

Nucleoside diphosphate kinase (NDPK) is a direct target of AMP-activated protein kinase (AMPK) and is inhibited by AMPK-mediated phosphorylation at a conserved serine residue. This serine residue in NDPK is mutated in neuroblastoma, making the enzyme constitutively active.
Abstract
AMP-activated protein kinase (AMPK) is a key energy sensor that regulates metabolism to maintain cellular energy balance. AMPK activation has also been proposed to mimic benefits of caloric restriction and exercise. Therefore, identifying downstream AMPK targets could elucidate new mechanisms for maintaining cellular energy homeostasis. We identified the phosphotransferase nucleoside diphosphate kinase (NDPK), which maintains pools of nucleotides, as a direct AMPK target through the use of two-dimensional differential in-gel electrophoresis. Furthermore, we mapped the AMPK/NDPK phosphorylation site (serine 120) as a functionally potent enzymatic “off switch” both in vivo and in vitro. Because ATP is usually the most abundant cellular nucleotide, NDPK would normally consume ATP, whereas AMPK would inhibit NDPK to conserve energy. It is intriguing that serine 120 is mutated in advanced neuroblastoma, which suggests a mechanism by which NDPK in neuroblastoma can no longer be inhibited by AMPK-mediated phosphorylation. This novel placement of AMPK upstream and directly regulating NDPK activity has widespread implications for cellular energy/nucleotide balance, and we demonstrate in vivo that increased NDPK activity leads to susceptibility to energy deprivation–induced death.
INTRODUCTION
AMP-activated protein kinase (AMPK) functions as a cellular energy sensor activated by hypoxia, low glucose, and other stressors that lower ATP levels and raise AMP levels (Hardie et al., 2006; Shaw, 2006). In response to AMP/ATP ratio–altering events, activated AMPK turns on ATP-generating pathways and inhibits ATP-consuming pathways, thereby restoring the AMP/ATP ratio (Williams and Brenman, 2008). AMPK was first discovered as a protein whose activity inhibited preparations of acetyl-CoA carboxylase (ACC1), a regulator of cellular fatty acid synthesis (Winder et al., 1997). AMPK is a heterotrimeric protein with a 63-kDa catalytic α subunit and two regulatory β and γ subunits (38 and 36 kDa, respectively), each of which is encoded by distinct genes (α1, α2; β1, β2; γ1, γ2, γ3; Davies et al., 1994; Mitchelhill et al., 1994; Gao et al., 1996; Stapleton et al., 1996; Nielsen et al., 2003), and AMPK is implicated in a number of signaling pathways (Hardie, 2004, 2007; Shaw, 2009).
Upstream activation of AMPK is mediated by the tumor suppressor liver kinase B1 (LKB1; Shaw et al., 2004) and Ca2+/calmodulin–dependent kinase kinase β (Hawley et al., 2005; Hurley et al., 2005). Although LKB1 has clear roles in metabolism, LKB1 is also known as Par-4 in Caenorhabditis elegans, a key regulator of cell polarity (Watts et al., 2000; Chartier et al., 2011). Nonetheless, all known AMPK upstream kinases phosphorylate AMPKα threonine 172 (Thr-172) in the “activation loop” of the catalytic α subunit (both α1 and α2 isoforms), and this phosphorylation event causes >100-fold increase in kinase activity (Hawley et al., 1996). Conversely, dephosphorylation of Thr-172 by phosphatases can turn AMPK activity off (Sanders et al., 2007; Rubenstein et al., 2008). In general, mammalian AMPK activity stimulates processes involved in ATP-producing, catabolic pathways (e.g., increasing the glucose transporter GLUT4 and mitochondrial biogenesis) and inhibits ATP-consuming anabolic pathways (e.g., gluconeogenesis, lipogenesis, and protein synthesis; Hardie and Hawley, 2001; Barnes et al., 2002; Holmes and Dohm, 2004; Hardie, 2007). The best-defined direct target of AMPK is the fatty acid synthesis and rate-limiting enzyme ACC, which AMPK phosphorylates and inhibits to subsequently lower malonyl-CoA levels and increase fatty acid uptake into mitochondria (Merrill et al., 1997; Winder et al., 1997; Hardie and Hawley, 2001).
Beyond lipid synthesis, AMPK can also switch off protein synthesis by using two different pathways. These pathways include activation of elongation factor‑2 kinase, which causes inhibition of the elongation step of translation (Winder et al., 1997; Horman et al., 2002), and inhibition of the target-of-rapamycin (TOR) pathway, which stimulates the initiation step of protein synthesis by the phosphorylation of multiple targets (Proud, 2004). TOR is directly activated by an upstream signaling pathway involving the TSC1–TSC2 (tuberous sclerosis complex) heterodimer. AMPK directly phosphorylates TSC2 and thereby activates the TSC (Inoki et al., 2003). There is also evidence suggesting that AMPK might directly target and inhibit TOR (Cheng et al., 2004). More recent studies identified MRLC, raptor, a clock-related gene, and ULK1 as direct targets of AMPK (Lee et al., 2007; Gwinn et al., 2008; Lamia et al., 2009; Egan et al., 2011).
In this study, we searched for potential new targets of AMPK activity. From these efforts, we identified nucleoside diphosphate kinase (NDPK) as a potential downstream target of AMPKα. NDPK is a ubiquitous enzyme that catalyzes the transfer of the γ-phosphate group from a nucleoside or deoxynucleoside triphosphate (NTP or dNTP) to a nucleoside or deoxynucleoside diphosphate (NDP or dNDP, respectively) involving a high-energy phosphoenzyme intermediate (Rosengard et al., 1989; Engel et al., 1995). Functionally, NDPK maintains pools of nucleoside and deoxynucleoside triphosphates for processes central to energy utilization, for example, DNA synthesis and translation, using diphosphate substrates (Engel et al., 1995). In addition, the Drosophila homologue of NDPK is required in vivo during normal development for guided cell migration (Rosengard et al., 1989; Randazzo et al., 1991; Dammai et al., 2003; Nallamothu et al., 2008). Here we suggest mechanisms by which AMPK normally inhibits NDPK activity through phosphorylation of a highly conserved serine within NDPK.
RESULTS
Two-dimensional differential in-gel electrophoresis analyses identify NDPK as a phosphoprotein
The overall goal of our study was the identification of potential new targets of AMPK—keys for understanding energy homeostasis and metabolic disease that might be mediated by AMPK signaling.
We performed two-dimensional differential in-gel electrophoresis (2D-DIGE) using cytosolic brain lysates derived from wild-type (WT) and AMPKα 1/2 double-knockout (KO) mice (Williams et al., 2011) devoid of all AMPK catalytic activity to identify proteins altered in abundance or posttranslational modification between the WT and KO. One particular protein spot showed a potential shift in migration when comparing WT- and KO-derived lysates but a similar total abundance (Figure 1A).
