) to predict both coaxial stacking arrangements and topologies of junctions at the secondary structure level. Our final results of ∼80% accuracy for three- and four-way junctions and 60% for higher-order junctions constitute a dramatic improvement over previous attempts.
We also analyze the contribution of each feature parameter and find that 8 out of the 15 variables for three-way junctions, and 8 out of 18 variables for four-way junctions provide essential contributions for coaxial stacking prediction, while 4–6 parameters are essential for junction family prediction. Recurring important feature parameters describe the size of loops within junctions and base-pair configurations at the end of helices. Applications to non-crystallized RNA junctions illustrate how our approach can be used to predict 3D configurations of junctions, namely the topology and coaxial stacking patterns. These predictions also demonstrate agreement with previous predictions and experimental FRET analysis (22,34,38–40).
MATERIALS AND METHODS
The random forests approach (Figure 3), first proposed by Breiman in 2001 (37), employs many random decision trees (200 used here for each random forest) which are built from the training set of input parameters such as loop length and number of consecutive adenines within loops (Table 1). Such parameters, which we define based on solved RNA structures (see below), define the ‘feature vectors’. Predictions are then made based on the majority votes among all decision tree results. The method uses a standard statistical method called 10-fold cross-validation to divide and test each tenth of the junction data at a time, and train the random forests classifier using the remaining nine pieces. The 10-fold cross-validation procedure is applied 75 times to ensure an unbiased selection of the data partition. The average prediction score over all random forests generated is then reported in Table 2. Below we describe our data and methodology.
Figure 3.

Sequence and loop length information from junctions is defined on the basis of secondary structure (a), to predict, by the random forests approach (b), the coaxial stacking as well as the junction family type (c). Color coding and symbols in the decision tree are defined as follows. Loop length features (|Li|, min(|L1|,|L2|), |L1′|) in blue, maximum number of consecutive adenines in loop (A(Li)) in orange, and free-energy associated with a pair of helices (ΔG(Hi,Hi+1), with i = 1, number of helices) in magenta (Table 1).
Table 1.
Parameter list used for coaxial stacking and junction family type prediction
| Feature | Description |
|---|---|
| |L1|, |L2|, |L3| | Loop lengths between junctions, labeled according to the 5′ to 3′ orientation of the entire RNA structure |
| Min(|L2|,|L3|), Min(|L1|,|L3|), Min(|L1|,|L2|) | Minimum loop lengths |
| |L1′|, |L2′|, |L3′| | Loop lengths sorted in ascending order (|L1′| ≤ |L2′| ≤ |L3′|) |
| A(L1), A(L2), A(L3) | Maximum number of consecutive adenines in loops L1, L2 and L3, respectively |
| ΔG(H1,H2), ΔG(H2,H3), ΔG(H3,H1) | Thermodynamic free-energy associated to the helical stacking between H1 and H2, H2 and H3, H3 and H1, respectively |
List of the 15 parameters used for 3-way junctions. Similarly, for 4-way junctions, there are 18 parameters. For higher-order junctions, there are 10 parameters because we use a divide and conquer approach as described in the text. At the nodes of every decision tree, 3 parameters are selected randomly out of the total for 3-way and higher-order junctions in order to grow the tree by partitioning the node into 2 new branches, and 4 parameters for 4-way junctions.
Table 2.
Prediction performances for coaxial stacking and junction family type
| Coaxial stacking prediction | 3-way junctions (%) | 4-way junctions (%) |
| No assumption on junction family | 81 | 77 |
| Junction family assumed known | 83 | 87 |
| Junction family prediction | 3-way junctions (%) | 4-way junctions (%) |
| No assumption on coaxial stacking | 85 | 74 |
| Coaxial stacking assumed known | 86 | 81 |
| Coaxial stacking prediction | 5 to 10-way junction (%) | |
| 60 | ||
| Two-step coaxial stacking prediction | 3-way junctions (%) | 4-way junctions (%) |
| (junction family, stacking) | 82 | 80 |
| Two-step junction family prediction | 3-way junctions (%) | 4-way junctions (%) |
| (coaxial stacking, junction family) | 86 | 71 |
Prediction performance is shown for each experiment by determining junction family type for 3 and 4-way junctions and coaxial helical stacking for all junctions. Prediction values improve when knowledge of the junction family type or coaxial stacking is provided.
Dataset of RNA junctions
We updated our previous non-redundant high-resolution data collection of 3D RNA junctions (23,27) by collecting recent structures from the RCSB Protein Data Bank, based on available structures as of November 2010. Our new dataset of 216 RNA junctions (Supplementary Table S1) containing coaxial stacking and junction family information is classified as before for junction order manually. Figure 1b shows a frequency histogram distribution of junctions arranged by degree of branching. More than half are three-way junctions, and the number decreases as the degree of branching increases. Junctions of higher order occur in RNAs of larger size such as the ribonuclease, group II intron and ribosomal RNA, whereas low-order junctions occur in a wide range of RNAs, from riboswitches to ribosomal RNAs. In many cases (128 out of the total 216 junctions), junctions appear within the RNA structure next to each other, separated only by a single helix. This occurs especially for large RNAs such as the ribosomal RNA, and ribonuclease P, where neighboring helices often align their corresponding coaxial helices to form a large helical element composed of two or more coaxial stacks. As previously noted (29), these large elements tend to segregate into domains, are planar in overall shape, and are stabilized by long-range interaction motifs.
