


Abstract
Mechanisms that preserve genome integrity are highly important during the normal life cycle of human cells. Loss of genome protective mechanisms can lead to the development of diseases such as cancer. Checkpoint kinases function in the cellular surveillance pathways that help cells to cope with DNA damage. Importantly, the checkpoint kinases ATR, CHK1 and WEE1 are not only activated in response to exogenous DNA damaging agents, but are active during normal S phase progression. Here, we review recent evidence that these checkpoint kinases are critical to avoid deleterious DNA breakage during DNA replication in normal, unperturbed cell cycle. Possible mechanisms how loss of these checkpoint kinases may cause DNA damage in S phase are discussed. We propose that the majority of DNA damage is induced as a consequence of deregulated CDK activity that forces unscheduled initiation of DNA replication. This could generate structures that are cleaved by DNA endonucleases leading to the formation of DNA double-strand breaks. Finally, we discuss how these S phase effects may impact on our understanding of cancer development following disruption of these checkpoint kinases, as well as on the potential of these kinases as targets for cancer treatment.
INTRODUCTION
Maintenance of genome integrity is essential to prevent development of diseases associated with genomic instability such as cancer and neurodegenerative disorders (1). The genome integrity of human cells is frequently threatened by DNA damage caused by exogenous or endogenous sources. To counteract such threat, human cells contain a network of DNA damage surveillance pathways that maintain genome integrity. These surveillance pathways control processes such as DNA repair, cell-cycle checkpoints, apoptosis and transcription. The checkpoint kinases ATR, CHK1 and WEE1 are key regulators of DNA damage surveillance pathways. ATR and CHK1 regulate the S and G2 checkpoints (2–5), replication initiation and replication fork stability (6–10) and homologous recombination repair (11–13). CHK1 also controls mitotic entry in unperturbed cells (14) and was reported to play a role in mitotic spindle checkpoint function (15), and in control of transcription (16). WEE1 has a major cell-cycle function in control of the G2/M transition (17,18).
ATR, CHK1 and WEE1 are in fact active during unperturbed normal cell-cycle progression, and all three kinases have roles besides their functions in the response to exogenous DNA damage. Deletion of ATR, CHK1 or WEE1 in mice causes embryonic lethality (19–23), demonstrating that these checkpoint kinases are essential for embryonic development. Importantly, recent work has revealed that the activities of these kinases are required during normal S phase to avoid deleterious DNA breakage, and thereby prevent loss of genome integrity in the absence of exogenous DNA damaging agents (24–27). Here, we review the progress in this area with focus on discussing the potential mechanisms involved in causing DNA breakage following inhibition of these checkpoint kinases. We also discuss the possible impact of these findings on our understanding of the roles of these checkpoint kinases in cancer development, and on the potential of these kinases as targets for cancer treatment.
Activation of ATR, CHK1 and WEE1
Before discussing the mechanisms how ATR, CHK1 and WEE1 are required to prevent loss of integrity in normal S phase, we briefly summarize the current knowledge about how these kinases are activated and how they are thought to control DNA replication.
Activation of ATR occurs upon the generation of lesions containing single-stranded DNA (ssDNA) (28), which evolve at stalled replication forks and following processing of DNA strand breaks (29,30). Coating of ssDNA by the single-strand binding protein RPA helps loading of ATR to DNA damage sites (31–33). ATR recognition of RPA-coated ssDNA is dependent on the ATR-interacting protein (ATRIP) (34), which binds RPA directly (35). ATR activation is also dependent on TOPBP1, RAD17 and the 9-1-1 (RAD9-RAD1-HUS1) complex (36–38). RAD17 is recruited by RPA-coated ssDNA and loads the 9-1-1 complex, which subsequently recruits TOPBP1 and brings it in close proximity to ATR so that TOPBP1 can activate ATR via direct interaction (39). Studies in Xenopus extracts suggest that ssDNA in itself is not sufficient to cause strong activation of ATR. While ATR activation by naked ssDNA pieces was low, high levels of ATR activation were observed at areas of ssDNA with 5′-primed ends (37). It has been suggested that these ends may be the loading site for the 9-1-1 complex (40). Thus, small pieces of ssDNA generated during normal replication do not lead to strong checkpoint activation, although they might potentially contribute to the low levels of ATR activation observed during normal unperturbed cell cycle.
CHK1 is a direct downstream target of ATR. ATR phosphorylates CHK1 on Ser 317 and Ser345 and stimulates its function (22,41,42). After the ATR induced phosphorylation, CHK1 undergoes autophosphorylation at Ser296 (43,44). The DNA damage-induced ATR phosphorylation most likely does not up-regulate CHK1 kinase activity per se. However, phosphorylated CHK1 can dissociate from chromatin (45,46), and ATR regulation of CHK1 may thereby control transition of DNA damage signals from chromatin to its targets.
The activity of WEE1 increases during S and G2 phases in parallel with its increased protein level (47), and can be stimulated by the CDK-interacting protein CABLES (48). In budding yeast, WEE1 activity is also stimulated by low levels of CDK activity in G2 phase (49). In Xenopus, activated XCHK1 phosphorylates the XWee1 kinase, contributing to increased Tyrosine 15 phosphorylation and inhibition of CDK activity following CHK1 activation (50). This regulatory mechanism has so far not been described in mammalian cells. At entry into mitosis WEE1 is inhibited both by phosphorylation and degradation allowing rapid increase in CDK activity (49,51–53). Subsequent CDK phosphorylation primes WEE1 for ubiquitylation via the β-TRCP SCF type of ubiquitin ligase, and this activity may be further supported by the Tome-1 SCF ubiquitin ligase (54).
