

Abstract
Introduction
MetMAb (OA-5D5) is a one-armed monoclonal antibody developed to bind to and inhibit c-MET receptor tyrosine kinase. Though early in clinical testing, this agent holds great promise in diseases thought to be driven by c-MET activation, as evidenced by the phase II results in non-small cell lung cancer, (NSCLC) where a benefit in overall survival was observed in patients with MET diagnostic positive disease. Thus far, both alone and in combination with other targeted agents, this drug has been well tolerated and no new significant safety signals have been identified.
Areas covered
The review summarizes the structure and function of the c-MET receptor and its ligand HGF, provides an overview of select targeted monotherapies developed to interfere in the MET-HGF signaling pathway, discusses pre-clinical and clinical data surrounding MetMAb, and concludes with an expert opinion regarding this novel agent.
Expert opinion
MetMAb has been well tolerated and based on phase II data testing it, in combination with erlotinib in advanced NSCLC, may have a role in improving survival in patients with disease driven by c-MET activation. However, phase III validation is underway and the results of these studies will help elucidate which patients will benefit most from this novel agent.
Keywords: MetMAb, c-MET, HGF, targeted therapy, monoclonal antibody, personalized medicine, non-small cell lung cancer
1. Introduction
With an understanding of the human genome and the technology to efficiently characterize genetic profiles of individual tumors, clinicians are now poised to match cancer therapy to the unique characteristics of malignant tumors [1]. The promise of molecular-targeted therapy is that dysregulated proteins are preferentially impacted, resulting in an improved therapeutic index over standard chemotherapy [2]. By identifying the patients that will benefit most from targeted therapies, personalized therapy is anticipated to improve treatment efficacy and reduce cost.
Though there are a number of challenges facing an individualized approach, successes such as matching treatment to the presence of the HER2 receptor in breast cancer [3] or BCR-ABL gene fusion in chronic myelogenous leukemia, have generated interest in identifying such a target in lung cancer. Despite advances in therapy, the 5-year overall survival for lung cancer still remains approximately 15% [4]. Major discoveries, such as EFGR receptor inhibition in EGFR mutant lung cancer, and the recent emergence of ALK inhibition in ALK translocation, are still limited in their impact, given that these two aberrations account for less than 20% of NSCLC. Several receptor tyrosine kinases have also been implicated in non-small cell lung cancer (NSCLC), and investigations into inhibiting one such receptor tyrosine kinase, c-MET, may be promising [5].
2. c-MET Receptor Tyrosine Kinase
Met was first identified as an activated oncogene, following the treatment of a human osteogenic sarcoma cell line with the carcinogen N-methyl-N′-nitro-N-nitrosoguanidine [6]. This resulted in a translocation placing a promoter region locus (TPR) on chromosome 1 adjacent to Met located on chromosome 7. The resultant TPR-MET fusion protein demonstrated constitutively activated MET TK activity [7]. Subsequent research has shown constitutive activation of c-MET to be implicated in a number of human cancers [For reviews see 7, 8]. In addition, c-MET can be activated following binding to its ligand, hepatic growth factor (HGF).
2.1 Structure of c-MET and HGF
c-MET is the prototypic member of a structurally unique subfamily of RTK [9]. The human Met gene is located on chromosome 7 band 7q21-q31 and spans 120kb. The Mr 170,000 precursor to c-MET is cleaved into a Mr 50,000 extracellular α chain and a Mr 140,000 membrane-spanning β chain [10] which are linked by disulfide bonds. The extracellular portion of the β chain of c-MET contains a semaphorin (Sema) domain, a 500 amino acid cysteine-rich sequence near the N-terminus [7, 11]. In addition, it contains a PSI domain (in plexins, semaphorins, and integrins) and four IPT repeats (in immunoglobulins, plexins, and transcription factors). c-MET also contains a transmembrane (TM) domain, a juxtamembrane (JM) domain, a tyrosine kinase (TK) domain, and a carboxy-terminal tail region [11] (Figure 1).
Figure 1.
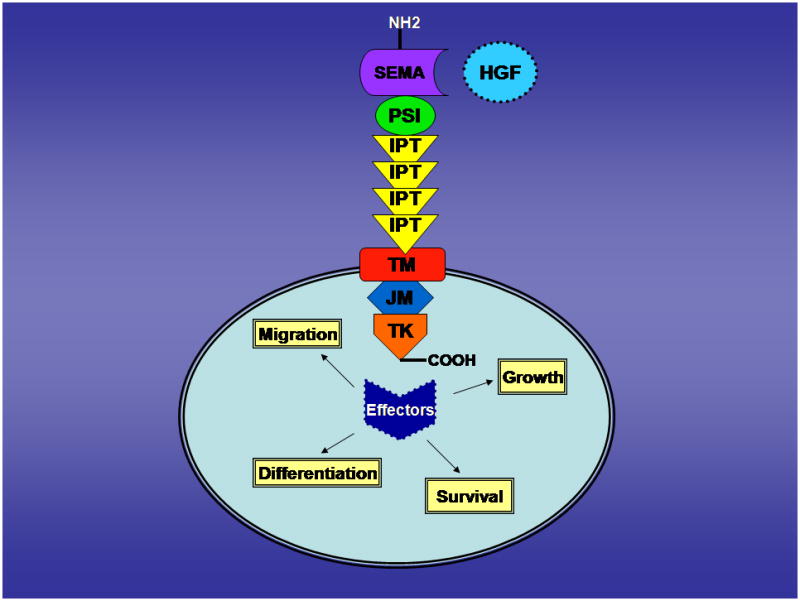
The extracellular domain serves as a high-affinity receptor for HGF, which is produced by stromal and mesenchymal cells. Binding of HGF induces autophosphorylation of tyrosine residues within the activating loop of the TK domain (Y1230/Y1234/Y1235). In turn, phosphorylation of Y1349 and Y1356, near the COOH terminus results in c-MET dimerization and the formation of a multifunctional docking site for adapter proteins such as Grb2, Gab1, PI3K, phospholipase C-γ, Shc, Src, Shp2, Ship1 [12, 13] thereby activating the intrinsic kinase activity of c-MET.