Description of the feature vector
To predict coaxial stacking and junction family types, we use information from RNA secondary structures such as the loop length within junctions, sequence content and free-energy associated to base-stacking interactions between the base pairs at the end of helices and their common loop region, as calculated by Mathews et al. (41,42). Table 1 lists the 15 parameters used for three-way junctions. Similarly, there are 18 parameters for four-way junctions and 10 for higher-order junctions (Supplementary Tables S2–S4). In each decision step, we consider for example, the loop length |Li| between any pair of consecutive helices Hi and Hi+1, and the minimum loop length between the neighboring loops (min(Li-1|, |Li+1|)); a smaller loop length from a neighboring loop (|Li−1|, |Li+1|) can compete in coaxial stacking formation. Loop lengths are incorporated in ascending order to improve prediction accuracy. In addition, the maximum number of consecutive adenines, A(Li), for each loop Li, is considered since it has been reported that adenines in loops often form A-minor motifs (43) in specific junction topologies (22,23,27).
To improve the prediction of coaxial stacking between helices in junctions, we include thermodynamic parameters, taken from the program RNAstructure (44), associated with each terminal base pair from contiguous helices and the loop Li intervening sequence (Figure 3a, ΔG parameter). If |Li| = 0, we use the free-energy values from the table of coaxial stacking for two helices with no intervening unpaired nucleotide. If the junction loop length |Li| = 1, we use the free-energy values from the table of coaxial stacking with one intervening mismatch, plus 2.1 kcal/mol for the terminal mismatch free-energy, as suggested by Tyagi and Mathews (34). As a terminal mismatch in Li can potentially form a non-canonical base-pair with a nucleotide in Li–1 or Li+1, we consider the minimum free-energy value for both cases. Since it is not possible to determine experimentally the thermodynamic parameters for loops of any length |Li|, the free-energies in junctions with loop lengths greater than one are estimated using a linear or a logarithmic function as follows. If 2 ≤ |Li| ≤ 6, we use formula 21 from (42) given by a+b|Li|+ch, where a, b and c are constants, and h denotes the number of helical elements. However, if |Li| > 6, we apply formula 22 from (42) given by a+6 b+1.1ln(|Li|/6) + ch. To restrict the free-energy to the coaxial stacking region of interest we define h, the number of helical elements, to be equal to 2. The value b = −0.3 is taken from (42), and the values a = 9.3 and c = −0.9 correspond to the recently optimized parameters (41).
Feature parameters for higher order junctions are defined ‘locally’. Specifically, to determine whether helices Hi and Hi+1 are coaxially stacked, we take into account the loop length |Li| between Hi and Hi+1 and their neighboring loop lengths |Li–1| and |Li+1|. We also use the number of consecutive adenines in Li–1, Li and Li+1, the minimum of the lengths of the neighboring loops (min(|Li–1|, |Li+1|)), and the thermodynamic free-energy associated to the coaxial stacking of Hi with Hi+1 as well as those of neighboring helices (Hi–1 and Hi, Hi+1 and Hi+2, respectively). In total we have 15 feature parameters for three-way junctions, 18 for four-way junctions, and 10 for higher order junctions.
Finally, the combined data of all these parameters are stored as feature vectors and then applied to the random forests classifier to train and then predict the most favorable junction family type and coaxial stacking (Figure 3b–c). Tables S2–S4 list in detail the feature parameters for all the junctions considered.
Prediction using random forests
In the training phase for each category, decision trees are built using 90% of the feature data at a time. Thus, we use 99 data elements for three-way junctions and about 58 for four-way junctions. For higher order junctions, we use data elements from 191 pairs of consecutive helices from higher order junctions (90%) plus 590 data elements from pairs of consecutive helices found in lower order junctions to augment the training data.
To build and grow each decision tree, we start at the top and select a number m of parameters at random, out of the total number of feature parameters M (Table 1), to split each node into two new branches. The value of m, which is held constant for the in forest, corresponds to the integer part of 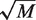 and is the default recommended value by (37). We set m = 4 for four-way junctions and m = 3 for three-way and higher order junctions. The best node partitioning, among all m parameters, is determined by the Gini criterion algorithm that optimizes the node splitting at each step (45).
and is the default recommended value by (37). We set m = 4 for four-way junctions and m = 3 for three-way and higher order junctions. The best node partitioning, among all m parameters, is determined by the Gini criterion algorithm that optimizes the node splitting at each step (45).
Each node split corresponds to a training data partitioning (Figure 3b). Each tree is grown by repeating the process recursively for each node until all the training data are considered. This tree-growing procedure is then repeated 150 000 times to generate 200 trees for each random forest, 10 random forests for each 10-fold cross-validation procedure and 75 times for each cross-validation implementation to ensure an unbiased selection of the data partition. The parameter choices for both the number of 10-fold cross-validation repeats (75) and the number of trees (200) per random forest were analyzed and optimized by testing several values. Supplementary Figure S1 shows the analysis of parameters for both coaxial stacking and junction family prediction on four-way junctions. Essentially, we inspect the convergence of the prediction accuracy of junction data, and select the smallest parameter values that produce approximately the same prediction performance. The parameters given by 75 for 10-fold cross-validation repeats and 200 trees per random forest are also optimal for 3 and higher order junction predictions.
In the prediction phase, we take the feature vectors associated with each tenth of the junction data from the 10-fold cross-validation partition, and apply the already built random forests from each category to make predictions. The average prediction accuracy over all 10 partitions is recorded. Finally, the average prediction score over all 75 tests is then reported (Supplementary Tables S2–S5).
