


Abstract
In this work we have studied the effect of chromatin structure on the base excision repair (BER) efficiency of 8-oxoG. As a model system we have used precisely positioned dinucleosomes assembled with linker histone H1. A single 8-oxoG was inserted either in the linker or the core particle DNA within the dinucleosomal template. We found that in the absence of histone H1 the glycosylase OGG1 removed 8-oxoG from the linker DNA and cleaved DNA with identical efficiency as in the naked DNA. In contrast, the presence of histone H1 resulted in close to 10-fold decrease in the efficiency of 8-oxoG initiation of repair in linker DNA independently of linker DNA length. The repair of 8-oxoG in nucleosomal DNA was very highly impeded in both absence and presence of histone H1. Chaperone-induced uptake of H1 restored the efficiency of the glycosylase induced removal of 8-oxoG from linker DNA, but not from the nucleosomal DNA. We show, however, that removal of histone H1 and nucleosome remodelling are both necessary and sufficient for an efficient removal of 8-oxoG in nucleosomal DNA. Finally, a model for BER of 8-oxoG in chromatin templates is suggested.
INTRODUCTION
Eukaryotic cells are constantly subjected to oxidative stress leading to a tremendous number of insults, which have to be efficiently repaired. In the DNA, 8-oxo-7,8-dihydroguanine (8-oxoG) is one of the major lesions induced upon oxidative stress. 8-oxoG is repaired by the Base Excision Repair (BER) pathway (1). BER uses a limited number of enzymes and how BER functions on naked DNA template is well characterized. 8-oxoG is recognized and removed by the enzyme 8-oxoguanine DNA glycosylase (OGG1), which exhibits both a glycosylase and an apurinic/apyrimidinic (AP) lyase activity (2,3). After 8-oxoG removal OGG1 nicks DNA at the site of the lesion and then the endonuclease (APE1) creates a free 3′-hydroxyl at the site of the nick. The resulting gap in DNA is further filled by polymerase β and the nick is finally ligated by DNA ligase III [reviewed in (4)].
In eukaryotic cells, DNA is organized into chromatin. Chromatin exhibits a complex organization. The first level of chromatin organization is the nucleosome, which consists of an octamer of core histones (two of each H2A, H2B, H3 and H4) around which ∼147 bp of DNA are wrapped into two left superhelical turns (5). Under physiological conditions the nucleosome forms a rather stable structure (6). Distinct nucleosomes, connected by linker DNA, form the 10-nm chromatin filament (5). The 30-nm chromatin fibre is generated at higher ionic strength upon interaction of the 10-nm chromatin filament with the linker histone H1 (7,8). The available data show that the globular domain of histone H1 binds to both the DNA minor groove located at the centre of the nucleosome and ∼20-bp DNA at the entry/exit of the nucleosome (9–11). The C-terminal domain of histone H1 (11,12) as well as the N-terminal tail of core histone H3 (13) interacts with the nucleosome linkers which leads to the formation of the stem structure, where the two linkers are brought in close vicinity to each other. The linker histones and the N-termini of the core histones as well as their post-translational modifications play a crucial role in the condensation and spatial organization of both the chromatin fibre and the mitotic chromosomes (14–16).
How BER functions on chromatinized templates is far from being clear. The different enzymes involved in BER appeared to act in distinct manners on nucleosomes. In general, the presence of nucleosomes strongly interferes with BER (17–22). For example, uracil DNA glycosylase (UDG) showed a strong reduction (down to 30 times) in accessibility in nucleosomal DNA (17–19). In addition, the UDG efficiency appeared to depend strongly on the rotational positioning of the inserted uracil within the nucleosomal DNA (22,23), i.e. when the uracil faces the solution it is removed as easily as in free DNA, while the removal of uracil facing histone octamer is inhibited by more than three orders of magnitude. The activities of OGG1, APE1 and polymerase β were strongly reduced for an 8-oxoG located near the dyad of strongly positioned nucleosomes and remodelling by SWI/SNF was required for efficient repair (24). In contrast, both the FLAP endonuclease (FEN1) and DNA ligase I were reported to have similar activities on both naked DNA and nucleosomally organized DNA (25,26).
In this work we have analysed how the BER-initiating enzyme OGG1 triggers repair of 8-oxoG in histone H1 assembled dinucleosomal templates. A single 8-oxoG was inserted either in the linker or within the core particle DNA. The data show that only the removal of histone H1 is required for efficient linker 8-oxoG repair, while both H1 removal and nucleosome remodelling are required for repair of nucleosomal 8-oxoG.
MATERIALS AND METHODS
DNA probes
We used the strongly nucleosome positioning sequence 601 (a kind gift from Drs J Widom and B Bartholomew) to construct our various dinucleosomal templates. To produce dinucleosomes carrying a single 8-oxoG in the linker DNA, DNA templates for nucleosome number one (N1) and nucleosome number two (N2) were produced by PCR, carrying respectively a DraIII site at 3′- and 5′-end to allow a directional ligation. The list of all primers used can be found in the Supplementary Materials. In order to vary the size of the linker DNA we used different set of primers hybridizing closer or further away from the 601 positioning sequence inserted in the plasmid pGEMT. A short DNA carrying the single 8-oxoG was produced by hybridization of two synthetic 20-mer oligonucleotides: the sense with the 8-oxoG and the anti-sense free of lesion. Annealing of these two strands creates two oriented cohesive ends compatible with the required DraIII sites of the nucleosomal templates N1 and N2. To insert the 8-oxoG lesion we digested the PCR products corresponding to nucleosome one and two by DraIII and ligated them sequentially with the short double-strand 20-mer oligonucleotide. The oligonucleotides were phosphorylated on the 5′-ends to avoid formation of nicked DNA.
