

Abstract
Peptide natural products exhibit broad biological properties and are commonly produced by orthogonal ribosomal and nonribosomal pathways in prokaryotes and eukaryotes. To harvest this large and diverse resource of bioactive molecules, we introduce Natural Product Peptidogenomics (NPP), a new mass spectrometry-guided genome mining method that connects the chemotypes of peptide natural products to their biosynthetic gene clusters by iteratively matching de novo MSn structures to genomics-based structures following current biosynthetic logic. In this study we demonstrate that NPP enabled the rapid characterization of >10 chemically diverse ribosomal and nonribosomal peptide natural products of novel composition from streptomycete bacteria as a proof of concept to begin automating the genome mining process. We show the identification of lantipeptides, lasso peptides, linardins, formylated peptides and lipopeptides, many of which from well-characterized model streptomycetes, highlighting the power of NPP in the discovery of new peptide natural products from even intensely studied organisms.
INTRODUCTION
Peptide natural products (PNPs) are ubiquitous chemicals found in all life forms where they exhibit diverse biological functions in development, protection, and communication1. Nature has evolved two orthogonal biosynthetic pathways to these highly modified peptides involving ribosomal and nonribosomal processes2. While nonribosomal peptides have limited distribution restricted mainly to microorganisms with large genomes3, ribosomally synthesized and posttranslationally modified peptides appear to have a much broader distribution throughout nature to also include humans4,5. The enormous diversity and distribution of PNPs and their associated biological functions, however, are only now being fully realized due to time-consuming discovery options. We report here a new mass spectrometry-guided genome mining method that quickly connects the chemotypes of expressed PNPs to their biosynthetic pathways, thereby enabling the rapid identification of transcriptionally active PNP biosynthetic gene clusters and the classification of their associated products in a streamlined discovery platform.
Among PNPs, ribosomally synthesized peptides encompass a rapidly expanding group of natural products6. Multiple classes of ribosomal peptide natural products (RNPs) of prokaryotic origin have been characterized via their biosynthetic pathways that entail diverse post-translational modification strategies to yield lantipeptides7, thiopeptides8, cyanobactins9, lasso peptides10, and other microcins11. Consequently, traditional RNP classification systems based on bioactivity, producer and structure11,12 have shifted towards a new classification based largely on biosynthesis (Supplementary Results, Supplementary Table 1). In RNP biosynthesis, the peptide sequence is encoded by a precursor gene directly translated by the ribosome to consist of leader peptide and core peptide regions13. The leader peptide serves as scaffold and recognition sites for processing enzymes that introduce post-translational modifications of the RNP biosynthetic machinery, while the core peptide constitutes the primary sequence of the produced peptide natural product that is modified. Post-translational modification of the core peptide by biosynthetic enzymes can often be extensive to provide a wealth of structural diversity rendering them, at first glance, unrecognizable as ribosomally synthesized molecular entities6 (Fig. 1). Nonribosomal peptides are conversely synthesized by multifunctional assembly line proteins that rather code for their amino acid precursors by an adenylating enzyme that selects and transfers its substrates to carrier proteins to facilitate peptide synthesis by the nonribosomal peptide synthetase (NRPS) machinery14. This process can capture a much wider array of substrates beyond the 20 proteinogenic amino acid building blocks that limit input into RNPs to yield notable examples such as the clinical agents vancomycin, daptomycin and cyclosporin2 (Fig. 1).
Figure 1.

Structural diversity of peptide natural products.
To estimate the extent of PNP chemical diversity in bacteria, we systematically queried the JGI database of 1035 completed genomes as of September 2010 for RNP and NRPS pathways. Searching for gene clusters harboring characteristic RNP biosynthetic Pfam (Protein family) domains15, we estimate that at least 71% of deposited bacterial genomes contain biosynthetic features that support common RNP classes (Supplementary Table 3). We identified 1966 candidate RNP gene clusters, 637 of which having two or more of the nine Pfam domains found most frequently in RNP gene clusters (Supplementary Table 2). In comparison, 69% of genomes we searched contained NRPS Pfam domains, while 53% had hybrid NRPS/PKS biosynthetic features (Supplementary Table 3). Since the training set for our algorithm contained only 24 known RNP gene clusters, the estimate of RNPs is not comprehensive. Nonetheless, this analysis clearly shows that the genetic capacity to produce RNPs is common in most microbial phyla and that RNPs represent one of the most underappreciated classes of bioactive molecules.
Given the sheer volume of predicted bacterial PNPs in publicly available genome strains, we set out to develop a method taking advantage of recent technological advances in mass spectrometry and genomics to streamline the discovery process. The recent development of genome mining has transformed natural product discovery by allowing for the targeting of new chemical entities predicted by bioinformatics16. In the case of RNPs, a produced peptide structure can be directly linked to the corresponding biosynthetic genes by identifying the core peptide sequence in the translated genome sequence4. Furthermore, large portions of NRPs often readily correlate to their predicted amino acid specificity found on their associated modular synthetases17 (Supplementary Fig. 1). This connection of PNP chemo- and genotype has been accomplished in numerous genome mining studies8–10,17–20. One of the major limitations with these approaches is that they only characterize one molecule at a time or require extensive genetic manipulations21. With an increasing number of available genome sequences, there is a growing need for new genome mining methods that can readily connect expressed natural products (chemotype) with their gene clusters (genotype) with the potential for automation.