The cell-cycle regulatory roles of ATR/CHK1 and WEE1 in S and G2 phases are mainly through regulation of CDK activity (Figure 1). WEE1 directly catalyses the inhibitory Tyrosine 15 phosphorylation of CDK1 and CDK2 and thereby inhibits CDK activity (18,47). Following its activation, CHK1 directly phosphorylates CDC25A (3), facilitated by 14-3-3γ (44). CHK1 activates NEK11 that also phosphorylates CDC25A directly (55), and the concerted action of the two kinases promotes the ubiquitin dependent degradation of CDC25A. The degradation of CDC25A leads to decreased removal of the phosphorylation of the CDK Tyrosine 15 residue. This results in inhibition of CDK activity that induces cell-cycle arrest (33,56). CHK1 can also phosphorylate CDC25B and CDC25C (5,57), which may also contribute to restrain CDK activity.
Figure 1.
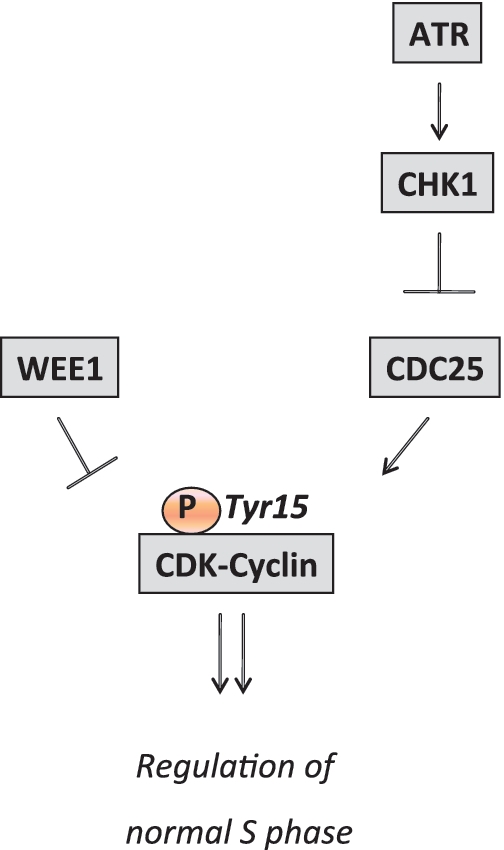
Regulation of CDK activity by ATR, CHK1 and WEE1 determines replication initiation during normal S phase. CDK activity is negatively regulated by phosphorylation of the Tyrosine 15 residue. This residue is phosphorylated by WEE1 and dephosphorylated by CDC25. CDC25 is negatively regulated by CHK1, which in turn is stimulated by the ATR kinase.
CDK mediated control of DNA replication initiation
When cells reach S phase, DNA replication is initiated from a large number of chromosomal locations known as replication origins. The precise activation of origins is a key part of replication control, because cells are thought to be unable to regulate the speed of the DNA polymerases. The initiation of DNA replication is triggered by the action of the CDK kinases and DBF4-CDC7 kinase (DDK) (58,59). CDK2 is considered a key CDK in control of DNA replication in S phase, although CDK1 can compensate if CDK2 is disabled (60). Once activated, CDK2 activity promotes origin firing through loading of CDC45L and AND-1/CTF4 at origins (61,62). The presence of CDC45L and AND-1/CTF4 at origins are required for origin unwinding by the MCM helicase, and thereby also required for the subsequent binding of the primase DNA polymerase (Pol) alpha that initiates DNA synthesis (61,63). Recent data suggest that the novel replication factors Treslin and GEMC1 are key phosphorylation targets of CDK2 that promote CDC45L loading (64,65).
In addition to promoting origin firing, CDK activity also seems to regulate the activation of individual replication clusters and the overall timing of replication through S phase, although this regulation is not well understood (66–69). A substantial number of origins are fired at distinct times through S phase to allow coordinated execution of DNA replication. In fact, more origins are licensed than are ever used, and in a normal S phase most origins are replicated passively by replication forks coming from neighbouring activated origins (70). When replication fork stalling or replication stress occurs, local dormant origins will fire to compensate for the lack of replication. Under conditions of exogenous DNA damage, checkpoint pathways block the activation of origins that normally fire throughout S phase, which constitutes the basis for the S phase checkpoint (70–72).
ATR, CHK1 and WEE1 control CDK activity to support genome integrity during DNA replication
Recent studies have uncovered that ATR, CHK1 and WEE1 are critical for coordinated duplication of the genome and preventing a catastrophic outcome of the sensitive replication process. In response to CHK1 inhibition by the drugs UCN-01 or CEP-3891, or siRNA mediated depletion of CHK1, the genome is rapidly destabilized and accumulates a massive amount of DNA double-strand breaks (27). This damage is preceded by massive accumulation of ssDNA near the replication fork. All these critical downstream effects of CHK1 inhibition are dependent on CDK activity (27). The deleterious effects of CHK1 inhibition can be blocked by depleting CDC25A, the CDK activator controlled by CHK1. Furthermore, cellular fitness of CHK1 depleted cells is markedly improved by co-depleting CDC25A (26). Consistent with these results, conditional CHK1 heterozygosity causes accumulation of DNA damage during DNA replication in mice in the absence of exogenous DNA damaging agents (24). WEE1 depletion by siRNA transfection also rapidly induces DNA damage in S phase in replicating areas, which is accompanied by a marked accumulation of ssDNA. Similar to the effects of CHK1 inhibition, these deleterious S phase effects of WEE1 depletion are highly dependent on CDK activity (26).