The training and prediction phases are implemented for all experiments using the random forests package on the R software (ver. 2.9.0) for statistical computing (46). The implementation uses a Linux Intel Pentium 4 3.0 Ghz processor. Single-step experiments take about 8 min of computing time, while two-step procedures require about 16 min. The training work took most of the time since it involves building the 200 decision trees, 75 times for each experiment. We tested the random forests procedure using other feature parameters, such as the difference between neighboring loop lengths, and the amount of adenines in loops, regardless of whether they appear consecutively or not. By measuring the significance of each parameter using the random forest protocol, we settled on the set of parameters used here that works best.
RESULTS
To simplify the analysis, we label the loop region between each pair of consecutive helices Hi and Hi+1 as Li. Each helix Hi and loop region Li is labeled according to the 5′ to 3′ orientation of the entire RNA (Figure 1a). The prediction results presented below are summarized in Table 2.
Coaxial stacking predictions for three- and four-way junctions
Three-way junctions are the most abundant type of junctions, representing about 51% of the total junctions. Any three-way junction can form a coaxial stack between helices H1H2, H2H3, H3H1, or, rarely, no coaxial stack. A coaxial stacking almost never occurs (<5% of the 110 three-way junctions) for helices sharing the loop region with maximum length. Therefore, given the relative data wealth and the loop region restriction, a better prediction is expected for three-way junctions compared to higher order junctions. Indeed, the random forests approach yields a coaxial stacking score of 81% (see Supplementary Table S5 for details).
In some circumstances, structural knowledge from the junction family can be determined in advance by using experimental methods such as FRET (47), Small-angle X-ray scattering (SAXS) (48), and Cryo-electron microscopy (Cryo-EM) (49). If the junction family is known a priori, it can be added as an additional parameter in the feature vector. This additional experimental input improves the coaxial stacking prediction accuracy to 83% (Table 2).
For four-way junctions, the number of coaxial helical stacking interactions can be zero, one or two and there are seven possible types of coaxial helical stacking configurations: H1H2, H2H3, H3H4, H4H1, H1H2–H3H4, H2H3–H4H1, no-stacking. When we apply the random forests classifier to predict four-way junction stacking (Supplementary Table S6), we obtain a 77% accuracy, slightly lower than three-way junction prediction, mainly due to the reduction of available structures (65 four-way junctions versus 110 three-way junctions). However, by adding knowledge from the junction family as one additional feature parameter, the coaxial stacking prediction accuracy improves substantially to 87% (Table 2).
Junction family prediction for three- and four-way junctions
To determine the junction family type for three- and four-way junctions, we must distinguish among the three types for three-way and nine possible families of four-way junctions (Figure 2). The random forests approach produces a prediction accuracy for junction family type of 85% for three-way junctions and 74% for four-way junctions. Moreover, by assuming coaxial stacking information a priori, and including it as an additional feature parameter, the accuracy improves to 86% for three-way junctions and 81% for four-way junctions, respectively (see Table 2, and Supplementary Tables S5–S6 for prediction performance details).
Prediction of coaxial helical stacking for higher order junctions
Junctions with 5 or more helices are less common, and therefore much less data are available for training the random forests classifier. In addition, no junction family classification can be determined, and the set of coaxial stacking configurations grows rapidly. However, we aim to predict whether any two consecutive helices in a junction stack coaxially or not, by including as training data the coaxial/no-coaxial stacking information for all pairs of consecutive helices found in lower-order, as well as higher-order, junctions. We then apply the 10-fold cross validation procedure only to the higher-order junctions. As Table 2 shows, the accuracy of our prediction is 60%. The usefulness of this method is also evaluated in the supplementary material using the sensitivity (60%) and positive predictive value (76%) (Supplementary Table S7). Essentially, these measurements show that we make fewer false positive predictions of coaxial stacks than false negatives.
Prediction using a two-step procedure
Coaxial stacking predictions improve when the junction topology information is included in the feature vector. While experiments might provide some additional information, these methods are expensive, time consuming and impractical to implement for every junction possibility. However, the random forests prediction for junction families provides an alternative. We thus propose a prediction protocol for coaxial stacking in two steps. First, we predict the junction family type using random forests. Second, we predict coaxial stacking by adding the prediction values as additional feature parameter, and evaluate the performance of this classifier. Although prediction accuracy from the first step is imperfect, applications to 3-way and 4-way junctions yield 82% and 80% overall prediction accuracy, respectively, with this 2-step protocol. These results represent a modest improvement (of about 1% and 3%, respectively) over the prediction of coaxial stacking with no assumptions on the junction family (Table 2). When we perform a two-step procedure to predict the junction family for 3 and 4-way junctions, by first predicting coaxial stacking, prediction accuracy increases to 86% for 3-way junctions and decreases (to 71%) for 4-way junctions (see also Supplementary Table S8 for prediction details).
Analysis on the importance of the feature parameters
To predict the helical arrangements of RNA junctions, the random forests method requires a training set built from feature parameters taken from solved RNA crystal structures, as described in ‘Materials and Methods’ section. These feature parameters are determined from the size and sequence loop, as well as base pairing configurations within junctions (Table 1). To determine the significance of each of our selected features on predicting coaxial stacking as well as junction topology, we apply the random forest protocol to evaluate each feature parameter independently, as well as their cumulative contribution for predicting coaxial stacking and junction topology on three- and four-way junctions. The results are presented on Supplementary Figure S2, where the prediction accuracy of each independent parameter is presented as a bar within a histogram and the cumulative prediction accuracy is shown as a polygonal graph.