Mass spectrometry is an important technique in the analysis of peptide natural products due to its high sensitivity, its easy implementation into automated processes such as metabolomic or proteomic platforms and its capability for de novo peptide structure elucidation by tandem MS (MSn)22. Peptides fragment in MSn experiments, e.g. collision-induced dissociation (CID), in a common way to yield fragment ions in the MSn spectrum that differ in mass by the amino acid monomers of the corresponding peptide sequence and, thus, enable de novo peptide sequencing. MSn is used in proteomic workflows to identify proteins by connecting peptide MSn data to protein sequence databases. Herein, one approach to link a proteolytic peptide to its database gene uses short de novo sequence tags for the database search23. However, automated de novo sequencing makes errors in one in every four amino acids, and this error rate is enhanced when PTMs are included. In addition, database proteomic tools still struggle to connect modified RNPs with their precursor genes in genomic databases due to scoring functions, which are limited in recognizing many PTMs per peptide24 and which allow for a specific percentage of false positives rates (FDR) without further confirmation of a spectrum-peptide match (Supplementary Table 4). Finally, there are no tools that connect MSn data of nonribosomally synthesized peptides to the corresponding NRPS genes. Given the advantage of mass spectrometry to automatically acquire data of partial peptide structures from small amounts of material, mass spectrometry could enable a more rapid connection of peptidic natural products with their biosynthetic genes if MSn data processing is effectively combined with genome mining of RNP and NRP biosynthetic pathways.
In this study, we establish the concept of MS-guided genome mining for peptide natural products, called Natural Product Peptidogenomics (NPP). We first highlight proof of concept experiments in which NPP characterizes the ribosomal lantipeptide AmfS from Streptomyces griseus IFO 13350 and the nonribosomal lipopeptide stendomycin I from Streptomyces hygroscopicus ATCC 53653 and their corresponding biosynthetic gene clusters. In all, we show that NPP can be applied to characterize many PNP chemo- and genotypes by introducing 14 new streptomycete PNPs in a very effective genome mining approach with the potential for automation.
RESULTS
The Natural Product Peptidogenomics concept
Natural Product Peptidogenomics (NPP) is an easy to implement and unbiased, mass spectrometry-based, chemotype-to-genotype genome mining approach to rapidly characterize ribosomal and nonribosomal peptide natural products and their biosynthetic gene clusters from sequenced organisms (Fig. 2). In short, Natural Product Peptidogenomics aims to match a series of mass shifts obtained from an MSn spectrum of a putative PNP to the genes that are responsible for its production. The NPP genome mining workflow has several iteration steps, which ensure that a match of peptide MSn data to a genomics-derived peptide structure makes sense biosynthetically. Herein, NPP takes advantage of the enormous wealth of knowledge of PNP biosynthesis gained over the past decade2. In practice, the NPP workflow starts with MALDI-TOF MS analysis of the organism or extract in order to detect unknown masses. We targeted the mass range of 1500–5000 Da as most masses in this window are not described from microbes and thus provided an opportunity to apply the NPP approach. However, there is no inherent limitation in size as long as the MSn data becomes a unique identifier for a biosynthetic pathway. MALDI-TOF MS analysis of crude butanol extracts or MALDI-imaging of agar cultures ensure that the compounds are actively expressed and captured on semi-solid media. Though not necessary for the PNP discovery process, MALDI-imaging links secreted metabolites directly to the morphology of microbial colonies25 and, thus, decreases potential media or extraction artifacts. Putative peptides are subsequently enriched using a MS-guided isolation via size-exclusion chromatography followed by enrichment and desalting steps. An enriched putative PNP is then analyzed in a sequence tagging step by MSn. In general, NPP sequence tagging is the formation of de novo sequence tags that are searchable in genome mining query space of PNPs (Fig. 2). This includes the generation of an amino acid sequence tag from a mass shift sequence in a MSn spectrum and subsequent processing of the MSn sequence tag into search tags. Herein, the mass shifts define the candidate amino acid residues from all possible monomers that could be encoded in a RNP-based precursor gene or that could be loaded by a corresponding NRPS. This processing of MSn mass shifts to genome mining monomers considers PTMs, nonribosomal substrates, fragmentation gas-phase behavior, and chemical modifications of amino acid residues during purification and MS analysis. NPP-based RNP genome mining interrogates the 6-frame translation of the genome for candidate precursor peptides that comprise any of the search tags. As there may be multiple matches to a 5–10 aa-long search tag, the correct RNP precursor gene is identified by applied biosynthetic knowledge in which the search tag should associate with the C-terminal half of a <100 aa-long ORF that clusters with RNP biosynthetic genes. NPP-based NRP genome mining, on the other hand, queries all predicted nonribosomal peptides of the target genome for the search tags. The effectiveness of NPP in connecting PNP structures with biosynthetic genes is its iterative approach in matching MSn-based structures to genomics-based candidate structures following biosynthetic logic as each search tag match has to be confirmed in mass, sequence and biosynthetic signatures with the MSn analysis (Fig. 2). This effectiveness was shown in a comparison of the NPP approach to current proteomic approaches in identifying precursor genes in RNP genome mining. None of the standard proteomic platforms such as Mascot26 or InsPecT23 could identify any of the NPP-characterized RNPs in a search with variable common RNP post-translational modifications (PTMs) or blind/unrestricted searches designed to find unknown PTMs (Supplementary Table 4). InsPecT was able to characterize two of the RNPs after predefining NPP-dissected PTMs in the analysis for each peptide.
Figure 2. General workflow of Natural Product Peptidogenomics (NPP).

NPP can be applied to characterize both ribosomal and nonribosomal peptide natural products in their genotype and chemotype from genome-sequenced organisms. Two proof of concept NPP experiments are outlined: Ribosomal peptides (RNPs) or nonribosomal peptides (NRPs) and their respective biosynthetic gene cluster can be characterized from a Streptomyces extract by MALDI-TOF MS detection, MSn sequence tagging and PNP genome mining. The iterative approach in matching MSn data to genomics-derived peptide structures is shown by dashed arrows. See Figures 3 and 4, respectively, for detailed NPP analysis of ribosomal and nonribosomal peptides.