Reduced ATR function can also cause a DNA damage response in S phase cells. Cultured MEFs and embryos from ATR mutated Seckel syndrome mice revealed high incidence of Cyclin A positive cells with strong pan-nuclear staining of γ-H2AX, consistent with increased replication stress (25). Induction of DNA damage, as assessed by increased γ-H2AX staining, was also observed in another mouse model hypomorphic for ATR (73). ATR depletion leads to deregulation of CDC25A levels in the absence of exogenous replication inhibitors and DNA damaging agents (74). In line with this, the DNA damage occurring after ATR inhibition is also blocked by depleting CDC25A, indicating that the loss of genomic integrity occurs as a consequence of deregulation of the ATR-CHK1-CDC25A-CDK pathway (75). These results suggest that similar to CHK1 and WEE1, ATR is also required to prevent formation of deleterious DNA lesions during normal S phase through control of CDK activity.
Increased CDK activity leads to unscheduled initiation of DNA replication origins
When ATR and CHK1 activity is lost, several cellular pathways are affected and could potentially contribute to the devastating cellular outcome. However, the loss of each of WEE1, ATR and CHK1 leads to increased CDK activity (3,26,74–76) resulting in a loss of control of the replication coordination. Inhibition of CHK1 leads to an increased loading of replication factor CDC45L onto chromatin, which is followed by a dramatic increase in replication initiation. Shortly after, there is increased ssDNA formation and RPA loading indicative of replication stress (27). In addition, CHK1 inhibition also causes slower replication fork speed (6,7), however, this is likely not directly due to the elevated CDK activity operating at existing forks. In fact, it was suggested that CHK1 indirectly promotes replication fork progression as a downstream consequence of the key role in control of replication initiation (7). Similarly to CHK1, WEE1 depletion causes increased ssDNA and RPA loading indicative of replication stress (26). Drug-based WEE1 inhibition also leads to a very rapid increase in origin firing and slower replication fork speeds (our unpublished observations). Importantly, DNA damage arising after both CHK1 and WEE1 inhibition, respectively, is ablated by partial suppression of DNA replication initiation obtained by depletion of CDC45L, CDT1 and MCM complex members (26,27). Based on our previous data and the common regulation of CDK activity by ATR/CHK1 and WEE1 kinases, we propose that the major cellular defect leading to DNA breakage in S phase following depletion of these kinases is CDK-driven unscheduled initiation of a large number of origins (Figure 2). Supporting the notion that CDK-driven events can lead to DNA breakage in S phase, other ways of enhancing CDK activity such as overexpression of Cyclin E, CDC25A or E2F1 also causes DNA damage in S phase (77). Interestingly, this chain of events has not been observed in yeast. Elevated CDK activity can induce chromosomal rearrangements in budding yeast by mechanisms rather involving inhibition of DNA replication licensing. However, these effects occurred only in G1 phase cells (78), and are therefore different from the S phase effects observed in human cells.
Figure 2.

Model describing potential mechanisms how loss of checkpoint kinases may cause DNA breakage during normal S phase. Loss of WEE1, ATR or CHK1 leads to increased CDK activity, which causes unscheduled replication initiation and subsequent DNA breakage. Following massive unscheduled initiation of replication, aberrant fork structures accumulate that are prone to breakage resulting in DNA DSBs. The high CDK activity may activate nucleases that directly catalyse the formation of DNA DSBs.
Unscheduled initiation is required for the generation of DNA lesions at the replication fork
Although unscheduled initiation is involved in the massive induction of DNA breaks after checkpoint kinase inhibition, it is not clear how it specifically contributes to break formation. A low but significant number of DNA breaks may be induced in several ways by the replication process itself (79). Importantly, unwound, single-stranded DNA at replication forks may be more susceptible to DNA breakage. This could for example occur after free radical attacks on the DNA backbone; however, checkpoint kinase inhibition is not expected to rapidly induce high levels of free radicals. In addition, ssDNA breaks existing in front of the replication fork in the template DNA can be converted to a double-strand break. During replication, the template strands on each arm of replication forks will no longer be base-paired to their original complementary template strands due to the actions of DNA helicases. Consequently, single-strand lesions within template DNA can cause double-strand breaks when the replication fork reaches such lesions. Thus increased initiation of DNA replication may in itself be sufficient to cause small amounts of DNA breakage if single-strand lesions are present. However, additional events are most likely required to achieve the massive induction of DNA breakage observed following CHK1 or WEE1 inhibition (26,27).
