

Abstract
The pathobiological role of p53 has been widely studied, however its role in normophysiology is relatively unexplored. We previously showed that p53 knock-down increased ploidy in megakaryocytic cultures. This study aims to examine the effect of p53 loss on in vivo megakaryopoiesis, platelet production and function, and to investigate the basis for greater ploidy in p53−/− megakaryocytic cultures. Here, we used flow cytometry to analyze ploidy, DNA synthesis and apoptosis in murine cultured and bone marrow megakaryocytes following thrombopoietin administration and to analyze fibrinogen binding to platelets in vitro. Culture of p53−/− marrow cells for 6 days with thrombopoietin gave rise to 1.7-fold more megakaryocytes, 26.1±3.6% of which reached ploidy classes ≥64N compared to 8.2±0.9% of p53+/+ megakaryocytes. This was due to 30% greater DNA synthesis in p53−/− megakaryocytes and 31% greater apoptosis in p53+/+ megakaryocytes by day 4 of culture. Although the bone marrow and spleen steady-state megakaryocytic content and ploidy were similar in p53+/+ and p53−/− mice, thrombopoietin administration resulted in increased megakaryocytic polyploidization in p53−/− mice. Although their platelet counts were normal, p53−/− mice exhibited significantly longer bleeding times and p53−/− platelets were less sensitive than p53+/+ platelets to agonist-induced fibrinogen binding and P-selectin secretion. In summary, our in vivo and ex-vivo studies indicate that p53 loss leads to increased polyploidization during megakaryopoiesis. Our findings also suggest for the first time a direct link between p53 loss and the development of fully functional platelets resulting in hemostatic deficiencies.
Keywords: megakaryocytes, megakaryopoiesis, p53, hemostasis, platelet function
Activation of the p53 tumor suppressor generally results in cell cycle arrest and/or induction of apoptosis [1]. Mounting evidence has implicated p53 as a critical regulator of hematopoietic stem cell proliferation, stress hematopoiesis following lethal irradiation and hematopoietic stem cell aging [2–4] (reviewed in [5]). Our recent studies [6–10] and earlier work by others [11–13] provided evidence that the p53 pathway, largely unexplored in megakaryopoiesis, plays an important role in megakaryocytic (Mk) differentiation. We have recently shown that knock-down of p53 increased ploidy in Mk cultures [7]. There are relatively few studies that specifically examine megakaryopoiesis or platelet production in the absence of p53. One such study indicated faster recovery of colony-forming Mk progenitors (CFU-Mk) after 5-fluorouracil (5-FU) treatment in p53−/− versus p53+/+ mice and a 3-fold increased bone marrow content of Lin−Sca-1+c-Kit+ cells in p53−/− mice following 5-FU treatment [14]. Platelet recovery after irradiation of p53−/− mice was 3 times faster [15], suggesting that p53 loss can result in faster platelet production. Despite the known propensity of p53−/− mice to succumb to cancer, mainly lymphoma and sarcoma [16], p53 loss has not been linked to abnormal megakaryopoiesis. On the contrary, a recent study demonstrated that intercrossing of p53−/− mice with mice engineered to contain an equivalent to the human deletion of the q arm of chromosome 5 (5q- syndrome), whose bone marrow (BM) cells express high levels of the p53 protein, reversed the low Mk/erythroid progenitor levels and low platelet levels encountered in the 5q– syndrome [17]. This study suggests that p53 loss can favor megakaryopoiesis and platelet production. Additionally, a study examining platelet recovery following myelosuppression in p53−/− and p53+/+ mice hypothesized that loss of p53 confers to hematopoietic stem cells and committed Mk precursors the ability to resist apoptosis to the same extent that administration of thrombopoietin (Tpo) supports survival [18]. However, none of these studies examined Mk differentiation in response to Tpo administration in the absence of p53 or Mk/platelet recovery in response to induced thrombocytopenia.
The first aim of this study was to characterize Mk polyploidization in the bone marrow of p53+/+ and p53−/− mice at steady state together with de novo platelet production. Additionally, we aimed to examine the regulation of Mk polyploidization and platelet production in response to (1) treatment of mice with Tpo to promote Mk differentiation and increased thrombopoiesis; and (2) administration of an anti-platelet antibody to specifically target the platelets and elicit a Mk differentiation and platelet production cascade to replenish the normal platelet levels in p53+/+ and p53−/− mice.
The next question we aimed to address was the impact of p53 on platelet functional responses. The presence of the p53 protein and the p53 inhibitor MDM2 in platelets has been previously documented [19]. However, the effect of the loss or functional inactivation of p53 on hemostasis is unknown. Here, we present findings postulating a reduced hemostatic response in p53−/− mice. Our study suggests a novel role for p53 in regulating hemostasis, perhaps by affecting components of the platelet granules or regulating platelet surface integrin activation.
MATERIALS AND METHODS
Mice
All procedures involving mice were approved by the University of Delaware Institutional Animal Care and Use Committee. Male p53−/− mice and p53+/+ (wild-type) littermates (B6.129S2.Trp53tm1Tyj/J colony) [16] were purchased from the Jackson laboratory (Bar Harbor, ME) and housed with free access to food and water.
Injection of thrombopoietin to stimulate in vivo megakaryopoiesis
p53−/− mice and age-matched p53+/+ littermates less than 3 months of age were injected once subcutaneously with 1.2 μg recombinant murine (rm) Tpo (Peprotech; Rocky Hill, NJ) diluted in saline or saline as control [20, 21]. On days 2 and 5 following Tpo treatment, the mice were bled retro-orbitally and sacrificed. Platelets were enumerated using the Unopette reservoirs (Becton-Dickinson; Franklin Lakes, NJ) and a hemacytometer (Hausser; Horsham, PA).
Injection of anti-CD41 antibody to induce thrombocytopenia
p53−/− mice and age-matched p53+/+ littermates less than 4 months of age were injected once intra-peritoneally with 0.5 μg rat anti-mouse CD41 (clone: MWReg30, BDPharmingen; San Diego, CA) per gram of body weight [22]. The antibody had been dialyzed prior to injection for 4 hours in 1 liter of ice-cold PBS to remove traces of sodium azide using the slide-a-lyzer mini dialysis units (Pierce; Rockford, IL). Daily blood counts were conducted for 5 days using a Hemavet (Drew Scientific; Waterbury, CT). Some of the mice were sacrificed on day 2 after anti-CD41 injection, while the rest were sacrificed on day 5 in order to harvest bone marrow.
Detection of reticulated platelets by staining with thiazole orange and measurement of CD41 expression on platelets
Reticulated platelets were measured as described previously [23]. Briefly, 1 μl of EDTA-anticoagulated blood was incubated in a total volume of 60 μl with 10 μg/ml thiazole orange (Sigma-Aldrich) and 1 μl anti-CD41-PE (clone MWReg30; BD-Pharmingen) for 15 min in the dark at room temperature. The samples were then fixed by addition of 1% para-formaldehyde for no more than 30 min at room temperature and acquired on a FACSAria. Measurement of CD41 mean fluorescence using flow cytometry was used to estimate αIIbβ3 expression on the platelet surface.
Thrombopoietin levels in serum
Blood was collected from the retro-orbital vein using plain Natelson capillaries and left to clot at room temperature in the absence of anti-coagulant. Serum was then collected by high-speed centrifugation. Thrombopoietin levels in the serum were measured using the Quantikine mouse Tpo ELISA (R&D Systems; Minneapolis, MN) [22]. The absorbance was read at 450 nm with the correction set to 595 nm using a DTX880 microplate reader (Beckman-Coulter; Brea, CA)
Murine Mk cell culture
Red blood cell-depleted bone marrow cells were processed as described in our previous study [7] and cultured in IMDM/ 10% FBS/ 1% penstrep media containing 50 ng/ml rmTpo [24]. Culture flasks were maintained in a fully humidified incubator at 37°C and 5% CO2.