NPP characterization of ribosomal peptide AmfS
As a proof of concept of the NPP workflow for RNPs, we targeted the known ribosomal peptide AmfS from Streptomyces griseus IFO 13350 because this is a well characterized lantipeptide with 4 post-translational modifications27 (Fig. 3). MALDI-imaging of S. griseus and MALDI-TOF MS analysis of an extract resulted in the detection of a secreted mass of 2212 Da. The peptide was subjected to CID fragmentation. In the MS2 spectrum, charge states of sequential fragment ions were assigned and the mass shift sequence 99-99-113-69-101 was identified (Fig. 3). The mass shifts were matched to all likely candidate amino acids to yield sequence tags by first substituting with proteinogenic amino acids where possible (Supplementary Table 5). Nonproteinogenic masses were next substituted with all possible RNP monomers arising from known PTMs. The shift of 69 Da was substituted to the nonproteinogenic amino acid dehydroalanine (Dha, Fig. 3, Supplementary Table 6). Dha is a candidate amino acid for ribosomal peptides because dehydrated Ser and Thr or dethiolated Cys are commonly observed in PNP MSn spectra either as a post-translational modification28 or an MSn gas phase rearrangement (Supplementary Fig. 3). From the resulting sequence tag VVI(L)S(C)T, a list of all possible search tags in both sequence directions was created to give eight putative PNP sequence tags for search against the S. griseus genome sequence (Fig. 3)29. Of the millions of possible peptide sequences based on a 6-frame translation, just one candidate 43 aa-long precursor peptide was identified by the search tag VVLCT (Fig. 3). This result fulfilled the RNP biosynthetic requirement of the search tag location in the C-terminal half of a <100 aa-long gene product. Next, a predicted core peptide sequence was compared in its calculated mass to the observed mass in the MS1 spectrum considering putative PTMs such as e.g. dehydrations. The calculated mass of the 22 aa-long core peptide 22T-43P differed by 4 putative PTM dehydrations from the observed mass (Fig. 3) in agreement with the formation of 2 Dha and 2 lanthionine bridges in AmfS30. In addition, the predicted core peptide sequence could be further verified at this step by comparison to the MSn data. Subsequent BLAST analysis of the neighboring genes identified the remainder of the AmfS biosynthetic gene cluster27 and, thereby, further verified the connection of RNP chemotype and genotype. Based on the gene cluster components, in particular the AmfS core peptide and the PTM introducing enzymes AmfA and AmfB, the analyzed peptide could be characterized as a class III lantipeptide from known RNP biosynthetic gene clusters (Supplementary Table 1). Finally, an AmfS structure could be verified based on the given core peptide sequence, the MSn data and the knowledge about AmfS-like lantipeptide PTMs30 (Fig. 3, Supplementary Fig. 2, Supplementary Table 1). The proof of concept characterization of AmfS and its gene cluster highlights the effectiveness of the NPP workflow by its iterative utilization of MSn data and genetic data to enable a peptidogenomic connection of a PNP chemotype with its genotype.
Figure 3. Peptidogenomic connection of a ribosomal peptide (RNP) chemotype with its biosynthetic genes (genotype) via sequence tagging and genome mining – Characterization of class III lantipeptide AmfS from Streptomyces griseus IFO 13350.

Iterative aspects in connecting MS data of the peptide chemotype to the genotype are highlighted in blue and in dashed arrows. Steps are as follows: (A) Detection of putative peptide mass signals by MALDI-TOF MS or Imaging MS, (B) determination of molecular weight, (C) MSn fragmentation (CID), (D) assignment of charge states, (E) identification of mass shifts, (F) substitution of proteinogenic mass shifts (Supplementary Table 5), (G) substitution of nonproteinogenic mass shifts with putative RNP monomers (Supplementary Table 6), (H) MSn sequence tag processing of putative biosynthetic or MS gas-phase modifications, (I) MSn sequence tag processing of sequence tag direction, (J) search in 6-frame translation of target genome, (K) identification of candidate precursor peptide via RNP biosynthetic rationale, (L) verification of precursor peptide sequence, (M) prediction of core peptide sequence based on observed mass and putative PTM mass shifts, (N) verification of core peptide sequence and mass, (O) prediction of biosynthetic gene cluster, (P) verification of putative PTMs, (Q) RNP classification, (R) structure prediction based on RNP class and MSn data, and (S) structure verification by MSn data.
NPP characterization of nonribosomal lipopeptides
With a minor adjustment to the NPP workflow, we can also discover NRPs (Fig. 4). This approach was exemplified with a set of lipopeptides detected by MALDI-imaging from a colony of S. hygroscopicus ATCC 53653 (Fig. 4). MSn analysis of SHY-1628 yielded the sequence fragment 99-99-83-83-71-113-99-57-115 (Fig. 4, Supplementary Fig. 4b) that was first processed into the RNP workflow since most of the mass shifts corresponded to proteinogenic amino acids via the sequence tag VV-83-83-A-I(L)-V-G. The masses of 83 Da were substituted with dehydrobutyrine (Dhb) whose biosynthetic precursor is Thr (Supplementary Table 6). Although we queried the 6-frame translation of S. hygroscopicus with the search tags VVTTAI(L)VG, no precursor peptides were detected based on the described biosynthetic requirements of RNPs. The inability to identify a precursor peptide from a long sequence tag suggested that the SHY-1628-based peptides could instead be a set of nonribosomal peptides. To explore this scenario, the original sequence tag was revised to include NRP-specific, nonproteinogenic residues for NRP genome mining (Supplementary Table 7). Hence, the 83 Da mass shifts could correspond to Dhb, NMe-Dha or homoserine lactone (HseL). HseL was excluded because of its common C-terminal NRP location. Dhb and NMe-Dha most likely derived biosynthetically from Thr and Ser, respectively, during or after NRP assembly due to enamine instability of putative Dha/Dhb monomers31. In addition, a Dhb mass shift could also derive from MS gas-phase induced ring-opening elimination of a Thr-macrolactone bond with the NRP C-terminus32. The VVT(S)T(S)AI(L)VG sequence tags (Fig. 4) were evaluated against NRP sequences predicted by NP.searcher33 