For the template DNA carrying 8-oxoG near the dyad of the first nucleosome we used a previously described DNA template for the PCR carrying two restriction sites AccB7I and BglI in the 601 positioning sequence for the insertion of the 8-oxoG (24). We produced the N1 templates with 8-oxoG according to Menoni et al. (24) and we ligated it via DraIII to the nucleosome number two template. Before labelling, each of the templates were agarose gel purified using crystal violet staining to avoid UV visualization and formation of any other DNA damage. For the probes carrying 8-oxoG in the linker DNA, templates were digested by Bst98I and the top strand was labelled by polymerase Klenow enzyme with [α-32P]dTTP in the presence of 50 μM dATP and gel purified. For the probe carrying 8-oxoG near the dyad, EcoRI digestion was performed and the bottom strand was labelled with the same procedure.
Proteins
Conventional recombinant Xenopus laevis full length core histone proteins were produced in bacteria and purified as described (27). The clones encoding the human H1.5 and the mouse NAP-1 (mNAP-1) were expressed in bacteria and the corresponding proteins were purified as described in (11). Briefly, the soluble H1 was purified first by SP sepharose and then by fractionation over a Resource S cation exchange column (Biorad). NAP-1 was purified by Resource Q anion exchange column. Purified proteins were analysed by 15% or 18% SDS–PAGE, stained with Coomassie blue. RSC was purified from yeast cells by using a standard TAP tag protocol (28). The recombinant murine enzyme OGG1 was purchased from Sigma-Aldrich. According to the producer, 1 U of enzyme cleaves 50% of 0.5 pmol of DNA substrate for 10 min at 37°C.
Reconstitution and characterization of positioned dinucleosomes
Nucleosomes were reconstituted by salt dialysis using ∼100 ng of the labelled DNA probe and 2.5 μg of nucleosomal size carrier DNA and recombinant histones (29,30). The appropriate histone:DNA ratio was carefully adjusted experimentally in order to reduce the content of free DNA in all nucleosomal preparations to <2%. Electrophoretic Mobility Shift Assay (EMSA) was carried out in 5% (w/v) polyacrylamide gel (29:1) or 1% agarose gel (w/v) in 0.3× TBE buffer. Details on Atomic Force Microscopy (AFM) characterization of our positioned dinucleosomes are provided in (31). The DNase I experiments were performed as described (32). The ratio of the amounts of DNase I used for dinucleosome and free DNA was 1:0.25 respectively. The micrococcal nuclease (MNase) digestion of the 230-bp 601 nucleosomes was done at room temperature for 6 and 12 min in the presence of 8 U/ml of the enzyme. The reaction buffer was 10 Mm Tris, pH 7.4, 1 mM DTT, 25 mM NaCl, 5% glycerol, 100 µg/ml BSA, 1.5 mM CaCl2 and 100 µg/ml of plasmid DNA. The reaction was stopped by addition of 20 mM EDTA and 0.1 mg/ml proteinase K, 0.1% SDS. DNA was isolated and run on a 10% native polyacrylamide gel.
Assay for OGG1 cleavage
The experiments were performed according to Menoni et al. (24). Briefly OGG1 was incubated with ∼0.5 pmol of dinucleosomal substrates, containing ∼20 fmol of 8-oxoG modified labelled DNA probe, either naked or dinucleosome reconstituted, in a repair buffer consisting of 10 mM Tris–HCl pH 7.4, 32 mM NaCl, 2.5 mM MgCl2, 200 μM EDTA, 8 mM KCl, 100 μg/ml BSA, 1 mM DTT, 0.02% NP40, 5% glycerol. When RSC was present, 1 mM ATP was added to the reaction mixture. To maintain the integrity of nucleosomes, all reactions were performed at 29°C (33). Reactions were stopped by 20 mM EDTA, 0.1% SDS and DNA was extracted by phenol/chloroform and ethanol precipitated prior to be analysed on 8% acrylamide–8M urea denaturing gel electrophoresis. Remodelling reactions were carried out with 2.5 U of RSC in presence of 1 mM ATP.
Curves were fitted by the equation: R = (Rmt/tc)/(1+t/tc), where Rm is the maximum relative cleavage (%) and tc is the apparent time constant. The experimental points were fitted better by this algebraic equation than an exponential growth function. Note, that neither the equation we used, nor a simple exponential equation describes the true complex enzymatic model of OGG1 cleavage that is still under debate. In fact, we have used this equation as a mathematically convenient way to compare apparent time constants by using a least square algorithm provided by OriginPro 8.5 software (OriginLab Corporation). The reported mean values and the respective relative standard deviations (RSD) were obtained by at least two independent experiments.
RESULTS
In this work we aimed to study the efficiency of BER to remove 8-oxoG lesions in dinucleosomal templates. To this end we have used a model system consisting of tandem repeats of two 601 sequences to reconstitute two distinct strongly positioned dinucleosomes with 20- and 75-bp length of internal linker DNA, respectively. A single 8-oxoG was inserted either in one of the nucleosomes in the vicinity of the dyad or in the centre of linker DNA between the two nucleosomes of the dinucleosomal template (see schematics of the dinucleosomal templates, Figure 1A). To understand how the presence of histone H1 affects BER, we have used dinucleosomes assembled with histone H1. The efficiency of the BER-initiating enzyme OGG1 to remove the 8-oxoG and cleave the DNA within the different templates was then measured.
Figure 1.