FIGURE 1:
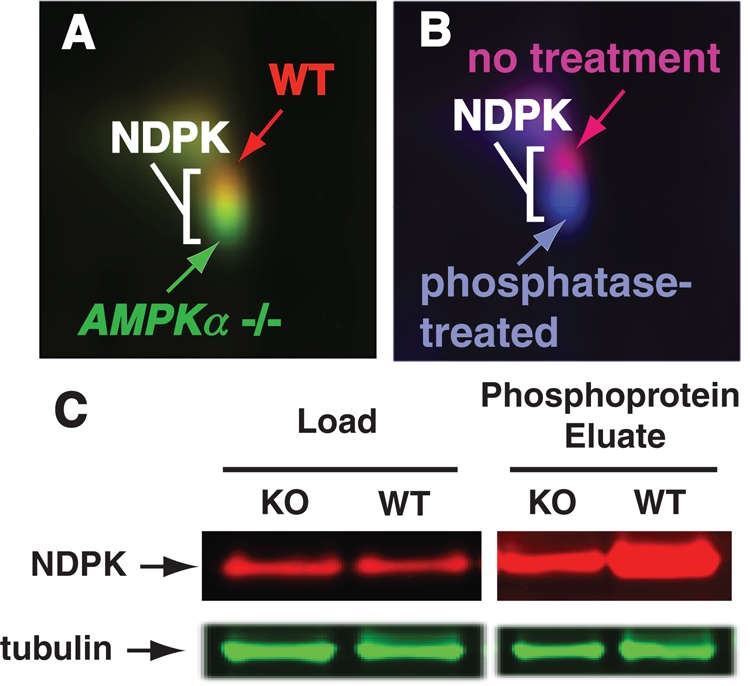
2D-DIGE identifies NDPK as a phosphoprotein with reduced phosphorylation in AMPKα1/2-targeted knockout brain. (A) 2D-DIGE identifies overlapping/adjacent protein spots with slower mobility in wild-type (red) compared with AMPKα-knockout (green) brain. (B) Phosphatase treatment of wild-type extracts (blue) results in greater mobility compared with untreated samples (magenta). All four protein spots were identified as NDPK. (C) Although total NDPK protein levels are similar, immobilized metal ion affinity chromatography purification of phosphoproteins demonstrates more phospho-NDPK in wild-type brain.
Given that AMPKα functions as a kinase and the WT spot migrated higher on the gel, this spot might contain a modified protein that was no longer phosphorylated in the KO and thus would migrate differently. A second 2D-DIGE experiment was performed with only WT lysate. However, the WT sample was divided into two samples; the first sample was treated with phosphatase, and the second sample was untreated. A qualitatively similar result to the WT/KO 2D-DIGE comparison was observed (Figure 1B).
After performing the 2D-DIGE experiments, all four adjacent spots of the gel corresponding to the WT or KO spots were excised and identified. All protein spots were confirmed to be nucleoside diphosphate kinase A (NDPK-A; EC 2.7.4.6), also known as Nm23 (nonmetastatic 23). Western blotting indicated that there was no significant difference in NDPK protein expression levels in the total WT versus KO lysate fractions (Figure 1C, Load, and Supplemental Figure S1A). However, a difference was noted when phosphoprotein enrichment was performed prior to Western blot analysis for NDPK, which indicated more phospho-NDPK exists in WT compared with KO brain lysate (Figure 1C, Eluate, and Supplemental Figure S1A).
Drosophila NDPK activity is inhibited by AMPK function, and loss of NDPK function can compensate for loss of AMPKα function
Although there are multiple genes that encode for NDPK activity in mammals (Boissan et al., 2009), there is only a single NDPK gene in Drosophila with previously characterized genetic loss-of-function mutations available for studies. We therefore used Drosophila genetics and a well-established biochemical NDPK activity assay (Timmons et al., 1995; Krishnan et al., 2001) to study NDPK activity in Drosophila. Using two genetically defined null alleles, we demonstrated gene-dosage responsiveness for NDPK activity (Figure 2A). We subsequently confirmed that the decreased NDPK biochemical activity also roughly correlated with the NDPK protein levels detected in various genetic NDPK mutant genotypes by Western blot (Figure 2B and Supplemental Figure S1B).
FIGURE 2:
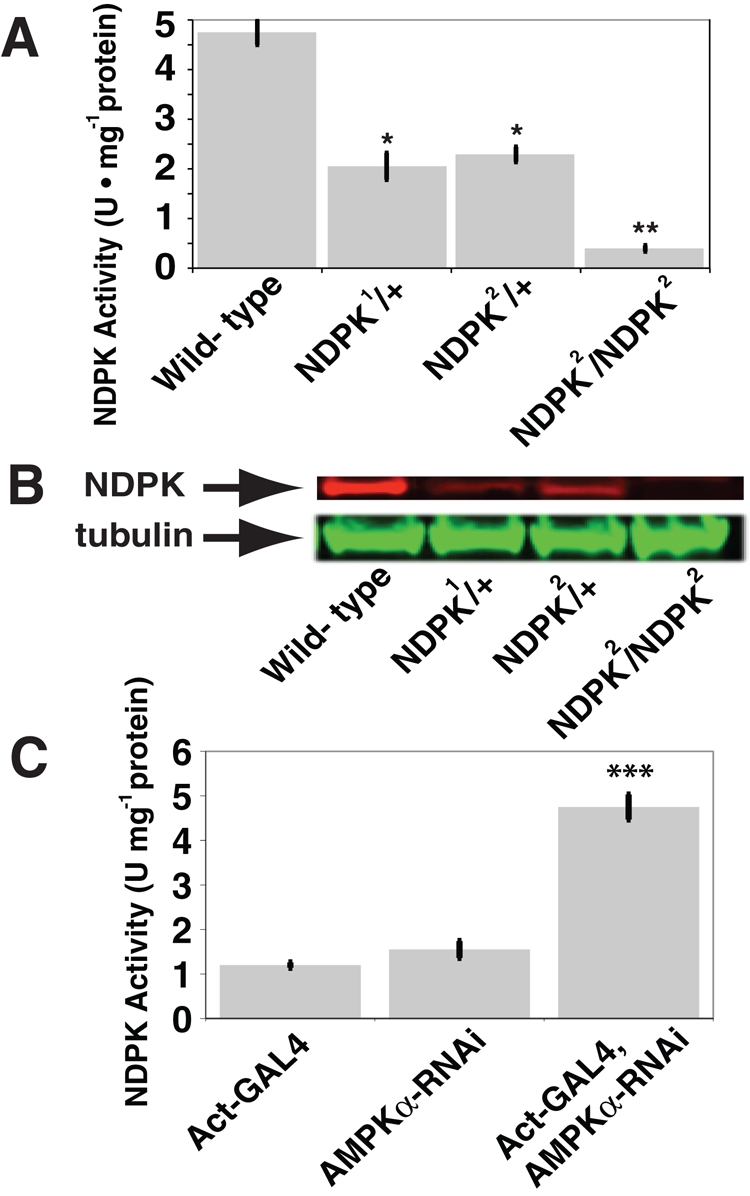
NDPK assay activity in Drosophila directly correlates with NDPK but inversely with AMPKα protein expression levels. (A) Quantification of the specific activities and (B) protein levels of the Drosophila NDPK alleles compared with wild-type show NDPK activity closely tracks NDPK protein levels (n = 5). **p < 0.001 and *p < 0.005 vs. the wild type. (C) The RNAi-mediated reduction of the AMPKα level in first-instar larvae leads to an increase in NDPK activity (n = 3). Actin-GAL4 was the driver for the expression of the AMPKα-RNAi transgenic element, and the RNAi-mediated reduction of AMPKα protein only occurs when the GAL4 is present. ***p < 0.0005. Data are shown as mean ± SEM.