and antiSMASH34, algorithms that predict NRPS gene clusters and their NRP products from the genome supercontig. This analysis matched the reduced and full 8-aa sequence tag to one candidate NRP sequence out of the five predicted NRPS sequences in the S. hygroscopicus genome (Fig. 4, Supplementary Fig. 5). Again through an iterative process, the corresponding gene cluster was inspected to contain an N-terminal acyl ligase domain associated with lipopeptide biosynthesis in full agreement with the observed 14 Da separation of the parent ions characteristic of lipopeptides. Further MSn (Supplementary Fig. 7b and Supplementary Tables 8–12) and NMR analysis (Supplementary Fig. 6 and Supplementary Table 13) identified the lipopeptides as members of the stendomycin antibiotic family of lipo-tetradecapeptides that contain a 7-membered macrolactone and a total of seven modifications35. Aside from stendomycin I, which was originally characterized from S. endus35, five new stendomycin analogs (II–VI) that differed in the acyl chain and in Val/Ile substitutions at positions 5 and 13 were characterized for the first time in S. hygroscopicus ATCC 53653. The biosynthetic features of the identified gene cluster matched the structure of stendomycin I in predicted NRPS substrates and modifications (Supplementary Fig. 5 and 8). Thus, as for RNPs, the iteration between MSn analysis and genome mining enabled the fast and reliable connection of a NRP chemo- and genotype (Fig. 4). For example, a low-resolution mass shift of 115 Da was detected in the MS2 spectrum of stendomycin I (Supplementary Fig. 4b) that was first assigned to Asp. However, the corresponding module of the putative stendomycin NRPS instead predicts N-Me-Thr (also 115 Da) at this position and, thus, the mass shift in the MSn spectrum could be explained. This example illustrates that in NRP sequence tagging, modifications such as N-methylations of proteinogenic masses and even nonproteinogenic masses should be considered if the first iterative round of NRP genome mining misses the assignment of the tag. We also successfully applied the NPP method to other NRPS-derived molecules such as the structurally diverse calcium dependent antibiotic36, surfactin37, plipastatin37, pyoverdine38, and daptomycin36 and in each case we identified the correct gene cluster (data not shown). Recently, the NPP workflow enabled the discovery of the arylomycin gene cluster with a sequence tag of just two amino acids39. This highlights the point that with NRPS-derived molecules, minimal sequence information can be sufficient to find a match in a NRP database of <10 predicted NRP sequences per genome despite >526 known NRP monomers40 because of the iterative nature of using biosynthetic knowledge in the workflow. To complete the structure analysis, additional analytical methods such as NMR and Marfey's analysis are needed to complement the wealth of tandem MS and biosynthetic information as done with the stendomycins. The characterization of five stendomycin derivatives and their biosynthetic gene cluster in S. hygroscopicus demonstrates that the NPP workflow can be readily accommodated to additionally discover modified NRPs.
Figure 4. Peptidogenomic connection of a nonribosomal peptide (NRP) chemotype with its biosynthetic genes (genotype) via sequence tagging and genome mining – Characterization of the lipopeptide stendomycin complex from Streptomyces hygroscopicus ATCC 53653.

Iterative aspects in connecting MSn data of the peptide chemotype to the genotype are highlighted in blue and in dashed arrows. Steps are as follows: (A) Detection of putative peptide mass signals by MALDI-TOF MS or Imaging-MS, (B) determination of molecular weight, (C) MSn fragmentation (CID), (D) Assignment of charge states, (E) identification of mass shifts, (F) substitution of proteinogenic mass shifts (Supplementary Table 5), (G) substitution of nonproteinogenic mass shifts with putative NRP monomers (Supplementary Table 7), (H) MSn sequence tag processing of putative biosynthetic and MS gas-phase modifications, (I) MSn sequence tag processing of sequence tag direction, (J) search predicted NRP sequences from target genome with all search tags (NP.searcher/antiSMASH), (K) biosynthetic gene cluster analysis, (L) verification of predicted NRPS assembly-line, (M) NRP structure prediction, (N) verification of predicted NRP structure, (O) full structure elucidation based on MSn and NMR data, and (P) verification of NRP structure. Abbreviations: Dha – dehydroalanine; Dhb – dehydrobutyrine; HseL – homoserine lactone; Orn – ornithine.
NPP characterization of new RNP chemo- and genotypes
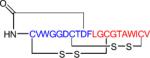
Next, we set out to interrogate several sequenced streptomycetes to explore the practicality of NPP to identify other uncharacterized RNPs. From eight Streptomyces strains, multiple previously uncharacterized RNPs and their gene clusters were identified by the NPP approach (Table 1). The first unknown RNP and its gene cluster that was characterized by NPP was a class I lasso peptide, SSV-2083, from S. sviceus ATCC 20983 (Table 1, Supplementary Fig. 9). The discovery and isolation of secreted SSV-2083 from sporulating colonies was guided by MALDI-imaging and MALDI-TOF MS of the ion at 2084 m/z. As MSn analysis of the unmodified compound provided no sequence information (Supplementary Fig. 9b). One of the main experimental challenges in the generation of the sequence tag is that many of these molecules are constrained by disulfide or thioether linkages, thereby providing poor to no fragmentation data (Supplementary Fig. 10). In such cases, samples are reductively dethiolated with NaBH4/NiCl2-treatment41 and re-subjected to tandem MS to reveal longer sequence tags for PNP genome mining. Deconstrained SSV-2083 yielded a 10-aa MSn sequence tag that we identified in the 6-frame translation of the S. sviceus genome in a 56-aa candidate precursor peptide. This observation enabled the identification of the SSV-2083 biosynthetic gene cluster containing conserved lasso peptide biosynthetic genes as well as a novel protein disulfide isomerase-encoding gene (Supplementary Fig. 9c). Alignment with known class I lasso peptides in combination with tandem MS data (Supplementary Fig. 9d) enabled the prediction of the SSV-2083 structure (Table 1) and represents the first class I lasso peptide gene cluster42.
Table 1.