HGF is secreted as an inactive monomer of 82kD, and is cleaved by urokinase type plasminogen activator (uPA) into a heterodimer of two disulfide-linked chains of 69 and 34 kD each [14]. The NH2-terminal fragment of HGF comprises the α-chain and contains the high-affinity c-MET receptor binding domain [Burgess]. High affinity binding has been shown to occur at IPT3 and IPT4 [15]. The COOH-terminal β-chain of HGF has a low-affinity binding domain which links to the Sema-domain of c-MET [16]. The HGF heterodimer has a high affinity for c-MET and is to date, the only identified ligand.
2.2 Function of HGF-MET
The receptor-ligand pair is involved in a wide variety of cellular signaling pathways and biological responses. The dyad promotes migration, cell growth, differentiation, angiogenesis, and survival — properties that are essential during normal processes such as morphogenesis and wound healing. c-MET plays a vital role in early development and Met knock-out in mice is embryonically lethal [17]. Furthermore, c-MET is upregulated after tissue injury to the kidney, liver, and heart, suggesting an important role in repair and regeneration [18].
These same functions play an essential role in malignancy [19]. c-MET activating mutations have been implicated in a number of solid tumors including papillary renal cell, ovarian, gastric, breast, hepatocellular, and head and neck squamous cell cancers [20]. High levels of c-MET expression have also been identified in both non-small cell lung cancer and small cell lung cancer [5,10]. One previous study showed that 67% of lung adenocarcinomas, 57% of large cell carcinomas, 57% of squamous cell carcinomas, and 25% of small cell lung cancers strongly expressed c-MET [5]. Missense mutations in the JM domain, such as R988C and T1010I, were identified in small cell lung cancer cell lines and these were associated with growth factor independence, increased cell motility, and enhanced tumorigenicity [10]. Specifically in the case of lung cancer, overexpression of c-MET may confer a poorer clinical prognosis and decreased overall survival [21].
Co-expression of c-MET and HGF in the same cell leads to activation of the autocrine loop and this has been found to be tumorigenic [19, 22]. Autocrine signaling is believed to generate deregulated cell growth and in turn, tumor formation [19]. Furthermore, HGF-MET signaling may induce uPA, leading to the production of more HGF and further stimulation of the pathway [19].
In addition to tumorigenesis, HGF-MET also has a role in invasion and metastasis. HGF was discovered as a stimulator of cell scattering by inducing the dissociation and mutual repulsion of epithelial cells [23]. Using in the NIH 3T3 cell line, HGF-MET signaling was found to result in more frequent lung metastasis than control cell lines using in vivo experimentation (19).
Furthermore, by acting directly on vascular endothelial cells, HGF-Met stimulation led to their proliferation and organization into capillary-like tubules, thereby inducing the angiogenesis necessary for tumorigenesis and metastasis [24]. In addition, HGF has been shown to promote the expression of angiogenic cytokines, IL-8 and VEGF, in head and neck cell lines [25].
3. Monoclonal antibody driven therapies
It is anticipated that interfering in the HGF-Met signaling pathway will impact the growth and spread of human cancers in which this pathway is influential. Targeted biological agents, specifically monoclonal antibodies (mAb), directed towards HGF-MET are being developed. These mAb have been developed in two varieties, anti-HGF and anti-MET. Select examples highlighting these two strategies will briefly be presented below.
3.1 Rilotumumab
Rilotumumab (AMG 102) is a fully human MAb (IgG2) developed by Amgen which specifically targets HGF. It binds to amino acid residues at the NH2-terminus of the β-chain of human HGF, with a preference for the mature, heterodimeric form [26]. Such binding led to the complete inhibition of c-MET autophosphorylation in vitro using human and monkey HGF [26]. Furthermore, cellular migration was inhibited with AMG102 in monkey HGF stimulated cells. Rilotumumab has been assessed in phase I and II studies, both as monotherapy and in combination. In a phase I study of patients with advanced solid tumors, there was evidence of good tolerability and a favorable pharmacokinetic profile, with a promising proportion of patients achieving stable disease in the setting of combination therapy [27]. However, in phase II testing in patients with recurrent glioblastoma, there was no significant OS or PFS benefit found with Rilotumumab monotherapy [28].