We next investigated the seemingly inverse relationship between NDPK and AMPK activities by reducing AMPKα function through the use of a transgenic RNA interference (RNAi)–based expression system that phenocopies genetic loss of AMPKα function and only allows Drosophila development to reach the late pupal/pharate adult stage without producing eclosing adults (Johnson et al., 2010). When larvae with decreased AMPKα function, through RNAi (Johnson et al., 2010; Supplemental Figure S2, A and B), were assayed for NDPK activity, a significant increase was observed (Figure 2C).
Although these results are suggestive that AMPK might inhibit NDPK activity, because no prior report described the regulation of NDPK activity by phosphorylation, it was unclear whether loss of NDPK would enhance or inhibit AMPK genetic function. Initially, we used the transgenic RNAi-based expression system to reduce AMPKα function and found that introducing a single NDPK loss-of-function mutation for either NDPK allele (Figure 2B and Supplemental Figure S1B) rescued otherwise lethal AMPKα knockdown animals to viability/eclosion (Table 1). In addition, heterozygous NDPK loss-of-function alleles were able to rescue previously published (Mirouse et al., 2007) null AMPKα lethal loss-of-function alleles to viability as well, an effect otherwise only seen by dominant-negative S6 kinase, which is known to antagonize AMPK function (Montagne et al., 1999). In support of these results, increased NDPK expression did not rescue AMPKα knockdown or loss-of-function animals but instead made the transgenic animals susceptible to energetic stress by accelerating starvation-induced death in a defined starvation paradigm (Johnson et al., 2010; Figure 3).
TABLE 1:
AMPKα RNAi and AMPKα loss-of-function rescue by NDPK.
| Transgene or loss-of-function mutation | AMPKα RNAi rescue (% expected) | AMPKα loss-of-function allele 1 rescue (number rescued/total scored) | AMPKα loss-of-function allele 2 rescue (number rescued/total scored) |
|---|---|---|---|
| UAS-S6k (DN) | Yes (11) | Yes (14/130) | Yes (23/122) |
| UAS-TOR (DN) | Yes (13) | No (0/68) | No (0/42) |
| NDPK1 | Yes (26) | Yes (7/91) | Yes (12/96) |
| NDPK2 | Yes (41) | Yes (5/101) | Yes (8/71) |
| UAS-AMPKα | Yes (100) | Yes (22/65) | Yes (36/75) |
| UAS-AMPKγ | No | No (0/63) | No (0/94) |
| UAS-GFP | No | No (0/52) | No (0/77) |
FIGURE 3:

NDPK transgenic overexpression in Drosophila leads to decreased survival under starvation conditions. Male flies (n = 30) were starved in empty food vials containing filter paper saturated with deionized H2O. Tubulin-GAL4 was the driver for overexpression of the NDPK transgenic element, and NDPK protein overexpression only occurs when the GAL4 element is present. Asterisk denotes statistically different values; for the 60-, 72-, 84-, and 96-h time points, p < 0.0072, <0.0005, <0.0001, and <0.013, respectively; n = 3. Data are shown as mean ± SEM.
AMPK directly inhibits NDPK activity through phosphorylation
The preceding results suggested that AMPK and NDPK genetically antagonize each other. Combination of these genetic results with the biochemical identification of decreased NDPK phosphorylation in AMPKα mutant brain suggests a potential model by which AMPK directly phosphorylates NDPK to inhibit NDPK function. The relationship between NDPK and AMPK was subsequently investigated in vitro using AMPK purified from a cell expression system (Dyck et al., 1996), which was subsequently added to NDPK protein. When these protein complexes were incubated together before performing NDPK assays, NDPK activity was significantly decreased and substantially further decreased upon additional AMPK activation (Figure 4A and Supplemental Figure S1C) with cobalt chloride (Lee et al., 2003; Supplemental Figure S3; CoCl2 treatment was the most effective AMPK activator of those tested), which demonstrates AMPK activity inhibits NDPK activity in vitro (Figure 4B).
FIGURE 4:

Activated AMPK activity decreases NDPK activity. Activated AMPK decreases NDPK activity. (A) CoCl2 addition to HEK293 cells transiently transfected with GST-AMPKα1, HA-β1, and HA-γ1 increases purified activated phospho-AMPKα and (B) incubating purified NDPK with AMPK decreases NDPK activity. **p < 0.005 and *p < 0.05 vs. GST alone (n = 5).
After establishing that AMPK can directly phosphorylate NDPK in vitro, we identified potential NDPK phosphorylation sites in vivo. The protein spot corresponding to NDPK from WT mouse brain was excised for phosphopeptide mapping (Yale Mass Spectrometry Core; 73.7% coverage, including all NDPK serines and threonines; Supplemental Figure S4). A single peptide (Figure 5A, underlined red) with three candidate serines was found to contain a single phosphoserine. Of importance, this sequence flanked the catalytic NDPK histidine residue and contained three total serine residues, only two of which were conserved from Drosophila to human.
FIGURE 5:

An identified in vivo phospho-NDPK peptide and identification of an AMPK-dependent NDPK phosphoserine inhibitory site. (A) Sequence alignment of the active-site region of NDPK (purple shading) and identified NDPK phosphopeptide (red underline). The active-site histidine and conserved serines (Ser-120 and Ser-125) are highlighted in green, red, and blue, respectively. (B) The specific activities of wild-type and each NDPK mutant with (dark-colored columns) and without (light-colored columns) the addition of activated AMPK. The decrease in activity for the S120A mutant protein was not statistically significant (p = 0.36, n = 3). *p < 0.05, n = 3. Data are shown as mean ± SEM.
To identify which serine residues may be phosphorylated by AMPK, the two conserved serine residues were mutated singly for in vitro AMPK phosphorylation/NDPK activity assays. Single mutations of either serine (S120 or S125) to alanine in NDPK had no significant effect on NDPK activity. Similarly, mutating S125 to the phosphomimetic amino acid glutamate also had no effect on NDPK activity (Figure 5B). However, mutating S120 to glutamate (E) resulted in insoluble protein under native protein purification conditions. In addition, when solubilized under denaturing/renaturation conditions, the S120E mutant protein was inactive even when WT NDPK that underwent the same treatment maintained significant activity (Supplemental Figure S5). We therefore added purified AMPK to purified NDPK protein—either WT or with the serine-to-alanine mutations to measure AMPK-mediated inhibition of NDPK activity. Indeed, adding the AMPK complex to S125A decreased NDPK activity; however, adding AMPK to S120A had no inhibitory effect on NDPK activity, suggesting that S120 is a residue that can be phosphorylated to inhibit NDPK activity (Figure 5B).