NPP characterization of nine new RNPs and associated gene clusters from seven genome-sequenced Streptomyces strains. Summary of diverse RNP chemo- and genotypes characterized by NPP in this study. For detailed analyses, see Supplementary results.
| Observed PNP | Class | Chemotype | Genotype |
|---|---|---|---|
| SSV-2083 | Class I lasso peptide |
|
|
| SRO15-2005 | Class II lasso peptide |
|
|
| SRO15-2212 | Class I lantipeptide |
|
|
| SAL-2242 | Class III lantipeptide |
|
|
| SRO15-3108 | Class II lantipeptide |
|
|
| SGR-1832 | Linaridin |
|
|
| SLI-2138 | N-formylated peptide |
|
|
| SCO-2138 | N-formylated peptide |
|
|
| SWA-2138 | N-formylated peptide |
|
|
 Precursor peptide
Precursor peptide
 Post-translationally modifying enzyme
Post-translationally modifying enzyme
 Protease
Protease
 Transporter
Transporter
 MreB
MreB
 Regulator
Regulator
NPP characterization of new RNP classes from Streptomyces
NPP also resulted in the discovery of two new RNP classes and their genetic origins from well scrutinized streptomycetes, namely SGR-1832 (Table 1, Supplementary Fig. 11) from S. griseus IFO 13350 and SCO-2138 (Table 1, Supplementary Fig. 12) from S. coelicolor A3(2)43. Based on the gene cluster and the MS fragmentation data, SGR-1832 was determined to be a linear 19-residue peptide with an N-terminal N,N-dimethylalanine, two dehydrobutyrines, and a rare C-terminal aminovinylcysteine (AviCys) residue. These unusual post-translational modifications are reminiscent of cypemycin, a related AviCys-containing linaridin from Streptomyces sp. OH-4156 whose biosynthesis was recently illuminated by genome mining18. Peptide SCO-2138, detected only in organic extracts, is also a previously unidentified 19-aa RNP from S. coelicolor A3(2) that produces a number of other peptide natural products44. The corresponding gene neighborhood containing a conserved unknown protein, a protease and a rod-shape determining protein45 is also found in other Streptomyces genomes (Supplementary Fig. 12c and d). Indeed, two SCO-2138 homologs were isolated and characterized by NPP from S. lividans TK24 (SLI-2138 – identical to SCO-2138, Table 1, Supplementary Fig. 12) and S. sp. E14 (SWA-2138 – isomeric to SCO-2138; Table 1, Supplementary Fig. 12). These RNPs have a 28 Da N-terminal modification, which we confirmed by FTMSn to be an N-formyl unit (Supplementary Fig. 12e). The SGR-1832 and SCO-2138 peptides represented undiscovered classes of RNPs at the time of this analysis and showcase that new RNP classes can be discovered by the NPP method.
Characterization of multiple PNPs in one NPP experiment
NPP analysis of the daptomycin-producing bacterium S. roseosporus NRRL 1599846 enabled the identification of three new RNPs and their gene clusters in a single NPP experiment (Supplementary Fig. 13). SRO15-2005 (Table 1, Supplementary Fig. 14) is a class II lasso peptide, SRO15-2212 (Table 1, Supplementary Fig. 15) is identical to the class III lantipeptide AmfS, which was previously uncharacterized from this strain, and SRO15-3108 (Table 1, Supplementary Fig. 16) is a class II lantipeptide that is predicted to undergo nine dehydrations during maturation. The detection of these three RNPs and their corresponding gene clusters in one NPP experiment demonstrates the potential of NPP as a high-throughput discovery methodology.
DISCUSSION
In this work, we introduce Natural Product Peptidogenomics as a chemotype-to-genotype genome mining approach for the characterization of ribosomal and nonribosomal peptide natural products and their respective biosynthetic gene clusters by identifying 14 peptides from well-known genome-sequenced streptomycetes. In contrast to global metabolomic47 and peptidomic24 strategies, NPP is a targeted approach in which MALDI-imaging or MALDI-TOF MS analysis of organic extracts is defined by a pre-selection of ions that are putative peptide natural products of expressed biosynthetic pathways. The innovation of NPP in efficiently linking these putative peptides to their gene clusters is firmly grounded in the connection of de novo MSn peptide sequence tags of modified peptides to precursor peptides or to predicted NRPS products by applying biosynthetic knowledge and iterative steps between MSn analysis and PNP genome mining for confirmation of putative chemotype-genotype matches. Because peptides are often structurally constrained, the generation of an MSn sequence tag is facilitated by structural deconstraining the peptide prior to MSn analysis. This yields simpler peptide structures and, thus, higher quality sequence tags as in the case of the class I lasso peptide SSV-2083 (Supplementary Fig. 9b). Deconstraining also aids in the elucidation of post-translational modifications such as the AviCys group of linaridin SGR-1832. In MSn sequence tag processing, the approach takes advantage of the degeneration of residues in the MSn sequence tag by mass, reactions in the mass spectrometer, biosynthesis or sequence directionality to ensure that the resulting search tags can be found in genomics-derived peptide sequences (Fig. 3 and 4). In PNP genome mining, the sequence tags are searched against a query space that is different for RNPs and NRPs. In RNP genome mining, the query space is the 6-frame translation of the target genome and, thus, large. The sequence tag for effective genome mining of a precursor peptide in this large query space should be at least 5 aa, otherwise too many candidate precursor peptides are obtained to be further differentiated based on RNP biosynthetic requirements. Several characterized precursor peptides that were identified in this study were not previously annotated in the NCBI database48 (i.e., peptides SCO-2138 and SGR-1832). These peptides were only found in the 6-frame translations of the S. coelicolor and S. griseus genome supercontigs. While the drawback of an extended database providing more candidate precursor peptides for a certain sequence tag is a potential concern, this larger protein inference problem, as it is known in global proteomics24, is effectively solved in NPP by the iterative matching of the candidate precursor peptides in mass, sequence and biosynthetic signatures to the MSn data.
MSn sequence tag processing and the iterative MSn and genomics analysis make the NPP de novo sequencing approach more effective in identifying precursor genes in RNP genome mining than current proteomic approaches. Neither Mascot26 nor InsPecT22 could identify any of the NPP-characterized RNPs in searches for unknown PTMs (Supplementary Table 4). InsPecT, which also relies on de novo sequence tagging, was able to characterize just two of the RNPs (SCO-2138 and SLI-2138) only after predefining NPP-characterized PTMs in the analysis. This is about what one would expect as proteomic tools typically annotate 5–15% of the collected data, although in rare cases this percentage can be higher. The main reason that these programs do not work for these peptides is because their scoring functions have been designed to work for protease-cleaved, water-soluble peptides. Proteomic programs require specific scoring functions for specific PTMs (e.g. specific for trypsin-cleaved ubiquitination tags or specific for phosphorylation) and simply have not been developed for RNP-based PTMs.