Detection of ploidy, DNA synthesis and apoptosis in Mk cells
To detect ploidy among Mk cells, red blood cell-depleted bone marrow cells or cultured Mk cells were stained with anti-CD41-FITC (BD-Pharmingen), fixed with 0.5% para-formaldehyde in PBS, permeabilized with 70% methanol, treated with 10 mg/ml RNAse and counterstained with Propidium Iodide (PI) [7, 25]. For DNA synthesis, p53+/+ and p53−/− Mk cell cultures were incubated for 12 hours with 10 μM bromo-deoxy-uridine (BrdU) (BD-Pharmingen) at 37°C, then stained with anti-CD41-FITC, fixed and permeabilized per the manufacturer's protocol, treated with 10 mg/ml RNAse and stained with PI [7]. For detection of Mk apoptosis, Hoechst 33342 was directly added at 0.01 mM into the Mk cell cultures and incubated for 2 hours at 37°C/ 5% CO2 [26]. At the end of the incubation, Mk cells were washed with PBS, stained with anti-CD41-FITC for 30 min at 4°C, washed again with PBS, resuspended in 1X Annexin binding buffer (BD-Pharmingen), stained with Annexin V-PE (BD-Pharmingen) and acquired.
Mouse tail-bleeding assays
Mice were anesthetized by isoflurane inhalation, tails were pre-warmed at 37°C for 5 min, 3-mm of the distal end was cut off using a scalpel, and the tail was immersed into fresh saline pre-warmed at 37°C. Primary bleeding time is defined as the time until bleeding cessation for a minimum of 10 sec. If bleeding resumed, the duration of the primary and subsequent bleeds were added up and reported as the total bleeding time. If the primary or total bleeding time exceeded 10 min, the assay was stopped and the tail was taken out of the saline and pressure was applied to stop the bleeding in order to avoid losing too much blood [27].
Flow cytometric and microscopical assessment of platelet stimulation with agonists
Heparinized blood was collected from the retro-orbital veins and platelet rich plasma (PRP) was isolated by low speed centrifugation. PRP was passed through a sepharose column as described previously [28], gel-filtered platelets were isolated and calcium was added to a final concentration of 1 mM. For flow cytometric assays, Alexa-Fluor-488-conjugated fibrinogen (Molecular Probes; Eugene, OR) or FITC-conjugated anti-P selectin (BD-Pharmingen) were added at 12.5 μg/ml or 1:100 dilution, respectively, together with a range of concentrations of AYPGKF (Kimmel Cancer Center of the Thomas Jefferson University; Philadelphia, PA, USA), a PAR-4 agonist peptide. The platelets were stimulated for 10 min at 37°C, then fixed for 10 min with 2% para-formaldehyde at room temperature. For microscopy, gel-filtered platelets were stimulated for 5, 15 or 30 min at 37°C with 50 μM AYPGKF or 10 μM PMA on glass slides pre-coated with 100 μg/ml human fibrinogen (Enzyme Research Laboratories; South Bend, IN, USA). Platelets were fixed and permeabilized as described [28], stained with Alexa Fluor-568-phalloidin and mounted. Slides were examined using an LSM 5 Live high Speed Confocal microscope (Zeiss; Oberkochen, Germany) equipped with a 100X oil objective.
Microarray analysis of p53 knock-down (KD) relative to control CHRF cells
CHRF is a wild-type p53+ human CD41+CD34+ megakaryoblastic cell line, which responds to PMA and undergoes polyploidization coupled with marked apoptosis and extension of proplatelet-like cytoplasmic protrusions. P53-KD and control human megakaryoblastic CHRF cells were cultured in IMDM/ 10% FBS and stimulated with 10 ng/ml PMA to induce terminal Mk differentiation, as described previously [7, 9, 29]. Cell samples taken from two independent biological replicate sets of p53-KD and control CHRF cell cultures either unstimulated (day 0) or on days 1, 3, 5 and 7 following stimulation with PMA were flash-frozen in liquid nitrogen to be used later for microarray analysis. Experimental methods for CHRF cell culture, RNA isolation, labeling and hybridization for the microarray analysis in CHRF cells and data normalization have been described [9, 30]. All data have been deposited in Gene Expression Omnibus as mandated by the (Minimum Information About Microarray Experiment) MIAME standards and have been assigned series record GSE30984. A list of platelet activation-related genes was curated based on the literature [6, 9, 31–33]. Differentially expressed genes were identified using statistical analysis of microarrays as implemented in MeV with a false discovery rate < 0.05 [34]. Validation of microarray findings was done with Real Time PCR as described in our previous work [7] using Taqman probes for ITGA2B (Hs01116228_m1), SELP (Hs00356351_m1) and MYH9 (Hs00159522_m1) from Applied Biosystems. Data were normalized against housekeeping gene GUSB (Hs99999908_m1).
Statistical analysis
Unless otherwise noted, all statistical comparisons were conducted using an unpaired 2-tailed Student's t-test.
Results
Increased polyploidization and cell size observed in cultured p53−/− Mk cells are caused by greater DNA synthesis and diminished apoptosis during megakaryopoiesis in the absence of p53
We have previously shown that knock-down of p53 in human megakaryoblastic CHRF cells led to increased Mk polyploidization in part due to greater DNA synthesis upon induction of Mk differentiation [7]. Additionally, we have shown that p53−/− culture-derived mouse Mk cells reach higher ploidy classes than p53+/+ Mk cells [7]. Here, we aimed to better characterize this ex-vivo-observed hyperploid murine p53−/− Mk phenotype. BM cells harvested from p53−/− mice and age-matched p53+/+ littermates were cultured ex-vivo in the presence of 50 ng/ml Tpo for six days to induce Mk differentiation. Cell size analysis of p53+/+ and p53−/− cultured CD41+ cells was performed using the Forward Scatter (FSC) area signal [35]. On day 6 of culture, the mean±SEM FSC signal for p53+/+ and p53−/− CD41+ cells was 121±4 and 151±9 (N=7, P=0.01), indicating that cell size was significantly increased in the highly polyploid p53−/− Mk cells. As we have previously observed [7], Mk cells derived from p53−/− BM progenitor cells always reached higher ploidy classes and exhibited higher percentage of highly polyploid cells. By day 6 of culture, there was a 1.7-fold increase in total CD41+ Mk cells for p53−/− Mk cells (N=7, P<0.02; Fig. 1A) and substantially increased polyploidy: 26.1±3.6% of p53−/− versus 8.2±0.9% of p53+/+ Mk cells reached ploidy classes ≥64N (N=7, P=0.0004; Fig. 1B).
Fig. 1.
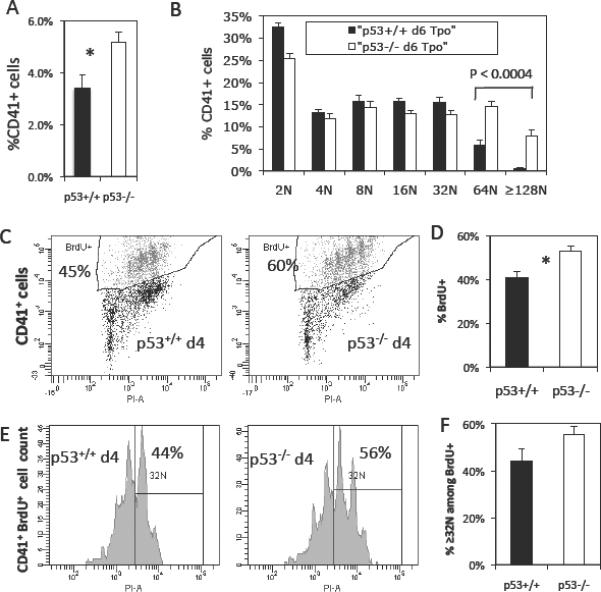
Ex-vivo culture of p53−/− progenitor cells with Tpo generates hyperploid megakaryocytic cells in part due to enhanced DNA synthesis. (A) Percent of CD41+ Mk cells on day 6 of culture of p53+/+ and p53−/− pronenitor cells with 50 ng/ml rm Tpo. Error bars: SEM, N=7, * represents P < 0.02, and (B) averaged ploidy data for p53+/+ and p53−/− CD41+ Mk cells on day 6 of Tpo culture. Error bars: SEM, N=7. (C) Representative DNA synthesis data from p53+/+ and p53−/− cultured Mk cells and (D) average of the DNA synthesis data for day 4. Error bars: SEM, N=3–4, * indicates P < 0.03. Polyploidization among the cycling CD41+ Mk cells (BrdU+) on day 4 of Tpo culture. (E) Representative ploidy histograms from BrdU+ p53+/+ and p53−/− cultured Mk cells and (F) average of the % ≥32N polyploid cells among BrdU+ Mk cells for day 4. Error bars: SEM, N=3–4.