In predicting coaxial stacking for three and four-way junctions, a minimum of eight parameters is required to make predictions efficiently, whereas at least 4–6 parameters are required for junction family prediction. Although the relevance of feature elements varies for each experiment, a recurring finding is the importance of the parameter describing the size of loops between helical elements (20,22,27): this is especially notable for coaxial stacking predictions of both three- and four-way junctions, where at least four of the most significant parameters are length dependent. Indeed, base stacking interactions involve London dispersion forces (50), hydrophobic forces (51), and interactions between partial charges within the nucleobase rings (52), all of which are related to the distance between the base pairs. Stacking forces on nucleic acids can also involve π–π interaction forces (53,54), although ab initio calculations by Šponer et al. (55) have disputed this fact. In addition to loop size, the base pairing configurations at the end of helices also emerge as crucial for predicting both coaxial stacking and junction families; for four-way junction families, three parameters related to base pairing, are among the top six parameters.
We also note that the feature parameters that represent the content of adenines is low in the ranking and often falls last; still, exceptions can be noted: for three-way junction predictions, an adenine feature ranks second for junction family prediction and fifth out of eight of the top features for coaxial stacking prediction. While adenine features are recognized as important in junction families (22,23), we suggest that their diverse structural roles within junctions blur the signal in our context.
Comparison with other methods
To compare our application to a previous approach by Tyagi and Mathews (34), we have pursued the comparisons below. Direct comparisons are difficult because while Tyagi and Mathews predict coaxial stacking for pairs of consecutive helices with one or none intervening mismatch loops by free energy minimization, we do not restrict the size of the strand between helical elements. Our definitions of junctions also differ: Tyagi and Mathews consider helical stems as those formed by at least one base pair, while we consider helical stems as those formed by at least two consecutive base pairs. Additional discrepancies between the coaxial stacking patterns reported by Tyagi and Mathews and our own observations exist.
To make a consistent comparison, we consider a junction dataset with elements from Table S1 of the Supplementary Material (34) that also agrees with our definition. We use the compiled list of three and four-way junctions in Supplementary Table S9 for testing and comparing both methods. To evaluate our method, we use a set of 91 three-way junctions for training our random forests and the same testing set as described in Supplementary Table S9. The coaxial stacking results from Tyagi and Mathews are reported in their article (34). While our method predicts coaxial stacking patterns from the compiled list with an 80% accuracy, results reported by Tyagi and Mathews showed that only 30% of the test elements from Supplementary Table S9 are accurately predicted. Similarly, we considered 49 four-way junctions for training our random forests and 27 four-way junctions for testing (Supplementary Table S9). Our results yield correct predictions of coaxial stacking patterns 92% of the time compared to 70% in (34).
Although the work of Lescoute and Westhof (22) provides a set of rules to predict coaxial stacking and junction families for three-way junctions, these rules depend both on expert decisions and knowledge from non-canonical base pairs and 3D contacts. For example, prediction of coaxial stacking is only limited to a special case where the continuous strand of the coaxial stack has no nucleotides, but this occurs for only 28 out of 110 of our three-way junctions. Furthermore, the prediction of junction families can only be determined for special cases of family B, and requires knowledge from non-canonical base pair contacts to discriminate between families A and C. This additional information is not easily recognized at the secondary structure level. Therefore, a comparison between both methods is not straightforward.
Predicting the topology adopted by unsolved RNA structures
Recently published works by the Westhof and Mathews groups (22,34) attempt to predict the topology and coaxial stacking patterns adopted by three-way junctions for RNAs whose structures have not yet been solved at atomic resolution. We similarly applied our random forest approach to determine the topology of these structures using only sequence and secondary structure information. The RNA structures considered are presented in Figure 4 and include the ‘Varkud’ satellite ribozyme (VS) (56), the Didymium group I-like intron ribozyme (DiGIR1) (57), a three-way junction formed between the U4 and U6 RNAs in the spliceosome (U4U6) (58), the hepatitis C virus (HCV) (59), and the recently solved RNase P (60). Figure 4 also shows our prediction results along with those of the Westhof and Mathews groups.
Figure 4.

Application to non-crystallized three-way junctions along with a comparison with the Westhof and Mathews groups (22,34). See text for details.
The VS ribozyme contains two three-way junctions determined by helices II–III–VI and III–IV–V (56). The Westhof group predicts a coaxial stacking between helices III and VI and family A for the first junction, and a coaxial stacking between III and IV and a family C for the second junction. Similarly the Mathews group predicts coaxial stacking between helices III with VI and between helices IV–V. By using secondary structure information, our method predicts a coaxial stacking between helices II and III with junction family C for the first junction, and a coaxial stacking between helices III and IV with junction family C for the second junction. Although no crystal structure is available, FRET analysis suggests (38,39) that our prediction for the first junction could be inaccurate, but that our prediction for the second junction can be correct both in the coaxial stacking pattern and junction family type.
The helices P3-P8-P15 of the DiGIR1 structure constitute a three-way junction. In agreement with the Westhof group, our random forest approach suggests a coaxial stacking between P3 and P8 and a junction family C, whereas Tyagi and Mathews predict a coaxial stacking between P8 and P15. The resemblance of this structure to the Tetrahymena group I intron also suggests the prediction is accurate (61).
Similarly, the three-way junction of the U4U6 snRNA in the spliceosome has also been considered for prediction. Our results propose a coaxial stacking between helices I and III and family C type. Lescoute and Westhof, however, predict a coaxial stacking between helices II and III and family B, while Tyagi and Mathews make no prediction.