In response to CHK1 inhibition, replication initiation is first increased, followed by subsequent fork stalling (6,7,27). ATR and CHK1 are known to directly support fork stability (79,80). It is therefore possible that lack of ATR/CHK1-mediated support of stalled forks will lead to fork collapse and DNA breaks. Data from budding yeast have clearly documented an important role for ATR (Mec1) and CHK1 (Rad53) in fork stability, however, this function is observed under conditions of massive fork stalling with hydroxyurea, an inhibitor of nucleotide metabolism (81,82). It is not clear if fork destabilization is an important contributor to the loss of genomic integrity after checkpoint kinase inhibition under unperturbed conditions in mammalian cells. Furthermore, it is important to note that the genome integrity protection mediated by CHK1 operates largely through control of the CDC25A-CDK pathway. Similarly, WEE1 also controls genome integrity via suppression of CDK activity (26). Given these observations, it is apparent that elevated CDK activity should destabilize existing replication forks if this was the major mechanism. It remains to be shown if this is the case. In addition, the massive unscheduled initiation could perhaps result in unbalanced or insufficient nucleotide pools that could potentially lead to fork stalling and eventual collapse, similar to that observed in cells treated with inhibitors of nucleotide metabolism such as hydroxyurea (83). However, this would likely not occur with the rapid kinetics observed after CHK1 and WEE1 inhibition, where inhibitors induce DNA damage within 2 h of treatment. In contrast, DNA damage upon hydroxyurea treatment alone is normally only observed after ∼24 h of treatment (84).
Checkpoint kinases or CDK may directly regulate nucleases
In addition to the unscheduled initiation of origins, additional possible explanations may more directly explain the occurrence of double-strand breaks after checkpoint kinase inhibition. It appears unlikely that such massive DSB accumulation can arise without the activity of enzymes that break the DNA backbone, which is a very solid structure. Stalled replication forks with exposed ssDNA are processed by a number of enzymes to promote genomic integrity. Homologous recombination pathways can salvage stalled forks (85), and the remodelling of the replication fork can generate structures that are resolved by endonucleases such as MUS81/EME1. The resulting double-strand break can be used to initiate fork restart (86). However, the DSBs may have additional roles since it can amplify checkpoint signalling to avoid premature mitotic entry, and if present in large amount they may serve to trigger cell death or senescence.
Nuclease substrate structures such as Holliday junctions could potentially be formed due to elevated activity of helicases that act upon stalled replication forks. Following ATR/CHK1 and WEE1 inhibition, increased CDK activity initiates excess origin firing, and this may lead to fork stalling due to shortage of important factors such as nucleotides or key replication proteins (Figure 3). The formation of nuclease substrates may also be promoted by high level of torsional stress, potentially occurring due to the elevated replicative helicase activities at origins. Torsional stress in front of the fork is thought to be able to promote reversal of the fork, since stress relief can occur via fork reversal (Figure 3). Massive fork reversal could potentially promote nuclease cleavage of the Holliday junctions and thereby cause high number of DNA double-strand breaks. Given that checkpoint inhibition leads to a large number of DNA replication fork lesions, it is likely that excellent substrates for such nucleases will appear in large quantity. The number of breaks following nuclease activity may overwhelm the cellular capacity to repair and process these lesions. The presence of breaks is further problematic to cells as CHK1 inhibition suppresses the HR pathway that would normally process the breaks (11).
Figure 3.

Model depicting how inhibition of checkpoint kinases may lead to double-strand breaks after replication fork reversal. When checkpoint kinases are inhibited CDK activity is up-regulated leading to increased origin firing. As a consequence of the enhanced origin activity, replication factors such as polymerase subunits may become limiting leading to fork stalling. Helicases can process stalled forks leading to fork reversal, which generates substrates for DNA endonucleases such as MUS81. Fork reversal may also occur due to increased initiation of replication that generates enhanced torsional stress due to helicase unwinding of the double-stranded DNA. This stress can be relieved by fork reversal, thereby forming substrates for endonuclease cleavage.
In fission yeast, it was shown that the CHK1 limits the effects of the endonuclease MUS81/EME1 at stalled forks (87). If CHK1 negatively regulates such endonucleases during normal DNA replication, loss of CHK1 could cause activation of the endonucleases and thereby lead to massive induction of DNA breaks. On the other hand, given that WEE1 depletion leads to a similar phenotype, it would rather be expected that increased CDK activity is the causative effect in activating such a DNA processing activity. Hence, it is possible that CDK has direct targets with enzymatic activity, i.e. a nuclease exhibiting aberrant activity, which could cause the massive induction of DNA damage (Figure 2). It remains to be determined to what extent such activities control genome integrity as well as the nature of the deregulated enzymes. Such nucleases should operate on the DNA structures generated by the excess CDK activity, and the elucidation of the contributions of such factors to genome integrity is an important task for future research.
DNA damage arising in S phase after acute inhibition of WEE1, ATR and CHK1 is likely dependent on nuclease activities. However, more subtle degrees of inhibition of these checkpoint kinases, as may more commonly occur in vivo, does not necessarily depend on nucleases. Fragile sites are areas in the genome that are prone to breakage after ATR and CHK1 inhibition, and recent evidence suggest that fragile sites can break in mitosis when sister chromatids are separating in anaphase (88). These sites are areas with few origins of replication, that critically depend on functional origins of DNA replication since neighbouring origins cannot compensate if forks stall and collapse (89). The dysfunctional replication upon reduced WEE1, ATR and CHK1 activity could lead to cells progressing into mitosis with unreplicated DNA. This is likely enhanced by G2 checkpoint deficiency upon reduced activity of these kinases. The outcome would be sister chromatid connections when chromosomes separate in anaphase, where breaks can occur by mechanical forces although nucleases may also cleave these structures.