To determine whether AMPK could phosphorylate NDPK, in vitro kinase assays were performed to monitor the incorporation of 32P into a modified version of the NDPK phosphopeptide (the NDPKtide, with all serines except S120 mutated to alanine; Figure 5A and Supplemental Figure S6) versus the SAMS peptide, a specific, well-established gold standard substrate for AMPK activity (Davies et al., 1989). The kinase assays revealed very similar calculated specific activities for the SAMS and the NDPKtide peptides, Vmax = 1.47 and 1.29 nmol min−1 mg−1, respectively, with the SAMS peptide being only a slightly better—but statistically insignificant—substrate (Figure 6, A and B, and Supplemental Figure S7).
FIGURE 6:

In vitro AMPK kinase assays demonstrate similar kinetics and affinity of AMPK for NDPK and SAMS peptide. (A) Kinase assays were performed with purified AMPK from untransfected (untrans.) HEK cell lysates, kinase-dead (KD) myc-tagged AMPK, and wild-type (WT) myc-tagged AMPK. A synthesized NDPK peptide (containing only one serine, Ser-120) and the SAMS peptide were compared as AMPK substrates (n = 3). (B) Kinase assays were performed with varying amounts of the substrate peptide and WT-AMPK to measure the kinetics of the SAMS and NDPK peptides (n = 3).
DISCUSSION
Elaborating targets for the cellular energy sensor AMPK is key for understanding the roles this molecule plays in energy homeostasis and metabolic disease. Through the use of proteomic techniques, we were able to identify proteins that are potentially regulated and/or phosphorylated by AMPK and further prioritize these proteins for study based on genetic evaluation in Drosophila. On the basis of these criteria and studies, the protein NDPK was identified as a good candidate for additional study.
Phosphopeptide mapping identified a peptide (Figure 5A, underlined in red) that contained a phosphorylated serine residue. Mutagenesis studies performed on the two conserved serine residues indicated that S120 is the critical residue for NDPK regulation (Figure 5B), which has been a speculated but, up to this point, not experimentally validated mechanism for NDPK regulation (Venerando et al., 2011). These results correlate well with previous studies indicating that NDPK is phosphorylated at serine residue(s) (MacDonald et al., 1993; Almaula et al., 1995; Treharne et al., 2009). In fact, two prior studies, one in Escherichia coli (Almaula et al., 1995) and a proteome-wide phospho-mapping study in Drosophila (Zhai et al., 2008), found this serine (S120) to be a candidate for phosphorylation. In addition, in vitro assays mixing protein kinase CK2 (CK2; formerly casein kinase 2) with NDPK demonstrated that serine phosphorylation of NDPK may be a mechanism to negatively regulate its activity (Biondi et al., 1996) and indicate that the serine phosphorylation of NDPK may inhibit NDPK's phosphotransferase function (Treharne et al., 2009), which are in agreement with our results. Therefore, NDPK activity, which normally has a high turnover rate, might be partially inhibited by serine phosphorylation (Biondi et al., 1996). Conversely, the need for large amounts of NTPs could be satisfied rapidly via phosphatase-catalyzed dephosphorylation of NDPK, thus increasing NDPK activity (Biondi et al., 1996). However, until now, the mechanistic details of NDPK inhibition due to a specific phosphorylation event in vivo have been unclear.
The critical serine identified in this work, S120, is strictly conserved in all prokaryotic and eukaryotic NDP kinases and has been linked to a direct involvement in the catalytic mechanism of stabilizing the NDPK catalytic site (His-118) through site-directed mutagenesis and crystal structure studies (Tepper et al., 1994; Giraud et al., 2006). A serine 120-to-glycine (S120G) mutation of nm23-H1 (NDPK-A) was even identified in several aggressive neuroblastomas (Chang et al., 1994). Of interest, biochemical studies indicated that this mutant S120G was still active, which is comparable to our results with our S120 mutants (Chang et al., 1994; Freije et al., 1997).
We verified serine 120 as a residue capable of being phosphorylated by AMPK through the use of in vitro kinase assays and comparisons to the SAMS peptide, which is a specific substrate for AMPK that contains the optimal AMPK-binding sequence (Gwinn et al., 2008). Although the NDPKtide does not contain the described optimal AMPK-binding sequence, several described AMPK substrates also do not contain this sequence, including histone H2B, myosin light chain, the tumor suppressor p53, and the cyclin-dependent kinase inhibitor p27 (Jones et al., 2005; Lee et al., 2007; Liang et al., 2007; Bjorklund et al., 2010; Bungard et al., 2010; Kim et al., 2011). Of importance, the NDPK peptide displayed kinetic characteristics similar to those of the SAMS peptide (Figure 6B), and these kinetic characteristics were completely abolished when S120 was individually mutated to an alanine (Supplementary Figure S7). Thus the NDPKtide sequence and serine 120 are a specific peptide region and residue targeted by AMPK for phosphorylation.
Although many studies have suggested phosphorylation may affect NDPK activity, a clear genetic link to a kinase in vivo has been missing to this point. A previous study (Treharne et al., 2009) provided 1) biochemical evidence to indicate a direct interaction between AMPK and NDPK, although other groups have been unable to reproduce these results (Annesley et al., 2011), and 2) data suggesting that AMPK activity decreases NDPK's phosphotransferase activity through the phosphorylation of NDPK serine residues, but was unable to mechanistically define the interaction and indicated that the AMPK/NDPK interaction may still be indirect. Other studies investigating the AMPK/NDPK interaction have also served to confound this interaction—see, for example, the retractions of several papers that had sought to directly address this interaction biochemically (Crawford et al., 2005, 2006a, 2006b, 2007). Nevertheless, these retracted papers claimed that NDPK regulated and activated AMPK, which is not the regulatory interaction we describe here.
In our proposed model, active NDPK helps produce nucleotides for various anabolic pathways in the presence of inactive AMPK, that is, a high-energy and/or low-stress cellular state (Figure 7A). However, when cellular ATP falls and AMP levels rise, AMPK is activated and phosphorylates the critical serine 120 residue of NDPK to decrease its activity and, thereby, save ATP stores (Figure 7B). Thus AMPK is able to fulfill its primary energy gauge function and balance the formation of nucleotides with the conservation of ATP under energetic stress. Such a model might also explain why a mutation of this critical serine to a nonphosphorylatable glycine residue would lead to more aggressive neuroblastomas (Chang et al., 1994); tumors could synthesize deoxynucleotides for DNA replication and use the increased biosynthetic products of NDPK for elevated levels of glycolysis (Green et al., 2011) even during energetic stress if this “off switch” was absent. This paradigm illustrates a potential direct link between tumor suppression and control of cellular metabolism. In addition, the activation of NDPK would also trigger an increase in the ADP/ATP ratio, which would further enhance AMPK activity by protecting AMPK from dephosphorylation. Neuroblastomas with the NDPK S120G mutation would then have elevated AMPK activities, as well as constitutive NDPK activation, which might increase glucose uptake/oxidation and help with tumor survival. Thus this AMPK-mediated NDPK inhibitory function is consistent with numerous studies suggesting that AMPK, as a molecule downstream of the human tumor suppressor LKB1, has tumor-suppressive activities (Jones et al., 2005; Shackelford and Shaw, 2009). Of course, it is unlikely that AMPK activity is the only activity capable of regulating NDPK activity; however, in brain lysates, it appears that AMPK activity is a major one.