Increased polyploidization observed in culture was partially due to 30% enhanced DNA synthesis among p53−/− Mk cells on day 4 of culture (N=3–4; P<0.03; Fig. 1C–D). Moreover, there was a tendency for increased polyploidization in ≥32N ploidy classes among actively cycling Mk cells (BrdU+) in p53−/− Mk cells on day 4 (N=3–4; Fig. 1E–F). This finding indicated that increased DNA synthesis largely occurred in the highly polyploid (≥32N) population of p53−/− Mk cells.
Increased polyploidization in ex-vivo-cultured p53−/− Mk cells can also be ascribed to diminished apoptosis in the absence of p53. Indeed, apoptosis, measured among nucleated CD41+ cells by assessing binding of Annexin V, was increased by an average of 31% in p53+/+ Mk cells on day 4 of culture with Tpo (Fig 2A–B).
Fig. 2.
Ex-vivo cultured p53−/− Mk cells exhibit diminished apoptosis compared to p53+/+ Mk cells on day 4 of Tpo culture. Cells were incubated with 0.01 mM Hoechst 33342, which serves to distinguish nucleated Mk cells from platelets and other CD41+ debris in the culture, stained with anti-CD41 and treated with Annexin V to identify apoptotic Mk cells. (A) Representative apoptosis data from p53+/+ and p53−/− cultured Mk cells and (B) average of the apoptosis data for day 4 of culture. Error bars: SEM, N=3, * indicates P < 0.05.
Tpo administration leads to increased in vivo Mk polyploidization in p53−/− mice
Mk cells in the BM were measured as the % of nucleated CD41+ cells. The numbers of BM-resident Mk cells were similar in p53+/+ and p53−/− mice (0.21±0.03% for both genotypes, N=7), while among p53+/+ Mk cells 29.7±3.1% reached ploidy of 16N or higher versus 30.2±3.9% among p53−/− Mk cells. Sternum BM and spleen histology sections did not reveal any abnormalities in the morphology or content of p53−/− Mk cells (data not shown). Additionally, p53−/− mice have normal serum Tpo levels and platelet synthesis and only slightly lower platelet counts compared to their p53+/+ counterparts (Table 1).
Table 1.
complete blood counts in untreated p53+/+ and p53−/− mice (N=11–14), reticulated platelets (N=19–22) and serum thrombopoietin (Tpo; N=4–6). WBC: white blood cells, NE: neutrophils, LY: lymphocytes, MO: monocytes, EO: eosinophils, BA: basophils, RBC: red blood cells, Hb: hemoglobin, PLT: platelets, MPV: mean platelet volume, %Retic: % reticulated platelets. Values ± SEM.
| p53+/+ | p53−/− | |
|---|---|---|
| WBC (×103/mm3) | 7.81±0.64 | 8.85±1.09 |
| NE (×103/mm3) | 1.89±0.30 | 2.30±0.33 |
| LY (×103/mm3) | 5.42±0.39 | 5.73±0.64 |
| MO (×103/mm3) | 0.41±0.06 | 0.62±0.12 |
| EO (×103/mm3) | 0.08±0.02 | 0.15±0.06 |
| BA (×103/mm3) | 0.02±0.01 | 0.04±0.02 |
| RBC (×106/mm3) | 8.56±0.22 | 8.45±0.28 |
| Hb (g/dL) | 12.1±0.3 | 11.7±0.4 |
| PLT (×103/mm3) | 907±54 | 789±49 |
| MPV (fL) | 4.6±0.1 | 4.6±0.0 |
| % Retic | 14.0±0.6% | 13.1±0.6 |
| Tpo (pg/ml) | 3,494±230 | 3,226±110 |
In order to examine the effect of p53 loss on in vivo Mk polyploidization and platelet formation under conditions of increased platelet production as a result of induced thrombocytopenia, we administered 0.5 μg anti-CD41 antibody per gram of body weight to p53−/− mice and p53+/+ age-matched littermates. Five days after anti-CD41 treatment, platelet counts were higher than their normal levels in p53−/− and p53+/+ mice (N=5; Fig. S1–A). Even though there was a tendency for increased platelet synthesis in p53−/− mice, we did not observe significant differences in the rate of platelet recovery between p53−/− and p53+/+ mice or in Mk content and polyploidization in the BM (Fig. S1).
To determine whether Tpo induces increased polyploidization in vivo, as we have seen in cultured p53−/− Mk cells, we administered 1.2 μg Tpo to p53−/− mice and p53+/+ age-matched littermates. Five days following Tpo treatment, in agreement with the ex-vivo data shown in Fig. 1, we found higher Mk ploidy in the BM of p53−/− mice: 37.7±5.4% p53−/− Mks reached ploidy of ≥16N compared to 20.7±3.2% p53+/+ Mks (P<0.06, N=3–4; Fig. 3A–B). In addition, five days following Tpo treatment, we found a tendency for increased BM content of p53+/+ Mk cells – 0.46%±0.14% of BM-resident p53+/+ Mks relative to 0.23±0.03% of p53−/− Mks (N=3–4; Fig. 3C). Even though steady-state platelet counts were similar (Fig. 3D), the increase in p53−/− Mk polyploidization was accompanied by a tendency for modestly increased platelet formation (N=3–4; Fig. 3E).
Fig. 3.
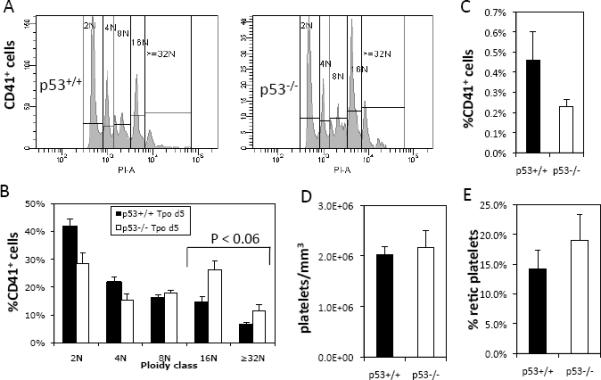
Megakaryopoiesis and platelet production in p53+/+ and p53−/− mice 5 days following Tpo administration. (A) Representative histograms of p53+/+ and p53−/− Mk cell ploidy indicate increased polyploidization in p53−/− Mk cells in vivo. (B) Averaged ploidy of CD41+ Mk cells in the bone marrow indicates increased percentage of p53−/− Mk cells in 16N and ≥32N ploidy classes, P < 0.06. (C) Percent of CD41+ Mk cells in the bone marrow, P = 0.13. (D) Platelet counts and (E) percent reticulated platelets in the circulation. Error bars: SEM, N=3–4.
p53−/− mice have longer bleeding times
Because we have shown a role for p53 in Mk development, we wondered whether p53 expression might affect hemostatic function. Indeed, wild-type platelets express p53 despite lacking a nucleus (Fig. S2). To determine whether p53 influences hemostasis, tail-bleeding times were assessed in p53−/− and p53+/+ mice [27, 36]. Bleeding data collected for mice younger than 10 weeks of age indicated hemostatic deficiencies in p53−/− mice. Primary bleeding times were prolonged in p53−/− mice (mean±SEM: 5.4±1.2 min) compared to p53+/+ mice (mean±SEM: 2.1±0.5 min) (N=10–11; P<0.02) and the duration of the primary bleed for 3/10 p53−/− mice exceeded 10 minutes (Fig. 4A). Total bleeding times were also prolonged in p53−/− mice (mean±SEM: 8.8±0.9 min) compared to p53+/+ mice (mean±SEM: 5.3±1.0 min) (N=10–11; P<0.02; Fig. 4B). Indeed, 8/10 p53−/− mouse tail bleeds exceeded the 10 min duration of the assay relative to 2/10 p53+/+ mouse tail bleeds.