We further demonstrated that NRPs are readily incorporated in the NPP workflow as in the case of the stendomycins (Fig. 4). Even though >50% of all amino acids in NRPs are L-/D-proteinogenic amino acids40, mass shift sequences obtained from a MSn spectrum defines the candidate monomers to be used for the generation of all possible sequences to be compared to the predicted sequences based on the amino acid specificity of the adenylation domains by programs such as NRPSpredictor249. In NRP genome mining, the query space consists of NRP megasynthetases predicted from the target genome by NP.searcher or antiSMASH and, thus, is relatively small, as most microbial genomes contain <10 NRPS gene clusters. Consequently, short sequence tags of just 2-aa can be sufficient to correlate the NRP to its cognate NRPS gene cluster39. In the case of stendomycin, even though we ultimately applied the 8-aa tag GVIATTVV, we could have functionally operated and would have obtained the similar results with just a 2-aa tag such as VV, VI, TT, IA, AT or GV since only one of the five S. hygroscopicus NRPS gene sets was appropriate in size and sequence. NRP sequences often contain modified and/or nonproteinogenic amino acid residues that can be addressed by including all appropriate nonproteinogenic monomers to a mass shift sequence and by considering their corresponding biosynthetic machineries during genome mining (Supplementary Table 7).
Since NPP is a mass spectrometry-guided approach, it is ultimately dependent on generating quality sequence tags. The challenge in NPP characterization of peptides <500 Da or 4 aa or less is in applying a limited sequence tag for genome mining rather than dealing with matrix background in the low m/z region during peptide detection by MALDI-TOF MS. The analysis of putative peptides in the mass range <1500 m/z will also increase the discovery of PNPs, in particular of NRPs. NRPs with curated gene clusters in the NORINE database have an average mass of ~950 Da and 8 monomers (Supplementary Fig. 1), whereas RNPs usually have a higher molecular weight and the tool is appropriate for all such peptides. NPP, however, in its current implementation, is challenged by NRPs with multiple heterocycles, such as thiopeptides,8 and hybrid NRPS-PKS products with a major polyketide portions. This will remain a challenge until the fragmentation rules are established. Another NPP restraint is the bioinformatics predictability of PNP sequences from inadequate genomic data in which poor sequence or annotation quality result in misassigned precursor and NRPS genes. Better genome assembly, improved gene annotation (especially of small ORFs), increased understanding of gas-phase fragmentation behaviors, and deeper knowledge of NRPS substrate specificity codes will further empower the tools described in this work.
In conclusion, NPP is a new, MS-based genome mining platform to guide the discovery of novel ribosomal and nonribosomal peptides. This approach enables streamlined screening of peptide chemotypes from multiple organisms and facilitates expanded studies on their isolation, complete structure elucidation, biological evaluation and pathway engineering that leads to an increased appreciation for the understanding of the biological roles and therapeutic potential of peptide natural products. With further automatization of the NPP workflow such as training for off-set functions of complex peptides, better understanding MS fragmentation behaviors and the expansion to smaller masses and additional organisms, NPP has the potential to open up new research directions in the (bio)chemistry of peptide natural products.
METHODS
MALDI-imaging of Streptomyces colonies
Streptomyces strains were grown on solid ISP2 medium (1 liter contains 4 g yeast, 10 g malt extract, 4 g dextrose, 20 g agar, pH 7) for 4–10 d at 28 °C until sporulation. Streptomyces spores from one plate were suspended in 1 ml sterile water:glycerol (3:1) and stored at 80 °C after inoculation. Thin layer ISP2 agar plates of sporulating Streptomyces colonies were prepared as described elsewhere25. The applied matrix was Universal MALDI matrix (Sigma-Aldrich). MALDI-imaging of Streptomyces samples on a Bruker MSP 96 anchor plate was performed on a Microflex Bruker Daltonics mass spectrometer outfitted with Compass 1.2 software suite (Consists of FlexImaging 2.0, FlexControl 3.0, and FlexAnalysis 3.0). Target plate calibration was done as described elsewhere25. The sample was run in positive reflectron mode, with 800 μm laser intervals in XY. After the target plate calibration was complete, the AutoXecute command was used to analyze the samples. The used FlexControl method had settings as described before25 with Detection parameters adjusted as follows: Mass Range - 800–4200 m/z and Detector Gain - Reflector 3.7–8.1. Mass calibration was accomplished using a Peptide standard mix (Bruker Daltonics) as external standard. After data acquisition, the data were analyzed using the FlexImaging software. The resulting mass spectrum was analyzed manually for mass signal >1500 m/z. Putative peptide mass signals >1500 m/z were assigned with individual colors for display of the distribution of the mass signal in the image.
MS analysis and sequence tagging
Peptide extraction, enrichment and preparation for MS analysis are described in the Supplementary Methods. Prepared peptide samples were injected for MS analysis by a nanomate-electrospray ionization robot (Advion) for consecutive electrospray into the MS inlet of a LTQ 6.4T FT-ICR mass spectrometer (Thermo Finnigan). MS and MSn data were acquired in positive ion mode. FTMS data were acquired in 400–2000 m/z scans. Selected peptide mass signals were manually isolated and fragmented by collision induced dissociation (CID). MSn data was collected either in IT or FT detection mode. All data were analyzed using QualBrowser, part of the Xcalibur LTQ-FT software package (ThermoFisher). FTMS masses were analyzed using Extract software (Thermo Electron Bremen). Peptide MSn sequence tags were assigned from MSn data by manual de novo sequencing within the mass accuracy of the mass spectrometer using mass shift list of proteinogenic aa monomers (Supplementary Table 5) and nonproteinogenic monomers (Supplementary Table 6 and 7). Sequence tagging emphasized on a correct assignment of 5–10 aa MSn sequence tags rather than longer, incorrect assignments for reliable genome mining. The MSn sequence tag was further manually processed into a set of search tags depending on the degree of degeneration of the MSn sequence tag. The MSn sequence tag processing included differentiation of positions with identical masses (e.g. Ile/Leu), positions with biosynthetic modifications (e.g. Dha derived from Ser or Cys in RNPs, Supplementary Table 6 and 7) and positions modified by MS analysis (e.g. Dha derived from Cys of a lanthionine-PTM or Dhb derived from Thr of a macrolactone linkage). In NaBH4/NiCl2-treated samples, positions were differentiated that might be chemically altered (e.g. Ala derived from Cys or Ala). The MSn sequence tag was also differentiated in its reversed direction.