ATR, CHK1 and WEE1 can affect malignant transformation
An important issue is whether loss of genomic integrity arising from deregulated replication caused by ATR, CHK1 and WEE1 disruption may contribute to cancer development. Heterozygous ATR and CHK1 mutations have been found in a subset of endometrial, colon, melanoma and stomach cancers (90–96). Several reports suggest that WEE1 function may also be compromised in cancer. Recently, it was shown that microRNA-155 (miR-155), which is frequently elevated in human cancers, cause down-regulation of WEE1 (97). Moreover, prostate epithelium, which is prone to prostate cancer development, expressed very low levels of WEE1 (98).
Studies of ATR and CHK1 heterozogosity in mice also suggest that compromised expression of these checkpoint kinases might contribute to cancer development. One report describes a modest increase in late tumour development in ATR heterozygous (+/−) mice (19), although increased tumourigenesis was not observed in other reports (25,99). However, on a mismatch repair-deficient (Mlh1 −/−) background, ATR heterozygozity caused a significant increase in tumourigenesis (100). CHK1 heterozygous (+/−) mice were prone to tumourigenesis on a WNT-1 transgenic background (22) and CHK1 heterozygosity induced in mouse mammary glands using a Cre/loxP system caused induction of mammary tumours in a p53 heterozygous background (101). It seems plausible that replication associated DNA damage due to insufficient CHK1 or ATR levels in S phase caused by hypomorphic mutations in these mice could contribute to promote genomic instability and tumour progression.
On the other hand, WEE1 and CHK1 have also been reported to be overexpressed in human cancers. WEE1 is overexpressed in human glioblastoma and a subset of breast cancers (102,103), and CHK1 mRNA expression was elevated in MYC-amplified neuroblastoma (104). Based on their role in restraining CDK activity, high levels of WEE1 and CHK1 would be expected to suppress rather than to promote cell growth. It may therefore seem like a paradox that high levels of CHK1 or WEE1 are found in human tumours where we rather would expect a selective pressure towards genetic alterations allowing uncontrolled growth. An explanation may be that these tumours contain other genetic alterations that promote increased CDK activity and replication stress, and if the CDK activity was too high, the replication associated damage would reach a level of severity resulting in cell death. These tumours may thus depend on the high levels of WEE1 or CHK1 to maintain CDK activity at sufficiently low levels for cell survival. Consistent with this hypothesis, inhibition of WEE1 leads to induction of DNA damage and cell death in tumours expressing high WEE1 levels (102). Furthermore, MYC is known to cause replication stress and elevated CHK1 expression was found selectively in MYC-amplified neuroblastoma (104). It is very possible that these cells depend on CHK1 to tolerate the high level of MYC expression.
Can the replicative roles of the checkpoint kinases be exploited for cancer treatment?
Checkpoint kinases guard against cancer-associated replication stress. In human tumours, replication stress induced by oncogenes or hypoxia leads to increased activation of ATR/CHK1 in S phase cells (105–107). Inhibitors of checkpoint kinases may therefore lead to selective killing of cancer cells with elevated replication stress (Figure 4). Currently, several inhibitors of CHK1 and WEE1 are in clinical trials (108–110); however this is largely based on their G2 checkpoint abrogation function and consequent induction of mitotic catastrophe. When used in combination with chemotherapeutic agents or radiation, inhibition of CHK1 or WEE1 can cause selective sensitization of p53 negative cells (111–116). It has been proposed that p53-negative cancer cells are particularly sensitive to G2 checkpoint abrogation because they lack the p53-dependent G1 checkpoint and therefore will depend more on the G2 checkpoint for DNA damage repair (117). On the other hand, p53-status does not always predict responses to CHK1 inhibition (118–121). Hence, additional effects of CHK1 inhibition, such as high levels of replication stress due to unscheduled initiation, likely contribute to cause cell death (11,27,104,122,123). Following inhibition of WEE1 or CHK1, DNA breakage in S phase (24,26,27) likely will cause killing of cancerous as well as normal tissue cells. Upon inhibition of these checkpoint kinases, loss of their function in restraining CDK activity might therefore contribute to cause unwanted normal tissue damage. However, the tumours would often contain a higher fraction of cycling cells than the surrounding normal tissue, as well as elevated replication stress due to genetic alterations (105,106) or hypoxia (107). The deleterious S phase effects in response to inhibition of these checkpoint kinases may therefore likely be more severe in the tumour compared to normal tissues, resulting in tumour selectivity. Moreover, treatments with checkpoint kinase inhibitors would benefit from a genetic profiling of the cancer, as this could allow pre-selection of patients with cancers that harbour marked replication stress.
Figure 4.

S phase effects during cancer treatment with inhibitors of ATR, CHK1 or WEE1 kinases may promote cancer specific killing. Cancer cells often contain elevated CDK activity due to genetic alterations, which may induce replication failures leading to DNA damage in S phase and subsequent genomic instability and tumour progression. Treatment with inhibitors of ATR, CHK1 or WEE1 further increases CDK activity leading to massive CDK-mediated DNA damage in S phase and subsequent cell death of S phase cancer cells.
In agreement with such tumour selective effects in S phase following checkpoint kinase inhibition, non-transformed mammary epithelial cells showed less γ-H2AX staining, less replication issues and less apoptosis compared to breast cancer cell lines in response to WEE1 inhibition (124). Inhibition of ATR can also sensitize cancer cells and ATR has also been suggested as a therapeutic target based on its role in restraining replication, although small-molecule ATR-inhibitors have only recently started to become available (75,125). The possibility of gaining tumour selective treatment based on inhibiting checkpoint kinases to induce deleterious DNA damage selectively in S phase tumour cells is an exciting task for future studies.