The 8-oxoG located in the linker DNA of the dinucleosome is removed by OGG1 as efficiently as in free DNA. (A) Schematics of the 601 dinucleosome constructs used for studying the repair of 8-oxoG. Tandem repeats of two 601 DNA sequences were used to reconstitute positioned dinucleosomes with linker DNA (between the two nucleosomes) of length of either 20 or 75 bp. The flanking free DNA arms at both ends of the dinucleosome were of 52 and 56 bp, respectively (for details see Supplementary Figure S1). 8-oxoG was inserted either in close vicinity to the nucleosomal dyad or in the centre of the linker DNA. Experiments were carried out either in the absence or in the presence of histone H1. (B) Eighteen percent SDS gel electrophoresis of the purified recombinant histones (left); right (oct), the equimolar mixture of the core histones used for the reconstitution of the dinucleosome. (C–E): Characterization of the reconstituted dinucleosome by EMSA (C), AFM (D) and DNase I footprinting (E). (F): Time course of the digestion of free DNA and dinucleosomes. Equal amounts of 32P-end labelled free DNA and dinucleosomes were incubated with 0.5 U of OGG1 for the times as indicated. The reaction was stopped and DNA was isolated by phenol–chloroform treatment. The products of the OGG1 cleavage reaction were separated on 8% denaturing PAGE and after exposure of the dried gel, data were quantified and expressed as a percentage of cut fractions versus incubation time. Solid curves are least squared fits of experimental points by the formula: R = (Rmt/tc)/(1 + t/tc). Rm values are 100% and 84 ± 4% for DNA and dinucleosomes respectively and tcDNA/tcdinuc = 0.94 ± 19%.
In the absence of histone H1 the linker DNA is as accessible as naked DNA to BER
Recombinant core histones were purified to homogeneity (Figure 1B) and used to reconstitute the dinucleosomal templates. The reconstituted dinucleosomes were then characterized by EMSA, AFM and DNase I footprinting (Figure 1). The EMSA shows a single band, corresponding to the reconstituted dinucleosomes with essentially no free DNA (Figure 1C). This indicates a complete association of the histone octamer with DNA under our experimental conditions for reconstitution. The AFM visualization shows, as expected, the presence of two nucleosomes within the dinucleosomal template (Figure 1D). The DNase I footprinting gives additional detail on the organization of the dinucleosome in solution. The reconstituted template exhibited two regions with strong protection, characteristic for well-positioned nucleosomes within the dinucleosomal template, separated by a region with a DNase I digestion pattern identical of naked DNA and corresponding to the linker DNA. All these data demonstrate that the reconstituted dinucleosomes are strongly positioned and represent a homogenous population of particles.
We next used these particles for studying how the inserted 8-oxoG within the linker DNA was cleaved and removed by OGG1 (Figure 1F). The same amount of either 32P-end labelled dinucleosomes (with linker length of 75 bp) or 32P-end labelled naked dinucleosomal DNA (that was used for reconstitution) were incubated for different times with 0.5 U of OGG1. The DNA was then analysed by an 8% denaturing PAGE and the cleavage efficiency of OGG1 was expressed as cut fraction versus the concentration (Figure 1F). As determined by curve fitting, half-maximal cleavage time (the cleavage time constant) for both free DNA and the dinucleosomal templates is essentially the same. We conclude that 8-oxoG within the linker DNA of the dinucleosome without histone H1 is recognized and processed by OGG1 as in naked DNA.
The presence of histone H1 interferes strongly with the removal of 8-oxoG from the linker DNA of the dinucleosomes
We next asked whether histone H1 affects the efficiency of OGG1 to cleave 8-oxoG in the linker DNA of dinucleosomal templates. We have used NAP-1 assisted deposition of H1 to generate high-quality dinucleosomes containing two histone H1 molecules per 32P-end labelled dinucleosome (34). Briefly, we first assembled NAP-1–histone H1 complex at a ratio 2:1 as previously described (11). Then we incubated the NAP-1–histone H1 complex with dinucleosomes (with linker DNA length of either 75 or 20 bp) at ratio ∼2.4:1 for 60 min at room temperature. The dinucleosomes were separated on either 5% native PAGE (Figure 2A) or 1% agarose gel (Figure 2B). Both samples run as a single band with electrophoresis mobility slightly slower than the respective control dinucleosomes without H1 (Figure 2A and B: lines 1, 5). In contrast, when only histone H1 was directly added to the dinucleosome solution, the dinucleosomes migrated as a smear throughout the length of the lane of the gel, which is indicative for the formation of aggregates (Figure 2A and B: lines 2–4, 7–9). These data illustrate, in agreement with previous data (11), that histone H1 is properly bound to the dinucleosome when NAP-1 was used as a chaperone for histone H1. Note that such dinucleosomes exhibit a specific •OH footprinting pattern, reflecting the binding of one H1 molecule at the entry/exit site of the nucleosome within the dinucleosomal template (11). Noteworthy, the proper NAP-1 deposition of histone H1 on nucleosomal DNA was also tested by using microccocal nuclease digestion of reconstituted mononucleosomes on the 230 bp 32P-body-labelled 601 fragments (Figure 2C). The data show that the micrococcal nuclease digestion pattern of H1 containing mononucleosomes, contrary to the control (without H1) mononucleosomes, showed a ‘chromatosome stop’ at 167 bp, further confirming the proper association of histone H1 with the nucleosomal templates [Figure 2C and (35)].
Figure 2.

NAP-1-mediated deposition of the linker histone H1 on dinucleosomes. (A and B) 32P-labelled dinucleosomes with linker DNA of either 75 or 20 bp were incubated with histone H1 alone or with NAP-1-H1 (2:1) complex at the indicated histone H1/dinucleosome ratios. The reaction mixture was then run on native 5% PAGE (A) or 1% agarose gel (B). (C) Miccrococal nuclease digestion of the mononucleosomes. Body-labelled mononucleosomes without H1 (lines 1, 2) and H1-containing (lines 3, 4) were digested with micrococcal nuclease and after arresting the reaction, the digested DNA was purified and run on 10% native PAGE. Lane 5 shows the DNA size marker in base pairs. The positions of the chromatosome band (167 bp) and the core particle (147 bp) are indicated by arrows.