Fig. 4.
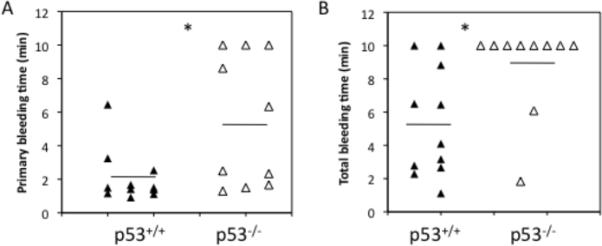
Tail-bleeding assay in less than 10-weeks-old p53+/+ and p53−/− mice following cutting 3-mm off the distal end of the tail. p53+/+ (black triangles) and p53−/− mice (open triangles). (A) Primary tail bleeding times. (B) Total bleeding times. N=10–11. Horizontal lines (drawn to scale) represent the means. * denotes statistical significance, P < 0.02.
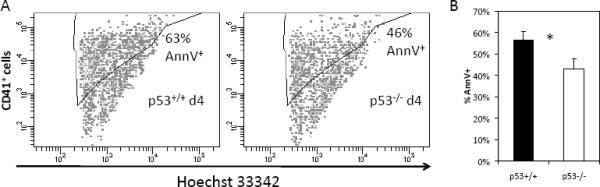
p53−/− platelets are less sensitive to a PAR-4 agonist-induced fibrinogen binding than p53+/+ platelets
We next examined fibrinogen binding of isolated platelets from p53−/− mice in response to AYPGKF, a specific agonist for the thrombin receptor PAR4 [37]. The affinity of the platelet integrin αIIbβ3 for fibrinogen is low in unstimulated platelets. Thrombin stimulation elicits inside-out signaling by the integrin, leading to a high affinity of αIIbβ3 for circulating fibrinogen. We found that platelets from p53−/− mice are less sensitive to PAR4 agonist-stimulated fibrinogen binding than p53+/+ platelets (Fig. 5). In particular, responses to 65–80 μM AYPGKF were significantly higher in p53+/+ relative to p53−/− platelets. The decreased sensitivity of p53−/− platelets to agonist-induced fibrinogen binding is not due to decreased expression of αIIbβ3, since this is equivalent in p53+/+ and
Fig. 5.
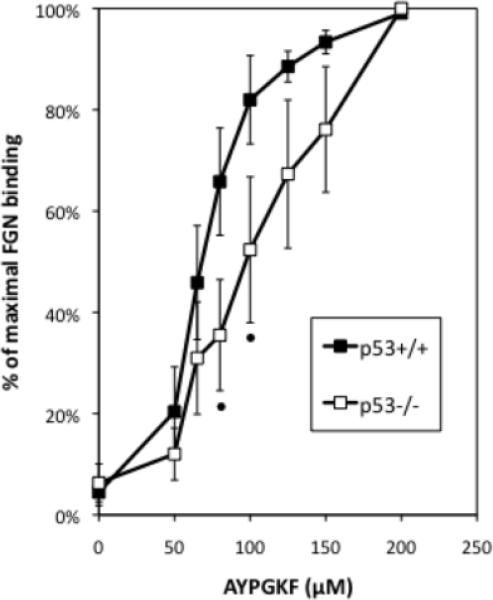
p53−/− platelets are less sensitive to PAR-4 agonist-induced fibrinogen binding in vitro. % of maximal binding to fluorescently-conjugated fibrinogen by p53+/+ and p53−/− platelets under a range of AYPGKF concentrations assayed by flow cytometry. N=5. Error bars: SEM. Statistical analysis was conducted using a 2-tailed paired Student t-test; • indicates P = 0.01.
p53−/− mouse platelets, as determined by flow cytometric measurement of αIIb expression [38] (p53+/+: 6,500±300 versus p53−/−: 6,900±300, N=9). Furthermore, in a static adhesion assay of AYPGKF- or PMA-stimulated platelets, p53+/+ and p53−/− platelet activation responses were similar judging by the extension of lamellipodia, and spreading of the flattened platelets on the fibrinogen-coated glass surface (Fig. S3). These findings suggest that there is no defect in outside-in signaling in p53−/− platelets, but that it is likely either a specific defect in inside-out signaling or granule secretion in p53−/− platelets that contributes to the hemostatic defect in these mice.
p53−/− platelets are less sensitive to a PAR-4 agonist-induced secretion of P selectin (CD62P) on their surface than p53+/+ platelets
Because some defects in sensitivity to agonist-induced fibrinogen binding are attributable to secretion defects, we examined the ability of isolated p53−/− platelets to undergo degranulation and expose P-selectin, the α granule component, on their surface following exposure to the PAR4 agonist AYPGKF [37]. p53−/− platelets are less sensitive than p53+/+ platelets to PAR-4 stimulated P selectin exposure (Fig. 6). This finding suggests a defect in PAR4-agonist-dependent α granule secretion in platelets lacking p53.
Fig. 6.
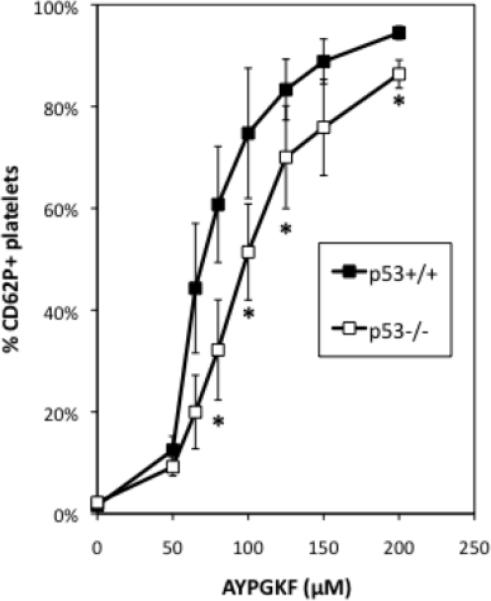
p53−/− platelets are less sensitive to PAR-4 agonist-induced P selectin (CD62P) secretion onto their surface. Display of secreted P selectin on the surface of p53+/+ and p53−/− platelets after stimulation with a range of AYPGKF concentrations assayed by flow cytometry. N=5, error bars: SEM. Statistical analysis was conducted using a 2-tailed paired Student t-test. * indicates P = 0.05.