CONCLUSION
The DNA replication process is highly regulated with a large number of control mechanisms securing correct timing and quality of the process. Recent work has revealed that the checkpoint kinases ATR, CHK1 and WEE1 are active during normal S phase progression in the absence of exogenous DNA damage. Their activities are critical to maintain genome integrity, and they largely control genome integrity by restraining CDK activity. Deregulated CDK activity will cause unscheduled firing of replication origins in S phase and thereby lead to the induction of DNA breaks in a not yet fully understood mechanism. Such replication-associated DNA lesions may contribute to promote loss of genome integrity and cancer progression following heterozygous mutations or other ways of inactivation of ATR, CHK1 or WEE1 during tumorigenesis. Furthermore, replication-associated DNA damage occurring in response to small-molecule inhibitors of ATR, CHK1 and WEE1 may be exploited to identify new potential strategies for tumour selective treatment.
FUNDING
The Danish Cancer Society, The Novo Foundation; The Lundbeck Foundation; The Danish Medical Research Council; The Norwegian Cancer Society (Grant# 41998871636-PR20060204); The Research Council of Norway (Grant# 177356/F20); and South-Eastern Norway Regional Health Authority (Grant# 2008081). Funding for open access charge: Internal funds, Copenhagen University and Oslo University Hospital.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors wish to thank Halfdan Beck and Viola Nähse-Kumpf for stimulating discussions and useful input.
REFERENCES
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl Acad. Sci. USA. 2002;99:14795–14800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 4.Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 6.Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol. Cell Biol. 2006;26:3319–3326. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl Acad. Sci. USA. 2010;107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedel AM, Pike BL, Gasser SM. ATR/Mec1: coordinating fork stability and repair. Curr. Opin. Cell Biol. 2009;21:237–244. doi: 10.1016/j.ceb.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair. 2007;6:953–966. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat.Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Wang H, Powell SN, Iliakis G, Wang Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004;64:7139–7143. doi: 10.1158/0008-5472.CAN-04-1289. [DOI] [PubMed] [Google Scholar]
- 13.Hu B, Wang H, Wang X, Lu HR, Huang C, Powell SN, Huebner K, Wang Y. Fhit and CHK1 have opposing effects on homologous recombination repair. Cancer Res. 2005;65:8613–8616. doi: 10.1158/0008-5472.CAN-05-1966. [DOI] [PubMed] [Google Scholar]
- 14.Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 2004;6:884–891. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- 15.Zachos G, Black EJ, Walker M, Scott MT, Vagnarelli P, Earnshaw WC, Gillespie DA. Chk1 is required for spindle checkpoint function. Dev. Cell. 2007;12:247–260. doi: 10.1016/j.devcel.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 17.Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 1993;74:463–474. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- 18.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 19.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 20.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 21.Tominaga Y, Li C, Wang RH, Deng CX. Murine Wee1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int. J. Biol. Sci. 2006;2:161–170. doi: 10.7150/ijbs.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 23.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 24.Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 25.Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beck H, Nähse V, Larsen MS, Groth P, Clancy T, Lees M, Jørgensen M, Helleday T, Syljuåsen RG, Sørensen CS. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell. Biol. 2010;188:629–638. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair. 2010;9:1249–1255. doi: 10.1016/j.dnarep.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 30.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 31.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997;385:176–181. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 32.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 34.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 35.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol. 2007;27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stokes MP, Van Hatten R, Lindsay HD, Michael WM. DNA replication is required for the checkpoint response to damaged DNA in Xenopus egg extracts. J. Cell. Biol. 2002;158:863–872. doi: 10.1083/jcb.200204127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 40.Majka J, Binz SK, Wold MS, Burgers PM. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J. Biol. Chem. 2006;281:27855–27861. doi: 10.1074/jbc.M605176200. [DOI] [PubMed] [Google Scholar]
- 41.Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clarke CA, Clarke PR. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem. J. 2005;388:705–712. doi: 10.1042/BJ20041966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasahara K, Goto H, Enomoto M, Tomono Y, Kiyono T, Inagaki M. 14-3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO J. 2010;29:2802–2812. doi: 10.1038/emboj.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 46.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu CL, Kirley SD, Xiao H, Chuang Y, Chung DC, Zukerberg LR. Cables enhances cdk2 tyrosine 15 phosphorylation by Wee1, inhibits cell growth, and is lost in many human colon and squamous cancers. Cancer Res. 2001;61:7325–7332. [PubMed] [Google Scholar]