By using these properly defined dinucleosomes, we next studied the accessibility of 8-oxoG towards OGG1 within the dinucleosomes with different linker lengths (Figure 3). The control dinucleosomes without H1 exhibited similar behaviour, i.e. no dependence on the length of the linker of the 8-oxoG accessibility towards OGG1 was observed (Figure 3A and B; the two upper panels). In contrast, the presence of H1 dramatically affects the cleavage efficiency of OGG1 in the linker DNA independently of its length (Figure 3A and B; the two middle panels). Quantification of the data showed that the rate of cleavage is ∼10-fold lower in the presence of H1 in nucleosomes with either linker length (Figure 3A and B; the lowest panels). Therefore, histone H1 strongly inhibits the accessibility of 8-oxoG in a manner independent of the length of the linker DNA.
Figure 3.

The presence of histone H1 strongly inhibits the efficiency of OGG1. 32P-end labelled control or H1 containing dinucleosomes with either 75 bp (A) or 20 bp (B) linker DNA with a single 8-oxoG inserted within the linker DNA were incubated with 0.2 U of OGG1 for the times indicated. The cleaved DNA was then isolated and separated on 8% denaturing PAGE. The positions of the full length (FL) and the cleaved (cut) DNA are indicated. The lower panels show the quantified data presented as percentage of cut fractions. Data were least square fitted as described in the experimental section and Figure 1F. The experimental points were fit to the same Rm = 83% that was obtained for the –H1 data and then fixed for the +H1 data point fitting. The mean values from two independent experiments for of T = tc−H1/tc+H1 were: 8.1 ± 12.4% and 10.3 ± 13.2% for the 75- and 20-bp linker DNA, respectively.
NAP-1 mediated removal of histone H1 restores the accessibility of linker DNA inserted 8-oxoG towards OGG1 within dinucleosomal templates
If histone H1 was really responsible for the observed inhibition of repair in linker-DNA inserted 8-oxoG, the removal of H1 should restore the efficient repair of the lesion by OGG1. To test this, we used NAP-1 to remove H1 from the dinucleosomes containing H1 (Figure 4A). Indeed, since NAP-1 is a chaperone of H1, the adding of increasing amount of NAP-1 to the dinucleosome solution would result in progressive removal of H1 from the dinucleosome and generation of H1-depleted particles with higher mobility and free NAP-1–H1 complexes. The experimental data show that this is really the case. As seen in Figure 4A, the increase of NAP-1 leads to dinucleosome with a faster mobility and finally, to the generation of a structure with electrophoretic mobility identical to a dinucleosome without H1. We attributed this effect to the NAP-1 induced removal of histone H1 from the dinucleosomes.
Figure 4.

NAP-1 mediated removal of histone H1 restores the efficiency of OGG1 to cleave 8-oxoG in the dinucleosomal linker DNA. (A) NAP-1 is able to remove histone H1 from the dinucleosomal template. 32P-end labelled 20-bp linker DNA dinucleosomes containing histone H1 were incubated with increasing excess of NAP-1 over nucleosomes as indicated and the mixture was run on a 1% agarose del (lanes 4–8). Nap1/nuc represents the molar ratio that does not take into account the 2-fold NAP-1 excess already present after H1 deposition. Lanes 1 and 2: dinucleosomes without histone H1. Note the increase of the electrophoretic mobility of the dinucleosomes upon increasing the concentration of NAP-1. (B) OGG1 Cleavage efficiency of 8-oxoG within the dinucleosome linker DNA. 32P-end labelled dinucleosomes without histone H1 (lines 1, 2, 6, 7) or with H1 (lines 3–5, 8–10) were incubated with an excess of NAP-1 over nucleosomes as indicated and then treated with either 0.5 (lanes 6–10) or with 5 U (lanes 1–5) of OGG1 for 90 min. The cleavage reaction products were separated on 8% denaturing gel. The dried gel was processed by a Phosphoimager (Fuji-Fla 5100) (upper panel). The position of the full length (FL) and cleaved (cut) DNA are indicated. The quantification of the data is shown on the lower panel. The relative SD is ±9% for line 8 and ±4–5% for all other lines.
We then studied how the presence of NAP-1, i.e. the removal of H1, affects the cleavage of the linker DNA 8-oxoG by OGG1. The different samples were treated with either 0.5 or 5 U of OGG1 for 90 min at 29°C and the products of the cleavage reaction were analysed by denaturing PAGE and quantified. The results presented in Figure 4B clearly show that in the presence of high concentrations of NAP-1, there is no difference in the accessibility of the 8-oxoG between dinucleosomes with and without histone H1. We conclude that the removal of histone H1 is responsible for this effect. In agreement with this, we found that NAP-1 has no influence on the cleavage efficiency of OGG1 on dinucleosome without H1.
The concerted action of NAP-1 and RSC is necessary and sufficient for efficient repair of 8-oxoG inserted in a core particles DNA within dinucleosomal templates
How could 8-oxoG (present in nucleosomal DNA) be removed from H1 containing dinucleosomal templates? In a previous study we have shown that 8-oxoG could be removed from nucleosomal templates lacking H1 only after ATP-dependent nucleosome remodelling (24). In addition, it was reported that the presence of H1 interferes with nucleosome remodelling (36,37). With this in mind, we hypothesized that the efficient repair of nucleosomal 8-oxoG requires both H1 release and remodelling of the dinucleosomal templates. We have studied this problem by using dinucleosome containing H1 with an 8-oxoG inserted in close vicinity to the dyad of one of the nucleosomes, since within this region the core histone–DNA interactions are quite strong and no significant unwrapping of the DNA ends is expected (Figure 5, see schematics). The samples were incubated with either NAP-1 alone or with chromatin remodeler RSC alone or with both (this allowed to either remove H1 or to remodel dinucleosomes with H1 and to remodel dinucleosomes stripped from H1 by NAP-1). At the same time the dinucleosomes were treated with OGG1 and the products of the cleavage reaction were analysed as previously described (see Figure 3 for detail). The removal of histone H1 has essentially no effect on the OGG1 cleavage of 8-oxoG (Figure 5, compare lane 1 with lane 4), while the treatment with RSC alone has increased the OGG1 cutting efficiency ∼5-fold (Figure 5, compare lane 3 with lane 6). Importantly, the simultaneous NAP-1 induced removal of histone H1 and remodelling of the dinucleosome by RSC result in a quasi complete (∼80–85%) cleavage of 8-oxoG by OGG1 (Figure 5, lane 5). Therefore, both histone H1 removal and dinucleosome remodelling are necessary and sufficient for efficient repair of nucleosomal 8-oxoG.