Genes coding for platelet integrin, cytoskeletal and α granule components are differentially regulated in p53-KD versus control CHRF cells
In order to shed more light into platelet components that could be affected directly or indirectly by p53 and participate in the hemostatic response, we carried out a microarray-based transcriptional analysis comparing p53-KD and control megakaryoblastic CHRF cells induced by PMA to undergo terminal Mk differentiation [7]. The decision to employ a human cell line rather than primary murine cells was based on several considerations. First, because we ultimately care to understand megakaryopoiesis and thrombopoiesis in humans, we argued that since the impact of p53 on megakaryopoiesis has been observed in both human and mouse Mk cells [7], any systemic effects of p53 on platelet-related genes would likely be preserved between the human and murine Mk cells. Second, there is a greater database of microarray data amassed for human megakaryopoiesis [6, 8, 9, 25]. Thus, given the extensive validation of the CHRF cell line as a sound model of human megakaryopoiesis [9], we chose to compare the transcriptional profile of p53-KD CHRF cells to that of control CHRF cells [7]. A list of platelet-related genes was curated based on published literature data [6, 9, 31–33] and differentially expressed genes were identified using statistical analysis of microarrays [34]. Thus, a number of genes coding for proteins important in hemostasis, but not known to carry p53-responsive sequences in their promoter regions, were differentially regulated (Fig. 7). Two genes coding for G-protein-coupled receptors (SUCNR1, P2RY1) and CCL5, coding for RANTES, a component of platelet α granules, were strongly up-regulated in p53-KD CHRF cells. Genes that were down-regulated in p53-KD CHRF cells code for platelet surface integrins (ITGB3 (CD61), GP5 (CD42d), ITGA2B (CD41)) and components of platelet α granules (SELP (P selectin), SPARC, LTBP, PPBP). Fibrinogen, von Willebrand Factor (VWF) and collagen receptors on the platelet surface are complexes with roles in mediating platelet adhesion and aggregation. The fibrinogen receptor, composed of integrins CD41 and CD61, assumes the activated conformation upon agonist stimulation, allowing ligand binding (eg., fibrinogen, VWF, fibronectin and vitronectin) [39].
Fig. 7.
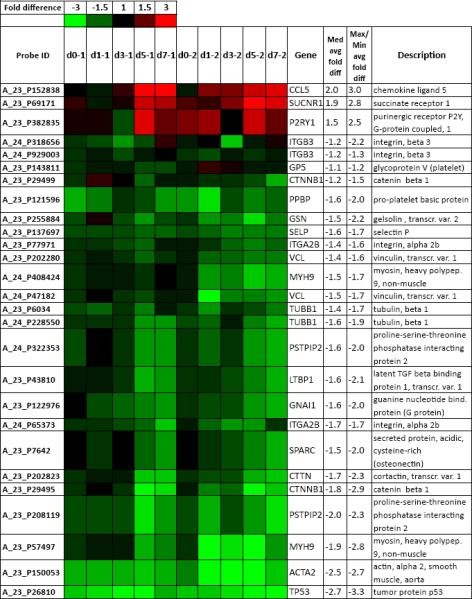
Differentially expressed Mk/platelet-related genes coding for components of platelet surface receptors, α granules and components of the platelet cytoskeleton. Rows show the probe ID on the Agilent 4x44K whole human genome microarray; expression ratios between p53-KD and control CHRF on day 0 (unstimulated) and days 1, 3, 5 and 7 after PMA stimulation for two biological replicates; the gene name; average median and average maximal/minimal fold difference; and gene description. Saturated red indicates 3-fold up-regulation in p53-KD cells relative to control cells, while saturated green indicates 3-fold down-regulation in p53-KD cells relative to control cells.
In addition, components of the platelet or Mk proplatelet cytoskeleton that are essential in platelet functions (MYH9, GSN, CTNNB1, ACTA2, VCL, TUBB1, CTTN, PSTPIP2) were found to be down-regulated in p53-KD CHRF cells (Fig. 7). Also found among the down-regulated genes were two p53 targets: a gene coding for a Gi-family subunit (GNAI1) and a gene coding for a protein kinase C isoform, essential in dense granule secretion (PRKCA) [40]. Microarray findings for ITGA2B, SELP and MYH9 were validated using Real Time PCR (Fig. S4). Our microarray findings suggest the possibility that genes coding for proteins important in platelet activation are indirectly regulated by p53. The differential regulation of these important platelet components in p53-KD compared to control CHRF cells is in line with the finding of altered platelet functionality leading to hemostasis defects in p53−/− mice. Future work will examine the possibility of regulation of these platelet components by p53 in megakaryocytes and platelets.
Discussion
High expression of the p53 protein in both diploid and polyploid primary human Mk cells has been previously reported [41], but the implications have remained unclear. Indeed, the process of polyploidization during terminal Mk differentiation could activate p53 potentially as a result of cellular stress and DNA damage, when the cells fail to complete cytokinesis and revert to G1 and S phases. Prompted by our preliminary findings on the activation of the p53 transcriptional network over the course of Mk differentiation in culture [6, 8–10] and the finding that p53 knock-down leads to enhanced polyploidization during Mk differentiation ex-vivo [7], we set out to characterize the effect of p53 loss on megakaryopoiesis and platelet production, and hemostasis in vivo.
In vivo versus in vitro megakaryopoiesis in the absence of p53
We were unable to find differences in Mk content in the BM or spleen, BM Mk ploidy or platelet counts between p53+/+ and p53−/− mice (Table 1 and data not shown). By contrast, when p53−/− BM cells were cultured with Tpo, they reached substantially higher levels of Mk polyploidy than p53+/+ Mk cells (Fig. 1), which was due in part to enhanced DNA synthesis, especially in the polyploid compartment and in part to diminished apoptosis (Figs. 1–2). Indeed, in several cases, p53−/− Mk cells were able to reach ploidy classes of up to 256N by day 6 of culture with Tpo (Fig. 1). A role for p53 as a regulator of DNA synthesis in Mk cells is consistent with an earlier finding that knock-down of p53 could increase Mk colony formation in cultures of peripheral blood mononuclear cells [42]. In an attempt to emulate in vivo the hyperploid p53−/− Mk phenotype observed in vitro, we supplied Tpo to induce Mk polyploidization and thrombocytosis [21, 43]. Five days following treatment, we observed increased p53−/− Mk polyploidy compared to p53+/+ Mk cells; however, platelet numbers were similar in p53+/+ and p53−/− mice (Fig. 3). We next evaluated the ability of p53−/− Mk cells to restore normal platelet levels in vivo following induced thrombocytopenia [44]. To our surprise, the kinetics of platelet production and the extent of Mk polyploidization were almost identical in p53+/+ and p53−/− mice (Fig. S1). However, when BM cells from p53−/− mice treated with either Tpo or anti-CD41 were put in culture, they always reached significantly higher ploidy classes than Mk cells derived from p53+/+ BM cells (data not shown).
The results above evoke the effects on Mk differentiation of p21, a p53-dependent cyclin-dependent kinase inhibitor, and survivin, a chromosome passenger protein that functions in the spindle assembly checkpoint and cytokinesis. p21−/− [41] and survivin-deficient [45] or -over-expressing [46] mice have normal steady state Mk development and platelet levels. Nevertheless, loss of p21 has been linked to enhanced polyploidization in vitro [47]. Furthermore, over-expression of survivin in vitro has been shown to arrest polyploidization [48], while its loss in vitro has been shown to promote polyploidization [45]. Considering that earlier studies in non-Mk cells have highlighted the necessity of deletion of both p53 and survivin for the spontaneous induction of polyploidization [49, 50], it is tempting to speculate that loss of both cell cycle regulators would effect a severely hyperploid Mk phenotype even in vivo. Additionally, pharmacological inhibition of Aurora B, another chromosome passenger protein, in Mk primary cell cultures, was recently shown to impact endomitosis leading to lower DNA synthesis mainly in 2N and 4N Mk cells, lower Mk progenitor proliferation and simultaneous induction of p53 [51]. Another possibility is that c-Myc, an oncogene whose over-expression is by itself sufficient to induce polyploidization in cancer cells in the absence of p53 [52], is repressed by p53 [53]. Notably, c-Myc−/− mice have pronounced Mk and platelets defects, such as increased BM Mk content, decreased Mk polyploidization and significantly increased thrombocytosis presumably due to the 4-fold increase of Mk progenitor cells in c-Myc−/− mice [54]. It is also possible that age-related [2] or gender-related [19] differences or systemic compensatory mechanisms, such as splenic sequestration of platelets [55], activation of p63 and p73 members of the p53 family in the absence of p53 [56] or even polyploid Mk cell progression into cytokinesis [57] could mask the effect of p53, p21, or other modulators on individual steps of megakaryopoiesis in vivo. Conceivably, the use of Mk/platelet-specific p53 knock-out mice might alleviate systemic compensation.