The HCV virus contains a three-way junction formed by helices IIIo-IIIabc-IIId. Our random forest approach proposes a coaxial stacking between helices IIIo and IIIabc along with a junction family C. This configuration agrees with one of the proposed possibilities by the Westhof group. The Mathews group predicts a coaxial stacking formed between helices IIId and IIIabc. Although no crystal structure is yet available, recent FRET studies support our IIIo-IIIabc coaxial stack configuration (40).
Finally, the ribonuclease P structure contains a three-way junction determined by the helices P5-P5.1-P7, and its crystal structure has now been solved (60). Both Mathews and Westhof groups predict a coaxial stacking pattern between P5 and P5.1. In addition, Lescoute and Westhof predict a junction family A type. By using only secondary structure information, our method correctly predicts a coaxial stacking between P5 and P5.1, and an incorrect junction family C type instead of A.
SUMMARY AND DISCUSSION
RNA junctions are important structural elements involved in the global architecture of RNA tertiary structure. The function of large RNAs requires complex coordinated motions among their components, and the flexibility of junctions contributes to the proper functioning of RNA. Predicting the 3D configurations of helices in junctions is thus an important step in determining RNA 3D structures. We have applied a data mining method to predict junction families (topologies) for three- and four-way junctions, as well as to predict coaxial helical stacking for any RNA junction order. This method relies on a set of parameters, or feature vector, obtained from known RNA 3D junctions taken from solved RNAs (Table 1), and uses a random forests protocol to make predictions. Predictions rely on RNA secondary structure information such as loop length, loop sequence content and thermodynamic parameters. Such secondary structure information can be predicted for a single sequence (44) or multiple sequences (62), or determined experimentally (63). Databases containing secondary structure information for RNA are now common (64).
Our results demonstrate the feasibility of using the data mining method to predict helical arrangements in junctions. More specifically, prediction of three- and four-way junction families yields an accuracy of about 85% and 74%, respectively. The difference in performance is due to the larger number of three-way junctions available (110 junctions) compared to four-way junctions (65 junctions). Three-way junctions are classified into three families, while four-way junctions have at least nine possibilities.
The accuracy for our predictions of coaxial stacking arrangements for three- and four-way junctions is about 81% and 77%, respectively; for higher-order junctions, it is around 60%, due to the lack of sufficient training data. It is also possible that prediction values for three- and four-way junctions are higher because the parameters depend on loop, sequence and thermodynamic features from the entire junction, while predictions of higher order junctions rely on local information (i.e. loop, sequence and thermodynamic parameters from pairs of consecutive helices and their nearest neighbors). In addition, most of the higher order junctions in our dataset belong to the ribosomal RNA, whose helical arrangements are often influenced by proteins.
We showed that predictions improve when knowledge from coaxial stacking or junction families is provided, for example from FRET, Cryo-EM and NMR data. By adding coaxial stacking interactions as an additional feature parameter, prediction of three-way junction family type increases to 86%, while prediction in four-way junction family type improves from 74 to 81%. Similarly, if the type of junction family is known, predicting coaxial stacking in three-way junctions increases from 81 to 83%, while prediction of coaxial stacking in four-way junctions increases dramatically from 77 to 87%. Experimental techniques such as FRET (47) and SAXS (48) can help provide such related information. An alternative approach consists of a two-step procedure, by initially using random forests to determine coaxial stacking (junction family, respectively) and then implementing random forests again to predict the junction family (coaxial stacking, respectively). Applying this two-step procedure to three- and four-way junctions improves all predictions except four-way junctions (Table 2).
Although our predictions improve upon previous work by Tyagi and Mathews (34), their approach differs from ours in both methodology and concept: Tyagi and Mathews predict coaxial stacking only when loop lengths between helices are one or zero, and we impose no such restriction on loop length. Another important difference is that we require helices in junctions to contain at least two consecutive canonical Watson–Crick base pairs, while Tyagi and Mathews consider helices formed by a single base pair as well. Still, our comparisons made over a common junction dataset (Supplementary Table S9) show that our method performs well for predicting coaxial stacking prediction for both three- and four-way junctions (80% and 92%, respectively) compared to previous attempts (30% and 70%, respectively) (34).
Overall, the statistical data-mining approach benefits from the modularity of RNA architecture and available training data. By analyzing the contribution of each feature parameter independently (Supplementary Figure S2), we found that 8 out of the total 15 and 18 parameters we formulated for three- and four-way junctions, respectively (Supplementary Tables S2–S3), provide essential contributions for coaxial stacking prediction, and 4–6 parameters are essential for junction family prediction. Our analysis highlights the importance of feature parameters built from the size of loops within junctions and base pair configurations at the end of helices. For instance, a short loop length between helical elements in junctions is correlated to coaxial stacking formation (22,23,27). As the loop length between helices increases, stacking forces decrease and the sequence content between helices may be less important. Indeed, the number of nucleotides in junction regions has been recognized as a factor for improving RNA secondary structure prediction (20,65). In addition, experiments that estimate thermodynamic parameters have shown that base stacking interactions in nucleic acids are sequence dependent. For instance, the stacking force associated with a Guanine-Cytosine (GC) stacked with a GC base pair is stronger than that associated with a GC stacked with a CG or AU base pair. Furthermore, adenines in the loop regions within junctions tend to stabilize junction topologies by forming A-minor interactions (43) that interact in cooperation with coaxial helical stacking (21–23,27). Therefore the presence of adenines can help predict both coaxial stacking as well as help in predicting junction family types. It follows that features built on these observations can capture some of the forces involved in the 3D structure of RNA junctions.