Figure 5.

Removal of histone H1 and RSC remodelling of the dinucleosome are necessary for efficient repair of 8-oxoG within the nucleosomal DNA. The 20-bp linker dinucleosomes, containing a single 8-oxoG in close vicinity to the nucleosome dyad, (see schematics at upper panel) with or without histone H1 were treated with either 2.5 U of RSC or 18× excess of NAP-1 or with both as indicated. At the same time the samples were treated with 1 U of OGG1 for 90 min and the cleaved DNA was run on 8% denaturing gel (middle panel). The position of the full length (FL) and cleaved (cut) DNA are indicated; 1/2, half of the amount of RSC; *indicates the position of the 32P radioactive label. Quantified data are shown on the lower panel as percentage of cut fractions. The relative SD is ±10–12% for lines 1–4 and ±3.5–5% for lines 5–10.
To further strengthen this conclusion we have carried out experiments with a solution containing an equimolar mixture of end-labelled dinucleosomes with 8-oxoG inserted either in the linker DNA or in the core particle DNA (see schematics in Figure 6). This sample was incubated either with NAP-1 or with both NAP-1 and RSC at 29°C together with 1 U of OGG1 for 90 min. The cleaved DNA was extracted and separated on 8% PAGE under denaturing conditions. This design of the experiment allows to study the accessibility of both linker and core particle located 8-oxoG within dinucleosomal templates in the same reaction conditions, thus, avoiding sample to sample variations. The obtained data are in complete agreement with the previous results. Indeed, the NAP-1 induced removal of H1 resulted in 80% cleavage of the linker 8-oxoG, while the core particle located one remained inaccessible to OGG1 (∼10% cut). In contrast, upon incubation with both RSC and NAP-1, both the linker and the nucleosomal 8-oxoG within the dinucleosomes became fully accessible to OGG1.
Figure 6.
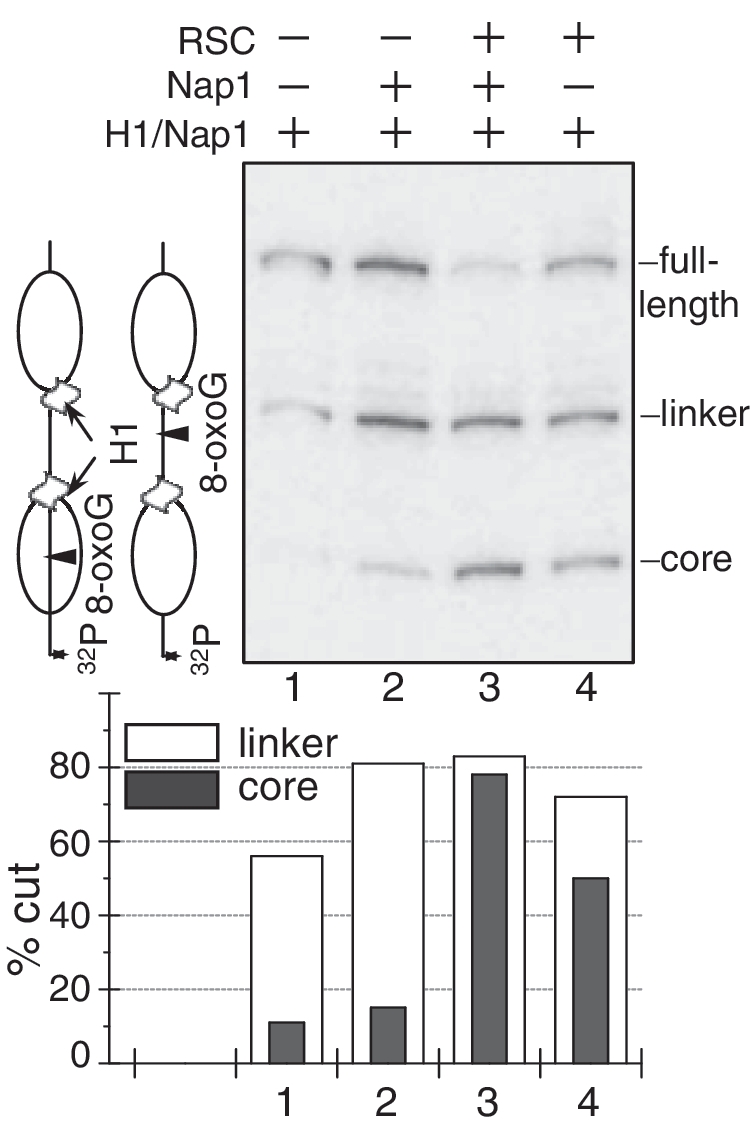
OGG1 cleavage of a mixture of two dinucleosome templates containing a single 8-oxoG either in the linker or within the core particle DNA. The 20-bp linker H1 containing dinucleosomal templates were mixed in equimolar amounts and incubated either with NAP-1 (18× excess) alone or with both RSC (2.5 U) and NAP-1 as indicated. The samples were simultaneously treated with 1 U of OGG1 and the cleaved DNA was run on 8% denaturing gel (upper panel). The arrows in the schematics (left) show the site of cleavage and indicate the mobility of the cleaved dinucleosomal template either in the linker (lower mobility) or in the core particle DNA (higher mobility). After exposure of the dried gel, the percentage of cut fraction within both the linker and the core particle DNA was measured (lower panel). The relative SD is ±11% for the two lowest values in lines 1 and 2 and ±3.5–4% for the remaining values.