Impact of p53 loss on platelet function and hemostasis
A very recent publication has provided an indirect link between impaired hemostasis and low levels of p53 in the context of Mk cells: it was demonstrated that mice lacking the PER2 gene exhibit increased bleeding times, delayed clot retraction and attenuated aggregation and secretion, while their Mk cells evade apoptosis and have lower p53 levels [58]. In our study, platelets from p53−/− mice appear to be less efficient in maintaining hemostatic plugs, as indicated by prolonged bleeding upon standardized injury (Fig. 4). Moreover, p53−/− platelets are less sensitive to agonist-mediated activation in binding fibrinogen (Fig. 5) or secreting P selectin from α granules to their surface (Fig. 6). Because spreading on fibrinogen-covered surfaces (Fig. S3) and αIIbβ3 expression were normal in p53−/− platelets, our data suggest a specific defect in inside-out signaling or degranulation. To our knowledge, this is the first study indicating a direct link between hemostatic deficiencies and the absence of p53. Our findings may in part be explained by p53-mediated direct or indirect regulation in the context of terminal Mk differentiation of a set of genes coding for platelet receptors and components of the platelet cytoskeleton and α granules. For instance, ACTA2, a p53 target coding for actin A2, and MYH9, coding for myosin IIA, are both down-regulated in p53-KD CHRF cells undergoing Mk differentiation. Actin associates with myosin to form actomyosin fibers participating in platelet contraction, which has been speculated to be responsible not only for platelet shape change but also for release of the platelet granule contents [59]. Moreover, p53-mediated down-regulation of other actin-related cytoskeletal components, such as GSN or CTTN, coding for gelsolin and cortactin, respectively, may be in part responsible for defects in platelet activation. Indeed, gelsolin participates in actin remodeling [60] and its loss leads to attenuated shape change of activated platelets and prolonged tail-bleeding times [61], while cortactin has roles in actin remodeling during platelet activation [62]. While p53−/− platelet shape change and actin polymerization during fibrinogen spreading appeared normal, we cannot exclude the possibility that the downregulation of actin and myosin-related components in p53-KD CHRF cells is associated with increased Mk polyploidization as CHRF cells grow adherent; actomyosin inhibition was recently linked to increased Mk polyploidization and proplatelet extension [63]. Additionally, low levels of Mk/platelet-specific β1 tubulin in p53-KD CHRF cells may suggest defects in Mk proplatelet formation resulting in attenuated packaging of granules and other organelles into the nascent platelets [64, 65].
In summary, we have demonstrated that p53−/− platelets exhibit an impaired response to agonist-induced activation of the fibrinogen receptor and secretion of P selectin on their surface. This finding may explain the longer tail bleeding times observed in p53−/− mice. Additional studies are required to determine if p53 loss affects the platelet structure and the composition of the α granules and to address the possibility of p53 affecting platelet function through non-transcriptional pathways.
Supplementary Material
Effects of induced immune thrombocytopenia in platelets and megakaryocytes of p53+/+ and p53−/− mice. (A) Platelet recovery (left axis, black lines) in p53+/+ and p53−/− mice following anti-CD41 induced thrombocytopenia. Percent reticulated platelets over the course of platelet recovery (right axis, grey lines) in p53+/+ and p53−/− mice. Error bars: SEM, N=11–14 for day 0, N=8 for days 1–2 and N=5 for days 3–5. Percent of bone marrow-resident megakaryocytes (B) and megakaryocytic polyploidization (C) in p53+/+ and p53−/− mice 5 days following anti-CD41 induced thrombocytopenia. Error bars: SEM, N=5.
Western blot showing expression of p53 protein in platelets. Platelet-rich-plasma from p53−/− mice and p53+/+ littermates was pelleted and lysed in 1× Laemmli buffer. The lysates were incubated overnight with anti-p53 (1:1000, clone: PAb421; Calbiochem; San Diego, CA). Staining with an antibody against pan-actin (Cell Signaling; Danvers, MA) was used for loading control.
Static adhesion assay of AYPGKF- and PMA-stimulated p53+/+ and p53−/− platelets on fibrinogen-coated glass coverslips. Gel-filtered p53+/+ and p53−/− platelets were stimulated with 50 μM AYPGKF or 10 μM PMA and layered on a fibrinogen-coated coverslips at 37°C for 5, 15 or 30 min and processed for staining with phalloidin. One biological replicate is shown, N=3.
Validation of microarray finding using Real Time PCR. Gene expression for ITGA2B (CD41), SELP (P selectin) and MYH9 (non-muscle myosin IIA) in p53-knockdown and control CHRF cells on days 1, 3, 5 and 7 after PMA stimulation. N=2, error bars: SEM.
Acknowledgements
P.A.A., D.S.W., W.M.M. and E.T.P. designed the study, wrote the manuscript and analyzed the data. P.A.A. and M.C. performed the research. This work was supported in part by funding to E.T.P. through the Delaware Biotechnology Institute and NIH grant RO1 HL081241 to D.S.W. P.A.A. acknowledges personal funding through the Delaware Biotechnology Institute and the A.S. Onassis foundation. The authors acknowledge use of the Bioimaging Center (Dr. Jeffrey Kaplan) and the Center for Translational Cancer Research, both at the University of Delaware, and the Histotechnology Core Facility at the A. I. duPont Hospital for Children (Wilmington, DE). Jinlin Jiang is acknowledged for help in image acquisition. The authors are thankful to Frank Warren, Sue Seta, Julie Mis and Jillian Hash from the University of Delaware Office of Laboratory Animal Medicine for expert animal care and help with various procedures. P.A.A. wishes to thank Prof. J.D. Crispino for helpful suggestions and Drs. Stephan Lindsey and J.C. Kostyak for helpful discussions over the course of the research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- [2].Dumble M, Moore L, Chambers SM, et al. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu Y, Elf SE, Miyata Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol. 2003;31:521–527. doi: 10.1016/s0301-472x(03)00072-9. [DOI] [PubMed] [Google Scholar]
- [5].Liu Y, Elf SE, Asai T, et al. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–3124. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen C, Fuhrken PG, Huang LT, et al. A systems-biology analysis of isogenic megakaryocytic and granulocytic cultures identifies new molecular components of megakaryocytic apoptosis. BMC Genomics. 2007;8:384. doi: 10.1186/1471-2164-8-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fuhrken PG, Apostolidis PA, Lindsey S, Miller WM, Papoutsakis ET. Tumor suppressor protein p53 regulates megakaryocytic polyploidization and apoptosis. J Biol Chem. 2008;283:15589–15600. doi: 10.1074/jbc.M801923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fuhrken PG, Chen C, Apostolidis PA, Wang M, Miller WM, Papoutsakis ET. Gene Ontology-driven transcriptional analysis of CD34+ cell-initiated megakaryocytic cultures identifies new transcriptional regulators of megakaryopoiesis. Physiol Genomics. 2008;33:159–169. doi: 10.1152/physiolgenomics.00127.2007. [DOI] [PubMed] [Google Scholar]