Examples where the method fails to predict the right stacking and/or family type due to external factors that promote or prevent coaxial stacking formation are shown in Figure 5. The first example shows a recently solved riboswitch molecule containing a three-way junction with loop lengths of 3, 3 and 6. Our statistical data show that coaxial stacking formation favors pairs of helices with a small loop length in between (27). However, coaxial stacking forms between the helices whose common loop has length 6 due to the interaction of the cyclic diguanylate (c-di-GMP) metabolite (Figure 5a in orange), which stacks with other nucleotides in the loop region. Interestingly, it has been reported (66) that the nucleotides that stack with c-di-GMP are more conserved than nucleotides that contact the c-di-GMP, thus emphasizing the importance of stacking for the riboswitch's function. A second example occurs in a four-way junction in the rous sarcoma virus with helices B (green) and C (magenta) having no nucleotides in between. While our approach predicts the formation of a coaxial helical stacking of B with C, no coaxial stacking occurs due to the presence of a nucleocapsid protein which pushes C away and prevents it from stacking with B (Figure 5b in orange). Experiments suggest that the high affinity RNA–protein binding in the RSV virus has an important role in genome packing. Supplementary Table S1 lists the proteins and metabolites that interact with every junction (proteins from 1NKW are not listed due to poor resolution in the protein structures). From this list we observe that about 80% of the junctions contain RNA–protein interactions. Thus although the presence of proteins can either prevent or promote coaxial stacking and junction topology conformations, the protein sequences involved are highly diverse and there is no obvious sequence signature to help determine feature parameters to make better predictions. The third example shows a five-way junction in the group I Azoarcus intron with a coaxial stacking formed between helices H2 (red) and H3 (blue) that this method fails to predict. The coaxial stacking interaction forms with the aid of a pseudoknot (Figure 5c in orange) which stacks with both H2 and H3. The method cannot predict coaxial stacking formation due to the presence of pseudoknots because the feature parameters do not include pseudoknot information. If pseudoknots are known a priori, this limitation can be removed by adding statistical parameters on the loop and stem lengths where coaxial stacking occurs, as presented in our statistical analysis of RNA 3D motifs (21).
Figure 5.

Examples of RNA junctions with coaxial stacking patterns formed by external factors that our method fails to predict. (a) The c-di-GMP metabolite (orange) promotes stacking between helices P1 (red) and P2 (blue) by favoring stacking interactions with the bases at the loop L1. (b) The rous sarcoma virus nucleocapsid protein (orange) prevents stacking between B (green) and C (magenta) even though there are no intervening nucleotides. (c) A pseudoknot (orange) stacks between helices P2 (red) and P3 (blue), allowing a coaxial stack to form. Helices are labeled as in Supplementary Table S1.
Many avenues for future improvement can be identified, especially regarding higher-order junctions. Because higher-order junctions are composites of sub-junctions of a smaller branching order (27), a divide and conquer approach could be envisioned by dividing the RNA into sub-junctions and predicting the topology of each sub-junction independently. Further improvements could arise from adding information from typical 3D motifs observed in junctions such as the U-turn (67) and A-minor/coaxial helix (15,21,40), for example, and by including information from loop–loop interactions. Although long-range interactions between loop regions near junctions are important for formation of a junction's topology (68), we have not yet incorporated this feature due to the lack of sequence specificity and the high diversity of such interactions. To achieve better predictions for higher order junctions, feature parameters may be improved based on these ideas.
Our junction prediction approach has applications to RNA 3D structure prediction. Figure 4 shows examples of both coaxial stacking patterns as well as junction topology predictions to non-crystallized three-way junctions. Our results agree for the most part with previous predictions (22) and experimental FRET analysis (38–40). In addition, our application of the random forest method to predict both coaxial stacking and junction topology can be used as a first approximation of the 3D structure of junctions, which can be provided to 3D folding programs such as NAST (69), as well as to atomistic MD models. Our predictions might also be implemented as restraints to the conformational space of RNA 3D structures. Work continues on this exciting front.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (EMT award # CCF-0727001 to T.S., grant # IIS-0707571 to J.W.); and the National Institutes of Health (grant # R01-GM081410 to T.S.). Funding for open access charge: National Science Foundation, National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Irnov I, Sharma CM, Vogel J, Winkler WC. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010;38:6637–6651. doi: 10.1093/nar/gkq454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ, Breaker RR. Identification of candidate structured RNAs in the marine organism ‘Candidatus Pelagibacter ubique'. BMC Genomics. 2009;10:268. doi: 10.1186/1471-2164-10-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinberg Z, Wang JX, Bogue J, Yang J, Corbino K, Moy RH, Breaker RR. Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 2010;11:R31. doi: 10.1186/gb-2010-11-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 6.