DISCUSSION
This work was focused on the effect of chromatin on BER of 8-oxoG lesion. As a model system we have used dinucleosomes with inserted single 8-oxoG either in the centre of the linker DNA or in vicinity to the dyad axis in one of the nucleosomes of the dinucleosomal template. Dinucleosomes with different linker length were analysed. The study was carried out with both dinucleosomal templates with or without histone H1. The efficiency of the BER enzyme OGG1 to remove 8-oxoG and concomitantly cut the DNA was analysed. We found that OGG1 was able to process 8-oxoG linker DNA lesion quite efficiently in dinucleosomal templates without histone H1 and that the length of the linker DNA had no effect on the efficiency of OGG1 action. These results are in agreement with Nakanishi et al. (38) showing that that linker DNA is as accessible as naked DNA to a glycosylase in an oliognucleosome template. Noteworthy, excision of linker DNA pyrimidine (6–4) pyrimidone photolesions by purified human NER factors was strongly inhibited (39). These differences in the activity between NER and BER might reflect the much larger size of the NER machinery, which would require a space greater than the space of the linker DNA to remove efficiently the photolesions.
The presence of histone H1 profoundly affects the accessibility of 8-oxoG to OGG1 within the linker DNA, i.e. the initial rate of 8-oxoG cleavage drops to ∼10-fold in the presence of H1. These data are in agreement with the recently reported reduction of the efficiency of removal of uracil by the glycosylase UDG at sites located in the vicinity where the globular domain of H1 is proposed to bind the nucleosomal DNA as well as within the extra-core DNA (22).
NAP-1 generated stripping of histone H1 from the dinucleosomes restores the efficiency of OGG1 to remove 8-oxoG from the linker DNA. Interestingly, both removal of histone H1 and remodelling of the nucleosomes by RSC were necessary for complete repair of the core particle located 8-oxoG. The requirement for efficient repair of the removal of linker histone H1 from the dinucleosomal templates might be associated with the inability of chromatin remodelling machines to strongly remodel dinucleosomes. Indeed, it was reported that linker histones completely inhibited the remodelling of chromatin arrays by CHD1 and to ∼50% by ACF (36,37,40). This inhibition might be associated with the presence of H1-induced stem structure, resulting in close juxtaposition of the exit/entry of the linker DNA (11). The stem structure would interfere with the remodelling process and only part of the nucleosomes can be efficiently remodelled.
We suggest that the NAP-1 induced stripping of H1 [which leads to disruption of the stem structure (11)] allows complete remodelling of the dinucleosomes by RSC and subsequently an efficient removal of 8-oxoG. In other words, in order to initiate repair of the 8-oxoG from the nucleosomes, they have to be remodelled, but this last process requires chaperone induced eviction of histone H1.
Based on our data we suggest the following mechanism for 8-oxoG repair (see schematics, Figure 7). BER initiation of 8-oxoG in the linker DNA requires only the removal of histone H1. This could be achieved either by a histone H1 chaperone or by the remodeller (40) or most probably by the concerted action of both of them. Although, histone H1 in contrast to core histones, is quite mobile in vivo and it is exchanged continuously between different chromatin regions (41–43). This high mobility of H1 would make the linker DNA localized 8-oxoG accessible to the BER enzymes and thus, easier to be repaired. The situation appeared to be more complex for the repair of 8-oxoG localized in the core particle. In this case, not only H1 has to be removed, but the nucleosome has to be remodelled (44). This suggests that in vivo the repair of nucleosomal 8-oxoG would require the concerted action of both H1 chaperone and a dedicated remodeler.
Figure 7.
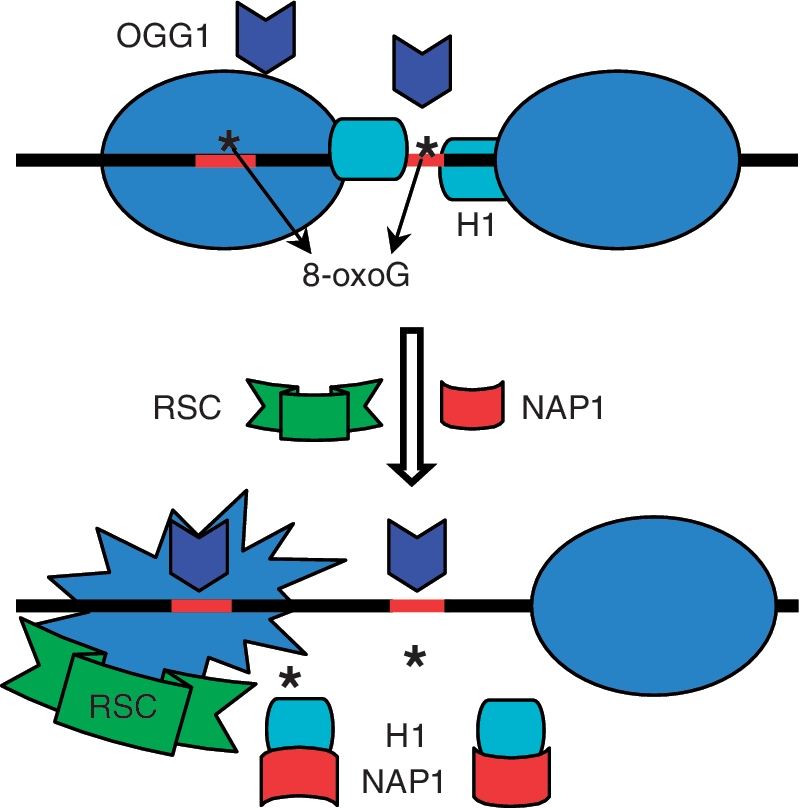
Model of the NAP-1 and RSC assisted initiation of BER within dinucleosomes. OGG1 is unable to repair 8-oxoG (denoted by Asterisk) in dinucleosomal templates. Removal of H1 is sufficient for 8-oxoG repair in the linker DNA, while both H1 removal and nucleosome remodelling are required for the excision of 8-oxoG located within nucleosomal DNA.