- 49.Harvey SL, Charlet A, Haas W, Gygi SP, Kellogg DR. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell. 2005;122:407–420. doi: 10.1016/j.cell.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 50.Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell. 2001;12:551–563. doi: 10.1091/mbc.12.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atherton-Fessler S, Liu F, Gabrielli B, Lee MS, Peng CY, Piwnica-Worms H. Cell cycle regulation of the p34cdc2 inhibitory kinases. Mol. Biol. Cell. 1994;5:989–1001. doi: 10.1091/mbc.5.9.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parker LL, Sylvestre PJ, Byrnes MJ, 3rd, Liu F, Piwnica-Worms H. Identification of a 95-kDa WEE1-like tyrosine kinase in HeLa cells. Proc. Natl Acad. Sci. USA. 1995;92:9638–9642. doi: 10.1073/pnas.92.21.9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGowan CH, Russell P. Cell cycle regulation of human WEE1. EMBO J. 1995;14:2166–2175. doi: 10.1002/j.1460-2075.1995.tb07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ayad NG, Rankin S, Murakami M, Jebanathirajah J, Gygi S, Kirschner MW. Tome-1, a trigger of mitotic entry, is degraded during G1 via the APC. Cell. 2003;113:101–113. doi: 10.1016/s0092-8674(03)00232-0. [DOI] [PubMed] [Google Scholar]
- 55.Melixetian M, Klein DK, Sorensen CS, Helin K. NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nat. Cell Biol. 2009;11:1247–1253. doi: 10.1038/ncb1969. [DOI] [PubMed] [Google Scholar]
- 56.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 57.Schmitt E, Boutros R, Froment C, Monsarrat B, Ducommun B, Dozier C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J. Cell Sci. 2006;119:4269–4275. doi: 10.1242/jcs.03200. [DOI] [PubMed] [Google Scholar]
- 58.Labib K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010;24:1208–1219. doi: 10.1101/gad.1933010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 61.Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, Dutta A. Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev. 2007;21:2288–2299. doi: 10.1101/gad.1585607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zou L, Stillman B. Formation of a preinitiation complex by S-phase cyclin-dependent loading of Cdc45p onto chromatin. Science. 1998;280:593–596. doi: 10.1126/science.280.5363.593. [DOI] [PubMed] [Google Scholar]
- 63.Kukimoto I, Igaki H, Kanda T. Human CDC45 protein binds to minichromosome maintenance 7 protein and the p70 subunit of DNA polymerase alpha. Eur. J. Biochem./FEBS. 1999;265:936–943. doi: 10.1046/j.1432-1327.1999.00791.x. [DOI] [PubMed] [Google Scholar]
- 64.Balestrini A, Cosentino C, Errico A, Garner E, Costanzo V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat. Cell Biol. 2010;12:484–491. doi: 10.1038/ncb2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumagai A, Shevchenko A, Dunphy WG. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell. 2010;140:349–359. doi: 10.1016/j.cell.2009.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gillespie PJ, Blow JJ. Clusters, factories and domains: The complex structure of S-phase comes into focus. Cell Cycle. 2010;9:3218–3226. doi: 10.4161/cc.9.16.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goren A, Cedar H. Replicating by the clock. Nat. Rev. Mol. Cell Biol. 2003;4:25–32. doi: 10.1038/nrm1008. [DOI] [PubMed] [Google Scholar]
- 68.Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomson AM, Gillespie PJ, Blow JJ. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J. Cell Biol. 2010;188:209–221. doi: 10.1083/jcb.200911037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karnani N, Dutta A. The effect of the intra-S-phase checkpoint on origins of replication in human cells. Genes Dev. 2011;25:621–633. doi: 10.1101/gad.2029711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- 73.Ragland RL, Arlt MF, Hughes ED, Saunders TL, Glover TW. Mice hypomorphic for Atr have increased DNA damage and abnormal checkpoint response. Mamm. Genome. 2009;20:375–385. doi: 10.1007/s00335-009-9195-4. [DOI] [PubMed] [Google Scholar]
- 74.Sørensen CS, Syljuåsen RG, Lukas J, Bartek J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle. 2004;3:941–945. [PubMed] [Google Scholar]
- 75.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janetka JW, Almeida L, Ashwell S, Brassil PJ, Daly K, Deng C, Gero T, Glynn RE, Horn CL, Ioannidis S, et al. Discovery of a novel class of 2-ureido thiophene carboxamide checkpoint kinase inhibitors. Bioorg. Med. Chem. Lett. 2008;18:4242–4248. doi: 10.1016/j.bmcl.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 77.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka S, Diffley JF. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002;16:2639–2649. doi: 10.1101/gad.1011002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lambert S, Carr AM. Checkpoint responses to replication fork barriers. Biochimie. 2005;87:591–602. doi: 10.1016/j.biochi.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 80.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 81.Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 82.Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003;22:4325–4336. doi: 10.1093/emboj/cdg391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–1083. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 84.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 86.Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 87.Froget B, Blaisonneau J, Lambert S, Baldacci G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol. Biol. Cell. 2008;19:445–456. doi: 10.1091/mbc.E07-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 89.Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 90.Bertoni F, Codegoni AM, Furlan D, Tibiletti MG, Capella C, Broggini M. CHK1 frameshift mutations in genetically unstable colorectal and endometrial cancers. Genes Chromosomes Cancer. 1999;26:176–180. [PubMed] [Google Scholar]
- 91.Kim CJ, Lee JH, Song JW, Cho YG, Kim SY, Nam SW, Yoo NJ, Park WS, Lee JY. Chk1 frameshift mutation in sporadic and hereditary non-polyposis colorectal cancers with microsatellite instability. Eur. J. Surg. Oncol. 2007;33:580–585. doi: 10.1016/j.ejso.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 92.Lewis KA, Mullany S, Thomas B, Chien J, Loewen R, Shridhar V, Cliby WA. Heterozygous ATR mutations in mismatch repair-deficient cancer cells have functional significance. Cancer Res. 2005;65:7091–7095. doi: 10.1158/0008-5472.CAN-05-1019. [DOI] [PubMed] [Google Scholar]