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 7.Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 8.Laing C, Schlick T. Computational approaches to 3D modeling of RNA. J. Phys. Condens. Matter. 2010;22:283101. doi: 10.1088/0953-8984/22/28/283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jossinet F, Ludwig TE, Westhof E. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics. 2010;26:2057–2059. doi: 10.1093/bioinformatics/btq321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez HM, Maizel JV, Jr, Shapiro BA. RNA2D3D: a program for generating, viewing, and comparing 3-dimensional models of RNA. J. Biomol. Struct. Dyn. 2008;25:669–683. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lilley DM, Clegg RM, Diekmann S, Seeman NC, Von Kitzing E, Hagerman PJ. A nomenclature of junctions and branchpoints in nucleic acids. Nucleic Acids Res. 1995;23:3363–3364. doi: 10.1093/nar/23.17.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott WG, Murray JB, Arnold JR, Stoddard BL, Klug A. Capturing the structure of a catalytic RNA intermediate: the hammerhead ribozyme. Science. 1996;274:2065–2069. doi: 10.1126/science.274.5295.2065. [DOI] [PubMed] [Google Scholar]
- 13.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–415. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 14.Kieft JS, Zhou K, Grech A, Jubin R, Doudna JA. Crystal structure of an RNA tertiary domain essential to HCV IRES-mediated translation initiation. Nat. Struct. Biol. 2002;9:370–374. doi: 10.1038/nsb781. [DOI] [PubMed] [Google Scholar]
- 15.Kim SH, Sussman JL, Suddath FL, Quigley GJ, McPherson A, Wang AH, Seeman NC, Rich A. The general structure of transfer RNA molecules. Proc. Natl Acad. Sci. USA. 1974;71:4970–4974. doi: 10.1073/pnas.71.12.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aalberts DP, Hodas NO. Asymmetry in RNA pseudoknots: observation and theory. Nucleic Acids Res. 2005;33:2210–2214. doi: 10.1093/nar/gki508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Walter AE, Turner DH. Thermodynamics of coaxially stacked helixes with GA and CC mismatches. Biochemistry. 1996;35:13753–13761. doi: 10.1021/bi960913z. [DOI] [PubMed] [Google Scholar]
- 19.Walter AE, Turner DH, Kim J, Lyttle MH, Muller P, Mathews DH, Zuker M. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc. Natl Acad. Sci. USA. 1994;91:9218–9222. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aalberts DP, Nandagopal N. A two-length-scale polymer theory for RNA loop free energies and helix stacking. RNA. 2010;16:1350–1355. doi: 10.1261/rna.1831710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xin Y, Laing C, Leontis NB, Schlick T. Annotation of tertiary interactions in RNA structures reveals variations and correlations. RNA. 2008;14:2465–2477. doi: 10.1261/rna.1249208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lescoute A, Westhof E. Topology of three-way junctions in folded RNAs. RNA. 2006;12:83–93. doi: 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laing C, Schlick T. Analysis of four-way junctions in RNA structures. J. Mol. Biol. 2009;390:547–559. doi: 10.1016/j.jmb.2009.04.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walter AE, Turner DH. Sequence dependence of stability for coaxial stacking of RNA helixes with Watson–Crick base paired interfaces. Biochemistry. 1994;33:12715–12719. doi: 10.1021/bi00208a024. [DOI] [PubMed] [Google Scholar]
- 25.Elgavish T, Cannone JJ, Lee JC, Harvey SC, Gutell RR. AA.AG@helix.ends: A:A and A:G base-pairs at the ends of 16 S and 23 S rRNA helices. J. Mol. Biol. 2001;310:735–753. doi: 10.1006/jmbi.2001.4807. [DOI] [PubMed] [Google Scholar]
- 26.Orr JW, Hagerman PJ, Williamson JR. Protein and Mg(2+)-induced conformational changes in the S15 binding site of 16 S ribosomal RNA. J. Mol. Biol. 1998;275:453–464. doi: 10.1006/jmbi.1997.1489. [DOI] [PubMed] [Google Scholar]
- 27.Laing C, Jung S, Iqbal A, Schlick T. Tertiary motifs revealed in analyses of higher-order RNA junctions. J. Mol. Biol. 2009;393:67–82. doi: 10.1016/j.jmb.2009.07.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz JA, Westhof E. The dynamic landscapes of RNA architecture. Cell. 2009;136:604–609. doi: 10.1016/j.cell.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Holbrook SR. Structural principles from large RNAs. Annu. Rev. Biophys. 2008;37:445–464. doi: 10.1146/annurev.biophys.36.040306.132755. [DOI] [PubMed] [Google Scholar]
- 30.Hohng S, Wilson TJ, Tan E, Clegg RM, Lilley DM, Ha T. Conformational flexibility of four-way junctions in RNA. J. Mol. Biol. 2004;336:69–79. doi: 10.1016/j.jmb.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 31.Lilley DM. Folding of branched RNA species. Biopolymers. 1998;48:101–112. doi: 10.1002/(SICI)1097-0282(1998)48:2<101::AID-BIP2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 32.Lilley DM. Structures of helical junctions in nucleic acids. Q. Rev. Biophys. 2000;33:109–159. doi: 10.1017/s0033583500003590. [DOI] [PubMed] [Google Scholar]
- 33.Klein DJ, Moore PB, Steitz TA. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 2004;340:141–177. doi: 10.1016/j.jmb.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 34.Tyagi R, Mathews DH. Predicting helical coaxial stacking in RNA multibranch loops. RNA. 2007;13:939–951. doi: 10.1261/rna.305307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Besseova I, Reblova K, Leontis NB, Šponer J. Molecular dynamics simulations suggest that RNA three-way junctions can act as flexible RNA structural elements in the ribosome. Nucleic Acids Res. 2010;38:6247–6264. doi: 10.1093/nar/gkq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bindewald E, Hayes R, Yingling YG, Kasprzak W, Shapiro BA. RNAJunction: a database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 2008;36:D392–D397. doi: 10.1093/nar/gkm842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breiman L. Random Forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 38.Lafontaine DA, Norman DG, Lilley DM. Structure, folding and activity of the VS ribozyme: importance of the 2-3-6 helical junction. EMBO J. 2001;20:1415–1424. doi: 10.1093/emboj/20.