Interestingly, in Tetrahymena cells the dynamics of binding of linker histones to chromatin depend on ATP-dependent process (45). Chromatin remodelers are able to remodel nucleosomes thanks to the freed energy by the hydrolysis of ATP. Thus, in living cells the remodelers themselves could play a role in the mobility of histone H1 and consequently in the repair of oxidative lesions by BER enzymes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institut National de la Santé et de la Recherche Médicale - INSERM; Centre National de la Recherche Scientifique - CNRS; the Association pour la Recherche sur le Cancer - ARC (Grant SFI20101201424); the Agence Nationale de la Recherche: ANR-08-BLAN-0320-02 ‘EPIVAR’ (to S.D.) and ANR-09-BLAN-NT09-485720 ‘CHROREMBER’ (to D.A. and S.D.). S.D. acknowledges La Ligue Nationale contre le Cancer (Equipe Labellisée La Ligue) to S.D. Funding for open access charge: Association pour la Recherche sur le Cancer - ARC, France.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr J. Workman for kindly providing us with the yeast strain expressing tagged RSC. The authors thank Drs Cendrine Faivre-Moskalenko and Fabien Montel for the help with the AFM experiment. The authors thank Drs Jean Cadet and Didier Gasparutto for providing us with the modified oligonucleotide used in the construction of the template DNA carrying 8-oxoG near the dyad of the first nucleosome.
REFERENCES
- 1.Lindahl T, Karran P, Wood RD. DNA excision repair pathways. Curr. Opin. Genet. Dev. 1997;7:158–169. doi: 10.1016/s0959-437x(97)80124-4. [DOI] [PubMed] [Google Scholar]
- 2.Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA. 1997;94:7429–7434. doi: 10.1073/pnas.94.14.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Kemp PA, Thomas D, Barbey R, de Oliveira R, Boiteux S. Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc. Natl Acad. Sci. USA. 1996;93:5197–5202. doi: 10.1073/pnas.93.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 5.Van Holde KE, Allen JR, Tatchell K, Weischet WO, Lohr D. DNA-histone interactions in nucleosomes. Biophys. J. 1980;32:271–282. doi: 10.1016/S0006-3495(80)84956-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claudet C, Angelov D, Bouvet P, Dimitrov S, Bednar J. Histone octamer instability under single molecule experiment conditions. J. Biol. Chem. 2005;280:19958–19965. doi: 10.1074/jbc.M500121200. [DOI] [PubMed] [Google Scholar]
- 7.Makarov VL, Dimitrov SI, Petrov PT. Salt-induced conformational transitions in chromatin. A flow linear dichroism study. Eur. J. Biochem. 1983;133:491–497. doi: 10.1111/j.1432-1033.1983.tb07491.x. [DOI] [PubMed] [Google Scholar]
- 8.Dimitrov SI, Russanova VR, Pashev IG. The globular domain of histone H5 is internally located in the 30 nm chromatin fiber: an immunochemical study. EMBO J. 1987;6:2387–2392. doi: 10.1002/j.1460-2075.1987.tb02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staynov DZ, Crane-Robinson C. Footprinting of linker histones H5 and H1 on the nucleosome. EMBO J. 1988;7:3685–3691. doi: 10.1002/j.1460-2075.1988.tb03250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crane-Robinson C. Where is the globular domain of linker histone located on the nucleosome? Trends Biochem. Sci. 1997;22:75–77. doi: 10.1016/s0968-0004(97)01013-x. [DOI] [PubMed] [Google Scholar]
- 11.Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, Bednar J, Dimitrov S. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl Acad. Sci. USA. 2010;107:9620–9625. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamiche A, Schultz P, Ramakrishnan V, Oudet P, Prunell A. Linker histone-dependent DNA structure in linear mononucleosomes. J. Mol. Biol. 1996;257:30–42. doi: 10.1006/jmbi.1996.0144. [DOI] [PubMed] [Google Scholar]
- 13.Stefanovsky V, Dimitrov SI, Russanova VR, Angelov D, Pashev IG. Laser-induced crosslinking of histones to DNA in chromatin and core particles: implications in studying histone-DNA interactions. Nucleic Acids Res. 1989;17:10069–10081. doi: 10.1093/nar/17.23.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makarov VL, Dimitrov SI, Tsaneva IR, Pashev IG. The role of histone H1 and non-structured domains of core histones in maintaining the orientation of nucleosomes within the chromatin fiber. Biochem. Biophys. Res. Commun. 1984;122:1021–1027. doi: 10.1016/0006-291x(84)91193-8. [DOI] [PubMed] [Google Scholar]
- 15.Scrittori L, Hans F, Angelov D, Charra M, Prigent C, Dimitrov S. pEg2 aurora-A kinase, histone H3 phosphorylation, and chromosome assembly in Xenopus egg extract. J. Biol. Chem. 2001;276:30002–30010. doi: 10.1074/jbc.M102701200. [DOI] [PubMed] [Google Scholar]
- 16.de la Barre AE, Angelov D, Molla A, Dimitrov S. The N-terminus of histone H2B, but not that of histone H3 or its phosphorylation, is essential for chromosome condensation. EMBO J. 2001;20:6383–6393. doi: 10.1093/emboj/20.22.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beard BC, Wilson SH, Smerdon MJ. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc. Natl Acad. Sci. USA. 2003;100:7465–7470. doi: 10.1073/pnas.1330328100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beard BC, Stevenson JJ, Wilson SH, Smerdon MJ. Base excision repair in nucleosomes lacking histone tails. DNA Repair. 2005;4:203–209. doi: 10.1016/j.dnarep.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 19.Jagannathan I, Cole HA, Hayes JJ. Base excision repair in nucleosome substrates. Chromosome Res. 2006;14:27–37. doi: 10.1007/s10577-005-1020-7. [DOI] [PubMed] [Google Scholar]