FIGURE 7:

AMPK mediates inhibition of NDPK activity during nutrient stress. Model for AMPK-mediated NDPK inhibition. (A) Under nutrient-rich conditions, AMPK remains inactive, and NDPK is active and uses ATP as an energy source to produce nucleotides/deoxynucleotides for several anabolic pathways. (B) During nutrient-limiting conditions, AMPK is active (and phosphorylated at Thr-172) and inhibits NDPK activity through the phosphorylation of Ser-120, thereby conserving cellular ATP stores. Dashed lines indicate decreased biochemical activities.
MATERIALS AND METHODS
Materials
All chemicals were of an analytical grade and, unless otherwise noted, from Sigma-Chemical (St. Louis, MO) or Fisher Scientific (Fair Lawn, NJ). [γ-32P]ATP (specific activity 3000 Ci/mmol) was from PerkinElmer (Boston, MA). The SAMS (HMRSAMSGLHLVKRR) and NDPK (RNIIHGSDAVKAKRR) peptides were synthesized by Abgent (San Diego, CA) and the University of North Carolina Microprotein Sequencing and Peptide Synthesis Facility (Chapel Hill, NC), respectively. The hemagglutinin (HA)-tagged human AMPKγ1 and rat AMPKβ1 constructs were gifts from R. Shaw. Glutathione S-transferase (GST)–tagged AMPKα1 was a gift from L. Witters. The antibodies used were as follows: anti-NDPK (C-20, sc-343; Santa Cruz Biotechnology, Santa Cruz, CA), anti–phospho-AMPKα (40H9) and anti-AMPKα (23A3; Cell Signaling, Beverly, MA), and anti–α-tubulin (clone B-5-1-2; Sigma-Aldrich). Anti-dNDPK (reactive to protein of Drosophila origin, dNDPK) was a gift from T. Hsu.
Transgenic animals and brain sample/lysate preparation
Three-month-old male mice were killed to obtain brain tissue (kindly provided by T. Williams, University of North Carolina). The genotypes used were as follows: wild type, human glial fibrillary acidic protein-Cre (hGFAP-Cre) mice (K. McCarthy, University of North Carolina), and conditional AMPKα 1/2 knockout mice (Williams et al., 2011) to produce AMPKα1−/−α2F/F-hGFAP-Cre mice. The animals were handled under protocols approved by the Institutional Animal Care and Use Committee (Institutional Animal Care and Use Committee ID 09-149.0) of the University of North Carolina–Chapel Hill and in accordance with National Institutes of Health guidelines.
For the preparation of brain lysate, a whole mouse brain (375–425 mg) was processed in 10 ml of ice-cold lysis buffer A (50 mM Tris-HCl, pH 7.5, protease [P2714; Sigma-Aldrich] and phosphatase [P5726; Sigma-Aldrich] inhibitor cocktails [note: for the preparation of the phosphatase-treated WT brain lysate for 2D-DIGE, the phosphatase inhibitor cocktail was omitted for 2.5 U/ml alkaline phosphatase (final concentration; P6774; Sigma) and the sample was incubated for 1 h with the addition of 1 mM MgCl2 (37°C) before the high-speed centrifugation step], and benzonase nuclease) using a Tissuemiser homogenizer (Fisher Scientific). The lysate was centrifuged at 1000 × g for 20 min (4°C). The supernatant was then centrifuged for a second time. The resultant clarified supernatant was centrifuged at 100,000 × g to produce the cytosol. The pellet was discarded, and the supernatant (cytosol) was directly used for Western blotting and then for 2D-DIGE analyses after it was cleaned using a 2D Clean-Up Kit (GE Healthcare, Piscataway, NJ).
2D-DIGE protocols and protein identifications
The 2D-DIGE experiments were performed by the University of North Carolina Systems-Proteomics Center using previously described methodologies (Osorio et al., 2007). Protein spot identification was performed by the Yale Mass Spectrometry and Proteomics Resource Core (New Haven, CT) using peptide mass fingerprinting tandem mass spectrometry data, as previously described (Osorio et al., 2007; Pinaud et al., 2008).
Purification of phosphoproteins by immobilized metal affinity chromatography
Phosphoproteins were purified from mouse brain lysates using a PhosphoProtein Kit (Qiagen, Valencia, CA), as described by the manufacturer. The brain lysates were prepared as described; however, 0.25% 3-([3-cholamidopropyl]dimethyl-ammonio)-1-propanesulfonate was included in the buffer and throughout the purification process, as recommended by the manufacturer's protocol.
Plasmid construction
A pET21b (Novagen, Gibbstown, NJ) construct containing the cDNA for wild-type NDP kinase A, inserted in the BamHI-NdeI site, was obtained from M.-L. Lacombe. The NDPK gene was PCR amplified from this construct using the primer pair F, 5′-AAAGGATCCGGCCAACTGTGAGCGTACCTTC-3′, and R, 5′-AAAGGATCCGGCCAACTGTGAGCGTACCTTC-3′), digested with BamHI and SalI, and ligated into a pET28b (Novagen) vector cut with the same restriction enzymes for expression as a histidine (His)-tagged protein.
Site-directed mutagenesis of NDPK was carried out using bridging PCR. The primer pairs for the construction of each NDPK variant are as follows: S120E (F, 5′-ATTATACATGGCGAGGATTCTGTGGAGAGTGC-3′; R, 5′-TCCACAGAATCCTCGCCATGTATAATGTTCCTG-3′), S125E (F, 5′-TTCTGTGGAGGAGGCAGAGAAGGAGAT-CGG-3′, R, 5′-CCTTCTCTGCCTCCTCCACAGAATCACTGCC-3′), S120A (F, 5′-ATTATACATGGCGCTGATTCTGTGGAGAGTGC-3′, R, 5′-TCCACAGAATCAGCGCCATGTATAATGTTCCTG-3′), and S125A (F, 5′-TTCTGTGGAGGCTGCAGAGAAGGAG-ATCGG-3′, R, 5′-CCTTCTCTGCAGCCTCCACAGAATCACTGCC-3′).
Purification of NDP kinase
Full-length His-tagged wild-type and mutant proteins were expressed in high yields using E. coli BL21-Gold (DE3) pLysS competent cells (Agilent Technologies, Palo Alto, CA) transformed with the appropriate construct using heat shock, as described by the manufacturer, and purified using batch/gravity-flow column purification with Talon IMAC resin (Clontech, Mountain View, CA) under native conditions (denaturing conditions, i.e., 4 M urea, were used throughout the purification process for the S120E mutant) following the manufacturer's instructions.
Cell culture and AMPKα/β/γ coimmunoprecipitation
For mammalian cell expression of AMPK, the AMPK subunits were used as previously described (Dyck et al., 1996). HEK293 cells were cultured in complete DMEM (Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) at 37°C in 5% CO2. For the transient expression of AMPK protein, the cells were plated 24 h before the experiments in 15-cm dishes and then transfected with the three plasmids using Lipofectamine 2000 (1 μg DNA per 2 μl; Invitrogen, Carlsbad, CA) following the manufacturer's protocols. Note: For some kinase assays, GST-AMPKα1 was replaced with WT or kinase-dead [KD] myc-AMPK (in a pCMV-myc vector; Clontech; Kazgan et al., 2010).