- [9].Fuhrken PG, Chen C, Miller WM, Papoutsakis ET. Comparative, genome-scale transcriptional analysis of CHRF-288-11 and primary human megakaryocytic cell cultures provides novel insights into lineage-specific differentiation. Exp Hematol. 2007;35:476–489. doi: 10.1016/j.exphem.2006.10.017. [DOI] [PubMed] [Google Scholar]
- [10].Giammona LM, Panuganti S, Kemper JM, et al. Mechanistic studies on the effects of nicotinamide on megakaryocytic polyploidization and the roles of NAD+ levels and SIRT inhibition. Exp Hematol. 2009;37:1340–1352. e1343. doi: 10.1016/j.exphem.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ritchie A, Braun SE, He J, Broxmeyer HE. Thrombopoietin-induced conformational change in p53 lies downstream of the p44/p42 mitogen activated protein kinase cascade in the human growth factor-dependent cell line M07e. Oncogene. 1999;18:1465–1477. doi: 10.1038/sj.onc.1202439. [DOI] [PubMed] [Google Scholar]
- [12].Ritchie A, Vadhan-Raj S, Broxmeyer HE. Thrombopoietin suppresses apoptosis and behaves as a survival factor for the human growth factor-dependent cell line, M07e. Stem Cells. 1996;14:330–336. doi: 10.1002/stem.140330. [DOI] [PubMed] [Google Scholar]
- [13].Sigurjonsson OE, Gudmundsson KO, Haraldsdottir V, Rafnar T, Agnarsson BA, Gudmundsson S. Flt3/Flk-2 ligand in combination with thrombopoietin decreases apoptosis in megakaryocyte development. Stem Cells Dev. 2004;13:183–191. doi: 10.1089/154732804323046783. [DOI] [PubMed] [Google Scholar]
- [14].Wlodarski P, Wasik M, Ratajczak MZ, et al. Role of p53 in hematopoietic recovery after cytotoxic treatment. Blood. 1998;91:2998–3006. [PubMed] [Google Scholar]
- [15].Horie K, Kubo K, Yonezawa M. p53 dependency of radio-adaptive responses in endogenous spleen colonies and peripheral blood-cell counts in C57BL mice. J Radiat Res (Tokyo) 2002;43:353–360. doi: 10.1269/jrr.43.353. [DOI] [PubMed] [Google Scholar]
- [16].Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- [17].Barlow JL, Drynan LF, Hewett DR, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med. 2010;16:59–66. doi: 10.1038/nm.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pestina TI, Cleveland JL, Yang C, Zambetti GP, Jackson CW. Mpl ligand prevents lethal myelosuppression by inhibiting p53-dependent apoptosis. Blood. 2001;98:2084–2090. doi: 10.1182/blood.v98.7.2084. [DOI] [PubMed] [Google Scholar]
- [19].Eidelman O, Jozwik C, Huang W, et al. Gender dependence for a subset of the low-abundance signaling proteome in human platelets. Hum Genomics Proteomics. 2010;2010:164906. doi: 10.4061/2010/164906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood. 2006;108:1509–1514. doi: 10.1182/blood-2005-11-011957. [DOI] [PubMed] [Google Scholar]
- [21].Arnold JT, Daw NC, Stenberg PE, Jayawardene D, Srivastava DK, Jackson CW. A single injection of pegylated murine megakaryocyte growth and development factor (MGDF) into mice is sufficient to produce a profound stimulation of megakaryocyte frequency, size, and ploidization. Blood. 1997;89:823–833. [PubMed] [Google Scholar]
- [22].Hitchcock IS, Fox NE, Prevost N, Sear K, Shattil SJ, Kaushansky K. Roles of focal adhesion kinase (FAK) in megakaryopoiesis and platelet function: studies using a megakaryocyte lineage specific FAK knockout. Blood. 2008;111:596–604. doi: 10.1182/blood-2007-05-089680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lindsey S, Papoutsakis ET. The aryl hydrocarbon receptor (AHR) transcription factor regulates megakaryocytic polyploidization. Br J Haematol. 2011;152:469–484. doi: 10.1111/j.1365-2141.2010.08548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kostyak JC, Naik UP. Calcium- and integrin-binding protein 1 regulates endomitosis and its interaction with Polo-like kinase 3 is enhanced in endomitotic Dami cells. PLoS One. 2011;6:e14513. doi: 10.1371/journal.pone.0014513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Giammona LM, Fuhrken PG, Papoutsakis ET, Miller WM. Nicotinamide (vitamin B3) increases the polyploidisation and proplatelet formation of cultured primary human megakaryocytes. Br J Haematol. 2006;135:554–566. doi: 10.1111/j.1365-2141.2006.06341.x. [DOI] [PubMed] [Google Scholar]
- [26].Gilles L, Guieze R, Bluteau D, et al. P19INK4D links endomitotic arrest and megakaryocyte maturation and is regulated by AML-1. Blood. 2008;111:4081–4091. doi: 10.1182/blood-2007-09-113266. [DOI] [PubMed] [Google Scholar]
- [27].Tucker KL, Sage T, Stevens JM, et al. A dual role for integrin-linked kinase in platelets: regulating integrin function and alpha-granule secretion. Blood. 2008;112:4523–4531. doi: 10.1182/blood-2008-03-148502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Prevost N, Woulfe DS, Jiang H, et al. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc Natl Acad Sci U S A. 2005;102:9820–9825. doi: 10.1073/pnas.0404065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang F, Jia Y, Cohen I. Fibronectin- and protein kinase C-mediated activation of ERK/MAPK are essential for proplateletlike formation. Blood. 2002;99:3579–3584. doi: 10.1182/blood.v99.10.3579. [DOI] [PubMed] [Google Scholar]
- [30].Yang H, Haddad H, Tomas C, Alsaker K, Papoutsakis ET. A segmental nearest neighbor normalization and gene identification method gives superior results for DNA-array analysis. Proc Natl Acad Sci U S A. 2003;100:1122–1127. doi: 10.1073/pnas.0237337100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mercher T, Cornejo MG, Sears C, et al. Notch signaling specifies megakaryocyte development from hematopoietic stem cells. Cell Stem Cell. 2008;3:314–326. doi: 10.1016/j.stem.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chang Y, Bluteau D, Debili N, Vainchenker W. From hematopoietic stem cells to platelets. J Thromb Haemost. 2007;5(Suppl 1):318–327. doi: 10.1111/j.1538-7836.2007.02472.x. [DOI] [PubMed] [Google Scholar]
- [33].Chen Z, Hu M, Shivdasani RA. Expression analysis of primary mouse megakaryocyte differentiation and its application in identifying stage-specific molecular markers and a novel transcriptional target of NF-E2. Blood. 2007;109:1451–1459. doi: 10.1182/blood-2006-08-038901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- [35].Muntean AG, Pang L, Poncz M, Dowdy SF, Blobel GA, Crispino JD. Cyclin DCdk4 is regulated by GATA-1 and required for megakaryocyte growth and polyploidization. Blood. 2007;109:5199–5207. doi: 10.1182/blood-2006-11-059378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jirouskova M, Shet AS, Johnson GJ. A guide to murine platelet structure, function, assays, and genetic alterations. J Thromb Haemost. 2007;5:661–669. doi: 10.1111/j.1538-7836.2007.02407.x. [DOI] [PubMed] [Google Scholar]
- [37].Nieswandt B, Schulte V, Bergmeier W. Flow-cytometric analysis of mouse platelet function. Methods Mol Biol. 2004;272:255–268. doi: 10.1385/1-59259-782-3:255. [DOI] [PubMed] [Google Scholar]
- [38].Goodall AH, Appleby J. Flow-cytometric analysis of platelet-membrane glycoprotein expression and platelet activation. Methods Mol Biol. 2004;272:225–253. doi: 10.1385/1-59259-782-3:225. [DOI] [PubMed] [Google Scholar]
- [39].Shattil SJ. Signaling through platelet integrin alpha IIb beta 3: inside-out, outside-in, and sideways. Thromb Haemost. 1999;82:318–325. [PubMed] [Google Scholar]