Genome mining of ribosomal peptides
A 6-frame translated supercontig was searched with all possible RNP search tags from a given MSn sequence tag in a standard text processing program. A candidate precursor peptide was defined in its N-terminus by pBLAST-search of its C-terminal partial sequence to find homologs or by reanalysis of the region in the supercontig in order to find missed alternative start codons that were not translated as a methionine in the 6-frame translation. A candidate precursor peptide was confirmed by (a) mass matching of putative core peptide sequence to the observed peptide mass by considering possible PTMs, (b) sequence matching of the putative core peptide to the MSn data, and (c) pBLAST analysis of the neighboring ORFs, i.e. gene cluster analysis. Based on the gene cluster components and observed PTMs, a RNP class could usually be characterized (Supplementary Table 1). In cases of unusual gene cluster components during the RNP gene cluster analysis, a putative new RNP gene cluster could be defined by search of homologous gene clusters (e.g. Supplementary Fig. 11c and 12c). Finally, a structure of the RNP could be predicted based on the characterized core peptide sequence and PTMs that were characterized or predicted from the MS- and bioinformatic analysis of the target peptide and its gene cluster, respectively.
Genome mining of nonribosomal peptides
A search tag that did not yield a candidate precursor peptide by 6-frame translation-based genome mining was subjected to genome mining of NRP gene clusters. Herein, the mass shift sequence was reanalyzed by applying NRP monomer mass shifts (Supplementary Table 7) to characterize all possible NRP search tags. The supercontig of the target organism (e.g. Streptomyces hygroscopicus ATCC 53653, Supplementary Fig. 5) was analyzed by NP.searcher33 and by antiSMASH,34 and NRP search tags were compared to the predicted NRP sequences in monomers and length. In case of a putative match, the corresponding NRP gene cluster was analyzed in its assembly line organization in the corresponding antiSMASH output and by InterPro50. The accessibility of NRP families to genome mining by the NPP approach was assessed by NP.searcher and antiSMASH-based analysis of GenBank files of characterized NRPS gene cluster families as described in Supplementary Methods.
Additional methods
Bioinformatic prediction of PNP pathways, proteomic analysis of characterized RNPs, and isolation and structure elucidation of Q027-1628 (stendomycin I) from marine Streptomyces strain CNQ-027 are described in Supplementary Methods.
Supplementary Material
Acknowledgments
We thank Natalie Castellana and Vineet Bafna for providing the algorithm to enable 6-frame translations of supercontigs. Financial support was provided by the National Institutes of Health (GM085770 to B.S.M. and GM086283 to P.C.D.) and the Beckman Foundation.
Footnotes
Author contributions R.D.K. designed and carried out experiments, analyzed data and wrote the paper. Y.L.Y., Y.X. and S.J.N. carried out experiments and analyzed data. P.C. and M.A.F. carried out the bioinformatic analysis and analyzed data. W.F. analyzed data. B.S.M and P.C.D. designed experiments, analyzed data and wrote the paper.
Competing financial Interests The authors declare no competing financial interests.
References
- 1.Daffre S, et al. Bioactive natural peptides. In: Rahman AU, editor. Studies in Natural Products Chemistry. 1st edn. Vol. 35. Elsevier; Oxford: 2008. pp. 597–691. [Google Scholar]
- 2.Nolan EM, Walsh CT. How nature morphs peptide scaffolds into antibiotics. ChemBioChem. 2009;10:34–53. doi: 10.1002/cbic.200800438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donadio S, Monciardini P, Sosio M. Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat. Prod. Rep. 2007;24:1073–1109. doi: 10.1039/b514050c. [DOI] [PubMed] [Google Scholar]
- 4.Velasquez JE, van der Donk WA. Genome mining for ribosomally synthesized natural products. Curr. Opin. Chem. Biol. 2011;15:11–21. doi: 10.1016/j.cbpa.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganz T. Defensins and host defense. Science. 1999;286:420–421. doi: 10.1126/science.286.5439.420. [DOI] [PubMed] [Google Scholar]
- 6.Moore BS. Extending the biosynthetic repertoire in ribosomal peptide assembly. Angew. Chem. Int. Ed. Engl. 2008;47:9386–9388. doi: 10.1002/anie.200803868. [DOI] [PubMed] [Google Scholar]
- 7.Willey JM, van der Donk WA. Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- 8.Li C, Kelly WL. Recent advances in thiopeptide antibiotic biosynthesis. Nat. Prod. Rep. 2010;27:153–164. doi: 10.1039/b922434c. [DOI] [PubMed] [Google Scholar]
- 9.Donia MS, Ravel J, Schmidt EW. A global assembly line for cyanobactins. Nat. Chem. Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duquesne S, et al. Two enzymes catalyze the maturation of a lasso peptide in Escherichia coli. Chem. Biol. 2007;14:793–803. doi: 10.1016/j.chembiol.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Duquesne S, Petit V, Peduzzi J, Rebuffat S. Structural and functional diversity of microcins, gene-encoded antibacterial peptides from enterobacteria. J. Mol. Microbiol. Biotechnol. 2007;13:200–209. doi: 10.1159/000104748. [DOI] [PubMed] [Google Scholar]