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lilley DM. The Varkud satellite ribozyme. RNA. 2004;10:151–158. doi: 10.1261/rna.5217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouellet J, Melcher S, Iqbal A, Ding Y, Lilley DM. Structure of the three-way helical junction of the hepatitis C virus IRES element. RNA. 2010;16:1597–1609. doi: 10.1261/rna.2158410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 43.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl Acad. Sci. USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathews DH. RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics. 2006 doi: 10.1002/0471250953.bi1206s13. Chapter 12, Unit 12 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and Regression Trees. Boca Raton, Republished 1993: Chapman & Hall/CRC; 1984. [Google Scholar]
- 46.Liaw A, Wiener M. Classification and regression by randomForest. R. News. 2002;2:18–22. [Google Scholar]
- 47.Walter F, Murchie AI, Duckett DR, Lilley DM. Global structure of four-way RNA junctions studied using fluorescence resonance energy transfer. RNA. 1998;4:719–728. doi: 10.1017/s135583829898030x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16:1357–1367. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spahn CM, Jan E, Mulder A, Grassucci RA, Sarnow P, Frank J. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA-based translation factor. Cell. 2004;118:465–475. doi: 10.1016/j.cell.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Hanlon S. The importance of London dispersion forces in the maintenance of the deoxyribonucleic acid helix. Biochem. Biophys. Res. Commun. 1966;23:861–867. doi: 10.1016/0006-291x(66)90567-5. [DOI] [PubMed] [Google Scholar]
- 51.Tazawa I, Koike T, Inoue Y. Stacking properties of a highly hydrophobic dinucleotide sequence, N6, N6-dimethyladenylyl(3′ leads to 5′)N6, N6-dimethyladenosine, occurring in 16–18-S ribosomal RNA. Eur. J. Biochem. 1980;109:33–38. doi: 10.1111/j.1432-1033.1980.tb04764.x. [DOI] [PubMed] [Google Scholar]
- 52.Sarai A, Mazur J, Nussinov R, Jernigan RL. Origin of DNA helical structure and its sequence dependence. Biochemistry. 1988;27:8498–8502. doi: 10.1021/bi00422a030. [DOI] [PubMed] [Google Scholar]
- 53.Mignon P, Loverix S, Steyaert J, Geerlings P. Influence of the π–π interaction on the hydrogen bonding capacity of stacked DNA/RNA bases. Nucleic Acids Res. 2005;33:1779–1789. doi: 10.1093/nar/gki317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saenger W. Principle of Nucleic Acid Structure. New York: Springer-Verlag; 1984. [Google Scholar]
- 55.Šponer J, Leszczyński J, Hobza P. Nature of nucleic acid−base stacking: nonempirical ab initio and empirical potential characterization of 10 stacked base dimers. Comparison of stacked and H-bonded base pairs. J. Phys. Chem. 1996;100:5590–5596. [Google Scholar]
- 56.Beattie TL, Olive JE, Collins RA. A secondary-structure model for the self-cleaving region of Neurospora VS RNA. Proc. Natl Acad. Sci. USA. 1995;92:4686–4690. doi: 10.1073/pnas.92.10.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Einvik C, Nielsen H, Westhof E, Michel F, Johansen S. Group I-like ribozymes with a novel core organization perform obligate sequential hydrolytic cleavages at two processing sites. RNA. 1998;4:530–541. doi: 10.1017/s1355838298971758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nottrott S, Urlaub H, Luhrmann R. Hierarchical, clustered protein interactions with U4/U6 snRNA: a biochemical role for U4/U6 proteins. EMBO J. 2002;21:5527–5538. doi: 10.1093/emboj/cdf544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Honda M, Beard MR, Ping LH, Lemon SM. A phylogenetically conserved stem-loop structure at the 5′ border of the internal ribosome entry site of hepatitis C virus is required for cap-independent viral translation. J. Virol. 1999;73:1165–1174. doi: 10.1128/jvi.73.2.1165-1174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kazantsev AV, Krivenko AA, Harrington DJ, Holbrook SR, Adams PD, Pace NR. Crystal structure of a bacterial ribonuclease P RNA. Proc. Natl Acad. Sci. USA. 2005;102:13392–13397. doi: 10.1073/pnas.0506662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johansen S, Einvik C, Nielsen H. DiGIR1 and NaGIR1: naturally occurring group I-like ribozymes with unique core organization and evolved biological role. Biochimie. 2002;84:905–912. doi: 10.1016/s0300-9084(02)01443-8. [DOI] [PubMed] [Google Scholar]
- 62.Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 63.Mortimer SA, Weeks KM. Time-resolved RNA SHAPE chemistry: quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat. Protoc. 2009;4:1413–1421. doi: 10.1038/nprot.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andronescu M, Bereg V, Hoos HH, Condon A. RNA STRAND: the RNA secondary structure and statistical analysis database. BMC Bioinformatics. 2008;9:340. doi: 10.1186/1471-2105-9-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levitt M. Detailed molecular model for transfer ribonucleic acid. Nature. 1969;224:759–763. doi: 10.1038/224759a0. [DOI] [PubMed] [Google Scholar]
- 66.Smith KD, Lipchock SV, Livingston AL, Shanahan CA, Strobel SA. Structural and biochemical determinants of ligand binding by the c-di-GMP riboswitch. Biochemistry. 49:7351–7359. doi: 10.1021/bi100671e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jucker FM, Pardi A. GNRA tetraloops make a U-turn. RNA. 1995;1:219–222. [PMC free article] [PubMed] [Google Scholar]
- 68.de la Pena M, Dufour D, Gallego J. Three-way RNA junctions with remote tertiary contacts: a recurrent and highly versatile fold. RNA. 2009;15:1949–1964. doi: 10.1261/rna.1889509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.