- 93.Menoyo A, Alazzouzi H, Espin E, Armengol M, Yamamoto H, Schwartz S., Jr Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res. 2001;61:7727–7730. [PubMed] [Google Scholar]
- 94.Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002;62:4095–4099. [PubMed] [Google Scholar]
- 95.Zighelboim I, Schmidt AP, Gao F, Thaker PH, Powell MA, Rader JS, Gibb RK, Mutch DG, Goodfellow PJ. ATR mutation in endometrioid endometrial cancer is associated with poor clinical outcomes. J. Clin. Oncol. 2009;27:3091–3096. doi: 10.1200/JCO.2008.19.9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kumar K, Rahman Q, Schipper H, Matschegewski C, Schiffmann D, Papp T. Mutational analysis of 9 different tumour-associated genes in human malignant mesothelioma cell lines. Oncol. Rep. 2005;14:743–750. [PubMed] [Google Scholar]
- 97.Tili E, Michaille JJ, Wernicke D, Alder H, Costinean S, Volinia S, Croce CM. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc. Natl Acad. Sci. USA. 2011;108:4908–4913. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kiviharju-af Hallstrom TM, Jaamaa S, Monkkonen M, Peltonen K, Andersson LC, Medema RH, Peehl DM, Laiho M. Human prostate epithelium lacks Wee1A-mediated DNA damage-induced checkpoint enforcement. Proc. Natl Acad. Sci. USA. 2007;104:7211–7216. doi: 10.1073/pnas.0609299104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fang Y, Tsao CC, Goodman BK, Furumai R, Tirado CA, Abraham RT, Wang XF. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J. 2004;23:3164–3174. doi: 10.1038/sj.emboj.7600315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fishler T, Li YY, Wang RH, Kim HS, Sengupta K, Vassilopoulos A, Lahusen T, Xu X, Lee MH, Liu Q, et al. Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene. 2010;29:4007–4017. doi: 10.1038/onc.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts D, Kaspers GJ, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18:244–257. doi: 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iorns E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A, Mein CA, Natrajan R, Savage K, Tamber N, et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLoS One. 2009;4:e5120. doi: 10.1371/journal.pone.0005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl Acad. Sci. USA. 2010;108:3336–3341. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010;29:5095–5102. doi: 10.1038/onc.2010.249. [DOI] [PubMed] [Google Scholar]
- 106.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 107.Hammond EM, Dorie MJ, Giaccia AJ. Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Res. 2004;64:6556–6562. doi: 10.1158/0008-5472.CAN-04-1520. [DOI] [PubMed] [Google Scholar]
- 108.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010;16:376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2010;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 Kinase Targeting Combined with DNA-Damaging Cancer Therapy Catalyzes Mitotic Catastrophe. Clin. Cancer Res. 2011;17:4200–4207. doi: 10.1158/1078-0432.CCR-10-2537. [DOI] [PubMed] [Google Scholar]
- 111.Ashwell S, Zabludoff S. DNA damage detection and repair pathways–recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 2008;14:4032–4037. doi: 10.1158/1078-0432.CCR-07-5138. [DOI] [PubMed] [Google Scholar]
- 112.Dixon H, Norbury CJ. Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle. 2002;1:362–368. doi: 10.4161/cc.1.6.257. [DOI] [PubMed] [Google Scholar]
- 113.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010;9:514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 114.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 115.Leijen S, Beijnen JH, Schellens JH. Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase results in sensitization of p53-deficient tumor cells to DNA-damaging agents. Curr. Clin. Pharmacol. 2010;5:186–191. doi: 10.2174/157488410791498824. [DOI] [PubMed] [Google Scholar]
- 116.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters JW, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H, Maitra A, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Russell KJ, Wiens LW, Demers GW, Galloway DA, Plon SE, Groudine M. Abrogation of the G2 checkpoint results in differential radiosensitization of G1 checkpoint-deficient and G1 checkpoint-competent cells. Cancer Res. 1995;55:1639–1642. [PubMed] [Google Scholar]
- 118.Petersen L, Hasvold G, Lukas J, Bartek J, Syljuasen RG. p53-dependent G(1) arrest in 1st or 2nd cell cycle may protect human cancer cells from cell death after treatment with ionizing radiation and Chk1 inhibitors. Cell Proliferation. 2010;43:365–371. doi: 10.1111/j.1365-2184.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29:6149–6159. doi: 10.1038/onc.2010.343. [DOI] [PubMed] [Google Scholar]
- 120.Hirose Y, Berger MS, Pieper RO. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001;61:5843–5849. [PubMed] [Google Scholar]
- 121.Tse AN, Carvajal R, Schwartz GK. Targeting checkpoint kinase 1 in cancer therapeutics. Clin. Cancer Res. 2007;13:1955–1960. doi: 10.1158/1078-0432.CCR-06-2793. [DOI] [PubMed] [Google Scholar]
- 122.Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–4981. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McNeely S, Conti C, Sheikh T, Patel H, Zabludoff S, Pommier Y, Schwartz G, Tse A. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9:995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 124.Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res. Treatment. 2010;122:347–357. doi: 10.1007/s10549-009-0571-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, Cliby WA, Sarkaria J, Beale G, Edmondson RJ, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer. 2011;105:372–381. doi: 10.1038/bjc.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]