- 20.Nilsen H, Lindahl T, Verreault A. DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 2002;21:5943–5952. doi: 10.1093/emboj/cdf581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odell ID, Newick K, Heintz NH, Wallace SS, Pederson DS. Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair. 2010;9:134–143. doi: 10.1016/j.dnarep.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole HA, Tabor-Godwin JM, Hayes JJ. Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J. Biol. Chem. 2010;285:2876–2885. doi: 10.1074/jbc.M109.073544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinz JM, Rodriguez Y, Smerdon MJ. Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl Acad. Sci. USA. 2010;107:4646–4651. doi: 10.1073/pnas.0914443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menoni H, Gasparutto D, Hamiche A, Cadet J, Dimitrov S, Bouvet P, Angelov D. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol. Cell Biol. 2007;27:5949–5956. doi: 10.1128/MCB.00376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chafin DR, Vitolo JM, Henricksen LA, Bambara RA, Hayes JJ. Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J. 2000;19:5492–5501. doi: 10.1093/emboj/19.20.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huggins CF, Chafin DR, Aoyagi S, Henricksen LA, Bambara RA, Hayes JJ. Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Mol. Cell. 2002;10:1201–1211. doi: 10.1016/s1097-2765(02)00736-0. [DOI] [PubMed] [Google Scholar]
- 27.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 28.Li B, Howe L, Anderson S, Yates JR, 3rd, Workman JL. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2003;278:8897–8903. doi: 10.1074/jbc.M212134200. [DOI] [PubMed] [Google Scholar]
- 29.Mutskov V, Gerber D, Angelov D, Ausio J, Workman J, Dimitrov S. Persistent interactions of core histone tails with nucleosomal DNA following acetylation and transcription factor binding. Mol. Cell. Biol. 1998;18:6293–6304. doi: 10.1128/mcb.18.11.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angelov D, Verdel A, An W, Bondarenko V, Hans F, Doyen CM, Studitsky VM, Hamiche A, Roeder RG, Bouvet P, et al. SWI/SNF remodeling and p300-dependent transcription of histone variant H2ABbd nucleosomal arrays. EMBO J. 2004;23:3815–3824. doi: 10.1038/sj.emboj.7600400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montel F, Castelnovo M, Menoni H, Angelov D, Dimitrov S, Faivre-Moskalenko C. RSC remodeling of oligo-nucleosomes: an atomic force microscopy study. Nucleic Acids Res. 39:2571–2579. doi: 10.1093/nar/gkq1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angelov D, Molla A, Perche PY, Hans F, Cote J, Khochbin S, Bouvet P, Dimitrov S. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol. Cell. 2003;11:1033–1041. doi: 10.1016/s1097-2765(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 33.Flaus A, Owen-Hughes T. Dynamic properties of nucleosomes during thermal and ATP-driven mobilization. Mol. Cell Biol. 2003;23:7767–7779. doi: 10.1128/MCB.23.21.7767-7779.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shintomi K, Iwabuchi M, Saeki H, Ura K, Kishimoto T, Ohsumi K. Nucleosome assembly protein-1 is a linker histone chaperone in Xenopus eggs. Proc. Natl Acad. Sci. USA. 2005;102:8210–8215. doi: 10.1073/pnas.0500822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simpson RT. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry. 1978;17:5524–5531. doi: 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- 36.Maier VK, Chioda M, Becker PB. ATP-dependent chromatosome remodeling. Biol. Chem. 2008;389:345–352. doi: 10.1515/BC.2008.040. [DOI] [PubMed] [Google Scholar]
- 37.Hill DA, Imbalzano AN. Human SWI/SNF nucleosome remodeling activity is partially inhibited by linker histone H1. Biochemistry. 2000;39:11649–11656. doi: 10.1021/bi001330z. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi S, Prasad R, Wilson SH, Smerdon M. Different structural states in oligonucleosomes are required for early versus late steps of base excision repair. Nucleic Acids Res. 2007;35:4313–4321. doi: 10.1093/nar/gkm436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y, Hanaoka F. ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J. 2001;20:2004–2014. doi: 10.1093/emboj/20.8.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maier VK, Chioda M, Rhodes D, Becker PB. ACF catalyses chromatosome movements in chromatin fibres. EMBO J. 2008;27:817–826. doi: 10.1038/sj.emboj.7601902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408:877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- 42.Kimura H, Cook PR. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gautier T, Abbott DW, Molla A, Verdel A, Ausio J, Dimitrov S. Histone variant H2ABbd confers lower stability to the nucleosome. EMBO Rep. 2004;5:715–720. doi: 10.1038/sj.embor.7400182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shukla MS, Syed SH, Montel F, Faivre-Moskalenko C, Bednar J, Travers A, Angelov D, Dimitrov S. Remosomes: RSC generated non-mobilized particles with approximately 180 bp DNA loosely associated with the histone octamer. Proc. Natl Acad. Sci. USA. 2010;107:1936–1941. doi: 10.1073/pnas.0904497107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dou Y, Bowen J, Liu Y, Gorovsky MA. Phosphorylation and an ATP-dependent process increase the dynamic exchange of H1 in chromatin. J. Cell Biol. 2002;158:1161–1170. doi: 10.1083/jcb.200202131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.