- [40].Konopatskaya O, Gilio K, Harper MT, et al. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Baccini V, Roy L, Vitrat N, et al. Role of p21(Cip1/Waf1) in cell-cycle exit of endomitotic megakaryocytes. Blood. 2001;98:3274–3282. doi: 10.1182/blood.v98.12.3274. [DOI] [PubMed] [Google Scholar]
- [42].Mahdi T, Brizard A, Millet C, Dore P, Tanzer J, Kitzis A. In vitro p53 and/or Rb antisense oligonucleotide treatment in association with growth factors induces the proliferation of peripheral hematopoietic progenitors. J Cell Sci. 1995;108(Pt 3):1287–1293. doi: 10.1242/jcs.108.3.1287. [DOI] [PubMed] [Google Scholar]
- [43].Luoh SM, Stefanich E, Solar G, et al. Role of the distal half of the c-Mpl intracellular domain in control of platelet production by thrombopoietin in vivo. Mol Cell Biol. 2000;20:507–515. doi: 10.1128/mcb.20.2.507-515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hu Z, Slayton WB, Rimsza LM, Bailey M, Sallmon H, Sola-Visner MC. Differences between newborn and adult mice in their response to immune thrombocytopenia. Neonatology. 2010;98:100–108. doi: 10.1159/000280413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wen Q, Leung C, Huang Z, et al. Survivin is not required for the endomitotic cell cycle of megakaryocytes. Blood. 2009;114:153–156. doi: 10.1182/blood-2008-11-190801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McCrann DJ, Yezefski T, Nguyen HG, et al. Survivin overexpression alone does not alter megakaryocyte ploidy nor interfere with erythroid/megakaryocytic lineage development in transgenic mice. Blood. 2008;111:4092–4095. doi: 10.1182/blood-2007-11-122150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mantel C, Braun SE, Reid S, et al. p21(cip-1/waf-1) deficiency causes deformed nuclear architecture, centriole overduplication, polyploidy, and relaxed microtubule damage checkpoints in human hematopoietic cells. Blood. 1999;93:1390–1398. [PubMed] [Google Scholar]
- [48].Gurbuxani S, Xu Y, Keerthivasan G, Wickrema A, Crispino JD. Differential requirements for survivin in hematopoietic cell development. Proc Natl Acad Sci U S A. 2005;102:11480–11485. doi: 10.1073/pnas.0500303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yang D, Welm A, Bishop JM. Cell division and cell survival in the absence of survivin. Proc Natl Acad Sci U S A. 2004;101:15100–15105. doi: 10.1073/pnas.0406665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Beltrami E, Plescia J, Wilkinson JC, Duckett CS, Altieri DC. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J Biol Chem. 2004;279:2077–2084. doi: 10.1074/jbc.M309479200. [DOI] [PubMed] [Google Scholar]
- [51].Lordier L, Chang Y, Jalil A, et al. Aurora B is dispensable for megakaryocyte polyploidization, but contributes to the endomitotic process. Blood. 2010 doi: 10.1182/blood-2010-01-265785. [DOI] [PubMed] [Google Scholar]
- [52].Wu Q, Xu FL, Li Y, Prochownik EV, Saunders WS. The c-Myc target Glycoprotein1balpha links cytokinesis failure to oncogenic signal transduction pathways in cultured human cells. PLoS One. 2010;5:e10819. doi: 10.1371/journal.pone.0010819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sachdeva M, Zhu S, Wu F, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Guo Y, Niu C, Breslin P, et al. c-Myc-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Blood. 2009;114:2097–2106. doi: 10.1182/blood-2009-01-197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tiedt R, Coers J, Ziegler S, et al. Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood. 2009;113:1768–1777. doi: 10.1182/blood-2008-03-146084. [DOI] [PubMed] [Google Scholar]
- [56].Talos F, Moll UM. Role of the p53 family in stabilizing the genome and preventing polyploidization. Adv Exp Med Biol. 2010;676:73–91. doi: 10.1007/978-1-4419-6199-0_5. [DOI] [PubMed] [Google Scholar]
- [57].Leysi-Derilou Y, Robert A, Duchesne C, Garnier A, Boyer L, Pineault N. Polyploid megakaryocytes can complete cytokinesis. Cell Cycle. 2010:9. doi: 10.4161/cc.9.13.12078. [DOI] [PubMed] [Google Scholar]
- [58].Zhao Y, Zhang Y, Wang S, Hua Z, Zhang J. The clock gene Per2 is required for normal platelet formation and function. Thromb Res. 2010 doi: 10.1016/j.thromres.2010.11.025. [DOI] [PubMed] [Google Scholar]
- [59].Leon C, Eckly A, Hechler B, et al. Megakaryocyte-restricted MYH9 inactivation dramatically affects hemostasis while preserving platelet aggregation and secretion. Blood. 2007;110:3183–3191. doi: 10.1182/blood-2007-03-080184. [DOI] [PubMed] [Google Scholar]
- [60].Sun HQ, Yamamoto M, Mejillano M, Yin HL. Gelsolin, a multifunctional actin regulatory protein. J Biol Chem. 1999;274:33179–33182. doi: 10.1074/jbc.274.47.33179. [DOI] [PubMed] [Google Scholar]
- [61].Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- [62].Huang C, Tandon NN, Greco NJ, Ni Y, Wang T, Zhan X. Proteolysis of platelet cortactin by calpain. J Biol Chem. 1997;272:19248–19252. doi: 10.1074/jbc.272.31.19248. [DOI] [PubMed] [Google Scholar]
- [63].Shin JW, Swift J, Spinler KR, Discher DE. Myosin-II inhibition and soft 2D matrix maximize multinucleation and cellular projections typical of platelet-producing megakaryocytes. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1017474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Schwer HD, Lecine P, Tiwari S, Italiano JE, Jr., Hartwig JH, Shivdasani RA. A lineage-restricted and divergent beta-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr Biol. 2001;11:579–586. doi: 10.1016/s0960-9822(01)00153-1. [DOI] [PubMed] [Google Scholar]
- [65].Patel SR, Richardson JL, Schulze H, et al. Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood. 2005;106:4076–4085. doi: 10.1182/blood-2005-06-2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of induced immune thrombocytopenia in platelets and megakaryocytes of p53+/+ and p53−/− mice. (A) Platelet recovery (left axis, black lines) in p53+/+ and p53−/− mice following anti-CD41 induced thrombocytopenia. Percent reticulated platelets over the course of platelet recovery (right axis, grey lines) in p53+/+ and p53−/− mice. Error bars: SEM, N=11–14 for day 0, N=8 for days 1–2 and N=5 for days 3–5. Percent of bone marrow-resident megakaryocytes (B) and megakaryocytic polyploidization (C) in p53+/+ and p53−/− mice 5 days following anti-CD41 induced thrombocytopenia. Error bars: SEM, N=5.
Western blot showing expression of p53 protein in platelets. Platelet-rich-plasma from p53−/− mice and p53+/+ littermates was pelleted and lysed in 1× Laemmli buffer. The lysates were incubated overnight with anti-p53 (1:1000, clone: PAb421; Calbiochem; San Diego, CA). Staining with an antibody against pan-actin (Cell Signaling; Danvers, MA) was used for loading control.
Static adhesion assay of AYPGKF- and PMA-stimulated p53+/+ and p53−/− platelets on fibrinogen-coated glass coverslips. Gel-filtered p53+/+ and p53−/− platelets were stimulated with 50 μM AYPGKF or 10 μM PMA and layered on a fibrinogen-coated coverslips at 37°C for 5, 15 or 30 min and processed for staining with phalloidin. One biological replicate is shown, N=3.
Validation of microarray finding using Real Time PCR. Gene expression for ITGA2B (CD41), SELP (P selectin) and MYH9 (non-muscle myosin IIA) in p53-knockdown and control CHRF cells on days 1, 3, 5 and 7 after PMA stimulation. N=2, error bars: SEM.