After 24 h, fresh medium containing CoCl2 (200 μM) was added to the cells for 1 h in the incubator to activate AMPK, as previously described (Lee et al., 2003). Cells were then harvested, lysed in 0.5 ml of lysis buffer A plus 1.0% Triton X-100 with shaking for 1 h (4°C), and centrifuged at 16,000 × g for 10 min (4°C). GST- and myc-tagged AMPK were purified from the supernatants via GST pull-down, using glutathione Sepharose 4B (Amersham, GE Healthcare), and immunoprecipitation performed, using anti–c-Myc antibody (9E10; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) at a 1:100 dilution for 1 mg/ml lysate and A/G agarose (Pierce Protein Research Products, Rockford, IL), respectively, according to the manufacturer's instructions and as previously described (Kazgan et al., 2010). Washed, bead-adsorbed GST-AMPK was used for NDPK assays, as previously described (Dyck et al., 1996), and both GST- and myc-tagged AMPK were used for kinase assays.
NDPK assays
A well-known procedure to assay NDP kinase activities was used (Timmons et al., 1993; Krishnan et al., 2001). In brief, 10 μl of diluted enzyme (lysate or purified NDPK; see further discussion) was added to a 990-ml reaction mixture containing 100 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 100 mM KCl, 0.4 mM NADH, 6 mM ATP, 0.7 mM TDP, 4 mM phosphoenolpyruvate (PEP), and 10 U of pyruvate kinase and lactate dehydrogenase each. The absorbance of NADH at 340 nm was then recorded. A unit of activity is defined as the amount required to convert 1 μmol of NADH to NAD+ in 1 min.
For NDPK assays with flies or fly larvae, 20 adult flies or 40–50 fly larvae were homogenized in 50 μl of ice-cold buffer (100 mM Tris-HCl, pH 7.5, 10 mM MgCl2, and 100 mM KCl). The lysates were centrifuged at 10,000 × g for 10 min (4°C). The clarified supernatant was then diluted 1:10 for inclusion in the described assay (0.5–2 μg of protein used). Purified NDPK was purchased from Sigma-Aldrich (N2635) and also produced as purified His-tagged versions and used at amounts of 10–50 and 50–200 ng, respectively, in NDPK assays. For the inhibition assays including AMPK, 3 μg of bead-adsorbed GST-AMPK was incubated with purified NDPK or purified His-tagged NDPK at room temperature for 2 h before executing the NDPK assays.
Kinase assays
Kinase assays were performed according to previously described methods (Davies et al., 1989). Briefly, AMPK activity assays with GST-AMPK were performed at room temperature (25°C) in a 25-μl reaction mixture containing 3–12 μg of protein in kinase buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.0, 75 mM NaCl, 5 mM sodium acetate, 5 mM magnesium chloride, 1 mM dithiothreitol, 8% glycerol, 0.1 mM EDTA, 200 μM AMP and ATP, and 2 μCi of [γ-32P]ATP) with or without the SAMS or NDPK peptide. After a 30-min incubation period, the reaction mixtures were counted in a scintillation counter. Kinase assays with myc-tagged KD-AMPK and myc-tagged WT-AMPK were performed in the same manner as described earlier, but 0.5 μg of protein was added to the reaction mixture. AMPK activity is expressed as picomoles of 32P incorporation into the peptide per minute per microgram of protein.
Western blotting
Fly protein lysates for immunoblotting were prepared by collecting equal numbers of male and female flies (50 total) of each genotype in a 1.5-ml microfuge tube. One milliliter of lysis buffer A was added to each sample. Flies were then ground to homogeneity, incubated for 1 h with shaking (4°C), and centrifuged at 16,000 × g for 10 min (4°C). Supernatants were collected, and protein concentrations were determined using the Bio-Rad DC protein assay (Richmond, CA). Note: The preparation of brain lysate samples was described earlier.
Proteins (50 μg) were then boiled in loading buffer and subjected to SDS–PAGE (Invitrogen), followed by Western analyses using 1:1000 dilutions of all primary antibodies, with the exception of anti–α tubulin (1:16,000). Secondary antibodies (IRDye infrared antibodies; LI-COR Biosciences, Lincoln, NE) were used at a dilution of 1:2000. Scanning, analyzing, and quantification of blots were performed via the Odyssey Infrared Imaging System (LI-COR Biosciences). Three or more independent experiments were performed for all immunoblotting data.
Fly stocks, crosses, rescue experiments, and lifespan measurements
Drosophila melanogaster strains obtained from the Bloomington Stock Center (Bloomington, IN) included the following: Act-GAL4, tub-GAL4, awdKRS6 (referred to as NDPK1 in this study), awdMSM95 (NDPK2), UAS-AMPKαRNAI, the SNF1A1 and SNF1A3 mutants, UAS-SNF1A, UAS-S6k.KQ, UAS-Tor.TED, and UAS-GFP. UAS-SNF4 and UAS-NDPK were gifts from D. Kretzschmar and T. Hsu, respectively. All flies were maintained at 25°C in yeast-cornmeal vials, and all crosses were also performed in cornmeal-yeasted vials.
For the AMPKα RNAi rescue experiments, males carrying a transgene or loss-of-function mutation on the second or third chromosome were mated to virgin females carrying a GAL4 (either tub-GAL4 or Act-GAL4, respectively). From these crosses, male progeny carrying the transgene or loss-of-function mutation and the GAL4 were mated to virgin females carrying UAS-AMPKαRNAI. The progeny from this second cross were then scored for rescue, that is, viable adult flies in spite of AMPKα RNAi knockdown.
For the AMPKα loss-of-function rescue experiments, males carrying a transgene or loss-of-function mutation on the second or third chromosome were mated in parallel to virgin females from both the SNF1A1 and SNF1A3 loss-of-function lines. The male progeny from both of these crosses were scored for rescue, that is, viable adult flies in spite of carrying the lethal loss-of-function mutation/phenotypically, non–bar-eyed males.
Measurements of lifespan have been widely used in Drosophila as a metric of stress sensitivity (Johnson et al., 2010). Thirty 3- to 5-d-old male flies were starved in empty food vials that contained pieces of filter paper saturated with deionized H2O. We assessed the percentage survival of at least three replicate vials three times daily.
Statistical analyses
Comparisons were made using the unpaired Student's t test with p < 0.05 considered significant. Values are presented as the mean ± the SE of the mean (SEM) and are represented as error bars. Indirect immunofluorescent detection of a secondary antibody (LI-COR Biosciences) was scanned and standardized to an internal standard to calculate and quantify arbitrary units using the Odyssey Infrared Imaging System, and a representative Western blot is shown in each figure.
Supplementary Material
Acknowledgments
We thank B. Viollet (Université Paris Descartes, Paris, France) and K. McCarthy (University of North Carolina–Chapel Hill) for AMPKα1−/−α2F/F-hGFAP-Cre mice, R. Shaw (Salk Institute, La Jolla, CA) for HA-tagged AMPKβ and AMPKγ constructs, L. Witters (Dartmouth Medical School, Hanover, NH) for the GST-tagged AMPKα construct, T. Hsu (Boston University, Boston, MA) for anti-dNDPK antibody and UAS-NDPK flies, M.-L. Lacombe (Université Pierre et Marie Curie, Paris, France) for the plasmid containing wild-type NDPK, and D. Kretzschmar (Oregon Health and Science University, Portland, OR) for UAS-AMPKγ flies. R.U.O. is supported through T32 National Cancer Institute Training Grant 5-T32-CA09156-35 to the University of North Carolina–Chapel Hill Lineberger Comprehensive Cancer Center. J.E.B. is funded by National Institutes of Health Grant R01NS063858 and through University of North Carolina–Chapel Hill University Funds.