- 12.Cotter PD, Hill C, Ross RP. Bacteriocins: developing innate immunity for food. Nat. Rev. Microbiol. 2005;3:777–788. doi: 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- 13.Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Challis GL, Ravel J, Townsend CA. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 15.Finn RD, et al. The Pfam protein families database. Nucl. Acids Res. 2010;38:211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winter JM, Behnken S, Hertweck C. Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol. 2011;15:22–31. doi: 10.1016/j.cbpa.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Lautru S, Deeth RJ, Bailey LM, Challis GL. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat. Chem. Biol. 2005;1:265–269. doi: 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- 18.Claesen J, Bibb M. Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16297–16302. doi: 10.1073/pnas.1008608107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li B, et al. Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10430–10435. doi: 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kodani S, et al. The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11448–11453. doi: 10.1073/pnas.0404220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gressler M, Zaehle C, Scherlach K, Hertweck C, Brock M. Multifactorial induction of an orphan PKS-NRPS gene cluster in Aspergillus terreus. Chem. Biol. 2011;18:198–209. doi: 10.1016/j.chembiol.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Ng J, et al. Dereplication and de novo sequencing of nonribosomal peptides. Nat. Methods. 2009;6:596–599. doi: 10.1038/nmeth.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol. 2005;23:1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 24.Duncan MW, Aebersold R, Caprioli RM. The pros and cons of peptide-centric proteomics. Nat. Biotechnol. 2010;28:659–664. doi: 10.1038/nbt0710-659. [DOI] [PubMed] [Google Scholar]
- 25.Yang YL, Xu Y, Straight P, Dorrestein PC. Translating metabolic exchange with imaging mass spectrometry. Nat. Chem. Biol. 2009;5:885–887. doi: 10.1038/nchembio.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Ueda K, et al. AmfS, an extracellular peptidic morphogen in Streptomyces griseus. J. Bacteriol. 2002;184:1488–1492. doi: 10.1128/JB.184.5.1488-1492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McIntosh JA, Donia MS, Schmidt EW. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat. Prod. Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohnishi Y, et al. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol. 2008;190:4050–60. doi: 10.1128/JB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willey JM, Willems A, Kodani S, Nodwell JR. Morphogenetic surfactants and their role in the formation of aerial hyphae in Streptomyces coelicolor. Mol. Microbiol. 2006;59:731–742. doi: 10.1111/j.1365-2958.2005.05018.x. [DOI] [PubMed] [Google Scholar]
- 31.Wilkinson B, Micklefield J. Biosynthesis of nonribosomal peptide precursors. Methods Enzymol. 2009;458:353–78. doi: 10.1016/S0076-6879(09)04814-9. [DOI] [PubMed] [Google Scholar]
- 32.Romano A, Vitullo D, Di Pietro A, Lima G, Lanzotti V. Antifungal lipopeptides from Bacillus amyloliquefaciens strain BO7. J. Nat. Prod. 2011;74:145–51. doi: 10.1021/np100408y. [DOI] [PubMed] [Google Scholar]
- 33.Li MH, Ung PM, Zajkowski J, Garneau-Tsodikova S, Sherman DH. Automated genome mining for natural products. BMC Bioinformatics. 2009;10:185. doi: 10.1186/1471-2105-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medema MH, et al. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters. Nucl. Acids Res. 2011;39:339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bodanszky M, Izdebski J, Muramatsu I. Structure of the peptide antibiotic stendomycin. J. Am. Chem. Soc. 1969;91:2351–2358. doi: 10.1021/ja01037a028. [DOI] [PubMed] [Google Scholar]
- 36.Strieker M, Marahiel MA. The structural diversity of acidic lipopeptide antibiotics. ChemBioChem. 2009;10:607–616. doi: 10.1002/cbic.200800546. [DOI] [PubMed] [Google Scholar]
- 37.Roongsawang N, Washio K, Morikawa M. Diversity of nonribosomal peptide synthetases involved in the biosynthesis of lipopeptide biosurfactants. Int. J. Mol. Sci. 2010;12:141–72. doi: 10.3390/ijms12010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Visca P, Imperi F, Lamont IL. Pyoverdine siderophores: from biogenesis to biosignificance. Trends in Microbiology. 2006;15:22–30. doi: 10.1016/j.tim.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 39.Liu WT, Kersten RD, Yang YL, Moore BS, Dorrestein PC. Imaging mass spectrometry and genome mining via short sequence tagging identified the anti-infective agent arylomycin in Streptomyces roseosporus. J. Am. Chem. Soc. doi: 10.1021/ja2040877. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caboche S, Leclere V, Pupin M, Kucherov G, Jacques P. Diversity of monomers in nonribosomal peptides: towards the prediction of origin and biological activity. J. Bacteriol. 2010;192:5143–5150. doi: 10.1128/JB.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawulka KE, et al. Structure of subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to alpha-carbon cross-links: formation and reduction of alpha-thio-alpha-amino acid derivatives. Biochemistry. 2004;43:3385–3395. doi: 10.1021/bi0359527. [DOI] [PubMed] [Google Scholar]
- 42.Knappe TA, Linne U, Xie X, Marahiel MA. The glucagon receptor antagonist BI-32169 constitutes a new class of lasso peptides. FEBS Lett. 2010;584:785–789. doi: 10.1016/j.febslet.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 43.Bentley SD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 44.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vats P, Rothfield L. Duplication and segregation of the actin (MreB) cytoskeleton during the prokaryotic cell cycle. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17795–17800. doi: 10.1073/pnas.0708739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miao V, et al. Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology. 2005;151:1507–1523. doi: 10.1099/mic.0.27757-0. [DOI] [PubMed] [Google Scholar]
- 47.Koal T, Deigner HP. Challenges in mass spectrometry based targeted metabolomics. Curr. Mol. Med. 2010;10:216–226. doi: 10.2174/156652410790963312. [DOI] [PubMed] [Google Scholar]
- 48.Warren AS, Archuleta J, Feng WC, Setubal JC. Missing genes in the annotation of prokaryotic genomes. BMC Bioinformatics. 2010;11:131. doi: 10.1186/1471-2105-11-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Röttig M, et al. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucl. Acids Res. 2011;39:347–352. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunter S, et al. InterPro: the integrative protein signature database Nucl. Acids Res. 2009;37:211–215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.