Abbreviations used:
- AMPK
AMP-activated protein kinase
- hGFAP-Cre
human glial fibrillary acidic protein-Cre
- LKB1
liver kinase B1
- NDPK
nucleoside diphosphate kinase
- 2D-DIGE
two-dimensional differential in-gel electrophoresis
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-08-0699) on November 23, 2011.
The authors declare no conflict of interest.
REFERENCES
- Almaula N, Lu Q, Delgado J, Belkin S, Inouye M. Nucleoside diphosphate kinase from Escherichia coli. J Bacteriol. 1995;177:2524–2529. doi: 10.1128/jb.177.9.2524-2529.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annesley SJ, Bago R, Mehta A, Fisher PR. A genetic interaction between NDPK and AMPK in Dictyostelium discoideum that affects motility, growth and development. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:341–349. doi: 10.1007/s00210-011-0615-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK) J Cell Sci. 2002;115:2433–2442. doi: 10.1242/jcs.115.11.2433. [DOI] [PubMed] [Google Scholar]
- Biondi RM, Engel M, Sauane M, Welter C, Issinger OG, Jimenez de Asua L, Passeron S. Inhibition of nucleoside diphosphate kinase activity by in vitro phosphorylation by protein kinase CK2. Differential phosphorylation of NDP kinases in HeLa cells in culture. FEBS Lett. 1996;399:183–187. doi: 10.1016/s0014-5793(96)01299-9. [DOI] [PubMed] [Google Scholar]
- Bjorklund MA, Vaahtomeri K, Peltonen K, Viollet B, Makela TP, Band AM, Laiho M. Non-CDK-bound p27 (p27(NCDK)) is a marker for cell stress and is regulated through the Akt/PKB and AMPK-kinase pathways. Exp Cell Res. 2010;316:762–774. doi: 10.1016/j.yexcr.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Boissan M, Dabernat S, Peuchant E, Schlattner U, Lascu I, Lacombe ML. The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol Cell Biochem. 2009;329:51–62. doi: 10.1007/s11010-009-0120-7. [DOI] [PubMed] [Google Scholar]
- Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Zhu XX, Thoraval DH, Ungar D, Rawwas J, Hora N, Strahler JR, Hanash SM, Radany E. Nm23-H1 mutation in neuroblastoma. Nature. 1994;370:335–336. doi: 10.1038/370335a0. [DOI] [PubMed] [Google Scholar]
- Chartier NT, Salazar Ospina DP, Benkemoun L, Mayer M, Grill SW, Maddox AS, Labbe JC. PAR-4/LKB1 mobilizes nonmuscle myosin through anillin to regulate C. elegans embryonic polarization and cytokinesis. Curr Biol. 2011;21:259–269. doi: 10.1016/j.cub.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem. 2004;279:15719–15722. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- Crawford RM, Treharne KJ, Arnaud-Dabernat S, Daniel JY, Foretz M, Viollet B, Mehta A. Understanding the molecular basis of the interaction between NDPK-A and AMPK alpha 1. Mol Cell Biol. 2006a;26:5921–5931. doi: 10.1128/MCB.00315-06. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Crawford RM, Treharne KJ, Arnaud-Dabernat S, Daniel JY, Foretz M, Viollet B, Mehta A. Protein kinase CK2 acts as a signal molecule switching between the NDPK-A/AMPK 1 complex and NDPK-B. FASEB J. 2007;21:3398. [PubMed] [Google Scholar]
- Crawford RM, Treharne KJ, Best OG, Muimo R, Riemen CE, Mehta A. A novel physical and functional association between nucleoside diphosphate kinase A and AMP-activated protein kinase alpha1 in liver and lung. Biochem J. 2005;392:201–209. doi: 10.1042/BJ20050269. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Crawford RM, Treharne KJ, Best OG, Riemen CE, Muimo R, Gruenert DC, Arnaud-Dabernat S, Daniel JY, Mehta A. NDPK-A (but not NDPK-B) and AMPK alpha1 (but not AMPK alpha2) bind the cystic fibrosis transmembrane conductance regulator in epithelial cell membranes. Cell Signal. 2006b;18:1595–1603. doi: 10.1016/j.cellsig.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Dammai V, Adryan B, Lavenburg KR, Hsu T. Drosophila awd, the homolog of human nm23, regulates FGF receptor levels and functions synergistically with shi/dynamin during tracheal development. Genes Dev. 2003;17:2812–2824. doi: 10.1101/gad.1096903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Carling D, Hardie DG. Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem. 1989;186:123–128. doi: 10.1111/j.1432-1033.1989.tb15185.x. [DOI] [PubMed] [Google Scholar]
- Davies SP, Hawley SA, Woods A, Carling D, Haystead TA, Hardie DG. Purification of the AMP-activated protein kinase on ATP-gamma-Sepharose and analysis of its subunit structure. Eur J Biochem. 1994;223:351–357. doi: 10.1111/j.1432-1033.1994.tb19001.x. [DOI] [PubMed] [Google Scholar]
- Dyck JR, Gao G, Widmer J, Stapleton D, Fernandez CS, Kemp BE, Witters LA. Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic beta and gamma subunits. J Biol Chem. 1996;271:17798–17803. doi: 10.1074/jbc.271.30.17798. [DOI] [PubMed] [Google Scholar]
- Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel M, Veron M, Theisinger B, Lacombe ML, Seib T, Dooley S, Welter C. A novel serine/threonine-specific protein phosphotransferase activity of Nm23/nucleoside-diphosphate kinase. Eur J Biochem. 1995;234:200–207. doi: 10.1111/j.1432-1033.1995.200_c.x. [DOI] [PubMed] [Google Scholar]
- Freije JM, Blay P, MacDonald NJ, Manrow RE, Steeg PS. Site-directed mutation of Nm23-H1. Mutations lacking motility suppressive capacity upon transfection are deficient in histidine-dependent protein phosphotransferase pathways in vitro. J Biol Chem. 1997;272:5525–5532. doi: 10.1074/jbc.272.9.5525. [DOI] [PubMed] [Google Scholar]
- Gao G, Fernandez CS, Stapleton D, Auster AS, Widmer J, Dyck JR, Kemp BE, Witters LA. Non-catalytic beta- and gamma-subunit isoforms of the 5′-AMP-activated protein kinase. J Biol Chem. 1996;271:8675–8681. doi: 10.1074/jbc.271.15.8675. [DOI] [PubMed] [Google Scholar]
- Giraud MF, Georgescauld F, Lascu I, Dautant A. Crystal structures of S120G mutant and wild type of human nucleoside diphosphate kinase A in complex with ADP. J Bioenerg Biomembr. 2006;38:261–264. doi: 10.1007/s10863-006-9043-0. [DOI] [PubMed] [Google Scholar]
- Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D, Tamburini J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: From metabolism to cancer cell biology. Cell Cycle. 2011;10:2115–2120. doi: 10.4161/cc.10.13.16244. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. The AMP-activated protein kinase pathway—new players upstream and downstream. J Cell Sci. 2004;117:5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase—development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Holmes B, Dohm GL. Regulation of GLUT4 gene expression during exercise. Med Sci Sports Exerc. 2004;36:1202–1206. doi: 10.1249/01.mss.0000132385.34889.fe. [DOI] [PubMed] [Google Scholar]
- Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Johnson EC, Kazgan N, Bretz CA, Forsberg LJ, Hector CE, Worthen RJ, Onyenwoke R, Brenman JE. Altered metabolism and persistent starvation behaviors caused by reduced AMPK function in Drosophila. PLoS One. 2010;5(pii):e12799. doi: 10.1371/journal.pone.0012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kazgan N, Williams T, Forsberg LJ, Brenman JE. Identification of a nuclear export signal in the catalytic subunit of AMP-activated protein kinase. Mol Biol Cell. 2010;21:3433–3442. doi: 10.1091/mbc.E10-04-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KS, Rikhy R, Rao S, Shivalkar M, Mosko M, Narayanan R, Etter P, Estes PS, Ramaswami M. Nucleoside diphosphate kinase, a source of GTP, is required for dynamin-dependent synaptic vesicle recycling. Neuron. 2001;30:197–210. doi: 10.1016/s0896-6273(01)00273-2. [DOI] [PubMed] [Google Scholar]
- Lamia KA, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278:39653–39661. doi: 10.1074/jbc.M306104200. [DOI] [PubMed] [Google Scholar]
- Liang J, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- MacDonald NJ, De la Rosa A, Benedict MA, Freije JM, Krutsch H, Steeg PS. A serine phosphorylation of Nm23, and not its nucleoside diphosphate kinase activity, correlates with suppression of tumor metastatic potential. J Biol Chem. 1993;268:25780–25789. [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE. LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J Cell Biol. 2007;177:387–392. doi: 10.1083/jcb.200702053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem. 1994;269:2361–2364. [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Nallamothu G, Woolworth JA, Dammai V, Hsu T. Awd, the homolog of metastasis suppressor gene Nm23, regulates Drosophila epithelial cell invasion. Mol Cell Biol. 2008;28:1964–1973. doi: 10.1128/MCB.01743-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JN, Mustard KJ, Graham DA, Yu H, MacDonald CS, Pilegaard H, Goodyear LJ, Hardie DG, Richter EA, Wojtaszewski JF. 5′-AMP-activated protein kinase activity and subunit expression in exercise-trained human skeletal muscle. J Appl Physiol. 2003;94:631–641. doi: 10.1152/japplphysiol.00642.2002. [DOI] [PubMed] [Google Scholar]
- Osorio C, Sullivan PM, He DN, Mace BE, Ervin JF, Strittmatter WJ, Alzate O. Mortalin is regulated by APOE in hippocampus of AD patients and by human APOE in TR mice. Neurobiol Aging. 2007;28:1853–1862. doi: 10.1016/j.neurobiolaging.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Pinaud R, Osorio C, Alzate O, Jarvis ED. Profiling of experience-regulated proteins in the songbird auditory forebrain using quantitative proteomics. Eur J Neurosci. 2008;27:1409–1422. doi: 10.1111/j.1460-9568.2008.06102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. Role of mTOR signalling in the control of translation initiation and elongation by nutrients. Curr Top Microbiol Immunol. 2004;279:215–244. doi: 10.1007/978-3-642-18930-2_13. [DOI] [PubMed] [Google Scholar]
- Randazzo PA, Northup JK, Kahn RA. Activation of a small GTP-binding protein by nucleoside diphosphate kinase. Science. 1991;254:850–853. doi: 10.1126/science.1658935. [DOI] [PubMed] [Google Scholar]
- Rosengard AM, Krutzsch HC, Shearn A, Biggs JR, Barker E, Margulies IM, King CR, Liotta LA, Steeg PS. Reduced Nm23/Awd protein in tumour metastasis and aberrant Drosophila development. Nature. 1989;342:177–180. doi: 10.1038/342177a0. [DOI] [PubMed] [Google Scholar]
- Rubenstein EM, McCartney RR, Zhang C, Shokat KM, Shirra MK, Arndt KM, Schmidt MC. Access denied: Snf1 activation loop phosphorylation is controlled by availability of the phosphorylated threonine 210 to the PP1 phosphatase. J Biol Chem. 2008;283:222–230. doi: 10.1074/jbc.M707957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton D, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- Tepper AD, Dammann H, Bominaar AA, Veron M. Investigation of the active site and the conformational stability of nucleoside diphosphate kinase by site-directed mutagenesis. J Biol Chem. 1994;269:32175–32180. [PubMed] [Google Scholar]
- Timmons L, Hersperger E, Woodhouse E, Xu J, Liu LZ, Shearn A. The expression of the Drosophila awd gene during normal development and in neoplastic brain tumors caused by lgl mutations. Dev Biol. 1993;158:364–379. doi: 10.1006/dbio.1993.1195. [DOI] [PubMed] [Google Scholar]
- Timmons L, Xu J, Hersperger G, Deng XF, Shearn A. Point mutations in awdKpn which revert the prune/Killer of prune lethal interaction affect conserved residues that are involved in nucleoside diphosphate kinase substrate binding and catalysis. J Biol Chem. 1995;270:23021–23030. doi: 10.1074/jbc.270.39.23021. [DOI] [PubMed] [Google Scholar]
- Treharne KJ, Best OG, Mehta A. The phosphorylation status of membrane-bound nucleoside diphosphate kinase in epithelia and the role of AMP. Mol Cell Biochem. 2009;329:107–114. doi: 10.1007/s11010-009-0118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venerando A, Pagano MA, Tosoni K, Meggio F, Cassidy D, Stobbart M, Pinna LA, Mehta A. Understanding protein kinase CK2 mis-regulation upon F508del CFTR expression. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:473–488. doi: 10.1007/s00210-011-0650-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JL, Morton DG, Bestman J, Kemphues KJ. The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development. 2000;127:1467–1475. doi: 10.1242/dev.127.7.1467. [DOI] [PubMed] [Google Scholar]
- Williams T, Brenman JE. LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 2008;18:193–198. doi: 10.1016/j.tcb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Williams T, Courchet J, Viollet B, Brenman JE, Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci USA. 2011;108:5849–5854. doi: 10.1073/pnas.1013660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW, Wilson HA, Hardie DG, Rasmussen BB, Hutber CA, Call GB, Clayton RD, Conley LM, Yoon S, Zhou B. Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J Appl Physiol. 1997;82:219–225. doi: 10.1152/jappl.1997.82.1.219. [DOI] [PubMed] [Google Scholar]
- Zhai B, Villen J, Beausoleil SA, Mintseris J, Gygi SP. Phosphoproteome analysis of Drosophila melanogaster embryos. J Proteome Res. 2008;7:1675–1682. doi: 10.1021/pr700696a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.