3.2 L2G7
Another anti-HGF mAb (IgG2a), L2G7, was developed by Galaxy Biotech. In in vitro studies, L2G7 was able to nearly completely inhibit and/or regress growth in U87 and U118 gliomas, cells which express both MET and secrete HGF. The same effect was not documented in U251 gliomas which do not secrete HGF [29]. Though predominantly studied in the context of brain cancers, studies have also been done to evaluate the effect of L2G7 in lung cancer. In HGF transgenic mice treated with tobacco carcinogen to induce lung cancer, mice treated with L2G7 developed statistically significant fewer lung tumors than control mice [30]. As there is compelling pre-clinical data for this agent, a phase 1 clinical trial is currently underway using the humanized form of L2G7 called TAK 701 (NCT00831896).
3.3 DN30
Conversely, research has also focused on developing mAb to target the c-MET receptor directly. DN30, developed by Metheresis, is a mAb targeting the extracellular domain of c-MET at a site distinct from the binding site of its HGF ligand [31]. Such binding leads to the proteolytic cleavage of the extracellular domain of the c-MET receptor, through a process known as shedding [32]. Subsequently, the intracellular domain is rapidly degraded by the proteosome. Treatment with DN30 led to significant c-MET down regulation, reduced signal transduction, decreased cell invasion, angiogenesis, and metastatic spread [32]. However, work with DN30 has been complicated by its bivalent structure, which results in partial agonism [31]. Additional efforts to isolate a monovalent form of DN30 to solely achieve receptor shedding have been successful. Treatment with a DN30-Fab fragment resulted in the reduction of MET phosphorylation and inhibition of tumor growth, suggesting that monovalency should be the focus of anti-MET therapy [33].
3.4 MetMAb
In contrast to the bivalent DN30, a one armed antibody (OA-5D5) was developed by Genentech, thereby preventing HGF-mediated activation of c-MET, and resulting in suppressed growth in multiple cancer models.
4. MetMAb Chemistry
MetMAb (PRO143966, OA-5D5) is a humanized, monovalent anti-c-MET antibody derived from the agonistic monoclonal antibody 5D5 [34]. 5D5 attaches to the c-MET receptor and thus, inhibits HGF ligand binding. However, attachment of bivalent 5D5 to the c-MET receptor induces receptor phosphorylation and downstream signaling. MetMAb is produced as a recombinant protein in E.coli and is engineered with monovalent Fab fragments with murine variable domains for the heavy and light chains fused with human IgG1 constant domains [20]. In this manner, the Fab fragments bind to c-MET, but do not agonize the receptor and instead function as antagonists.
5. MetMAb Pharmacokinetics
MetMAb has undergone both preclinical and clinical studies evaluating the pharmacokinetics of the drug. In order to gain entry into the clinic, preclinical dose efficacy studies were performed in KP4 xenograft models at a dose range of 0.825–120mg/kg. Single dose pharmacokinetic studies were performed in mice, rats, and cynomolgus monkeys at a dose range of 0.5–30mg/kg [35].
These efficacy studies demonstrated that the area under the serum-concentration-time curve (AUC) was the pharmacokinetic driver of efficacy in the KP4 xenograft model. Clearance in the mice, rats, and monkeys was 21, 19, and 13 mL/day/kg, respectively. Estimates of the kinetics of OA-5D5 in humans were made based on allometric and species-invariant time transformations of cynomolgus monkey data. These calculations yielded an estimate of human clearance of 5.5–10 ml/day/kg. In addition, a starting dose for humans was established at 1mg/kg with dosing indicated every 1–3 weeks [35]. Using the predictor of progression free response (<20% increase in pancreatic tumor mass), a dosing regimen of 12.5mg/kg weekly and 20mg/kg every three weeks increased the likelihood of treatment success in these preclinical models [36].
Subsequently MetMAb was studied in a phase I dose-escalation study. In it, the human half-life approximated 10 days with a clearance of 8mL/kg/day [37]. Based on preclinical modeling and phase 1 clinical data, a recommended phase II dose (RP2D) of 15mg/kg IV every three weeks was selected to maintain a minimum tumoristatic concentration of 15 μg/mL in > 90% of patients.
6. Efficacy
6.1 Preclinical Data
Two major preclinical efficacy studies have been published thus far. The first was performed in malignant glioblastoma cell lines. Glioblastoma is characterized by HGF and c-MET overexpression, with higher expression correlated with greater tumor grade and vascularity. U87 human glioblastoma cells (c-MET and SF/HGF positive) and G55 human glioblastoma cells (c-MET positive and SF/HGF negative) were used to generate intracranial orthotopic xenografts in nude mice [3].
In vitro, U87 cells treated with OA-5D5 demonstrated less phosphorylation of c-MET, less cellular proliferation, a dose dependent decrease in cellular motility, and twice the rate of cellular apoptosis [3]. In vivo, U87 mice treated with OA-5D5 demonstrated a tumor size reduction of 98.7% when treatment was initiated one week after tumor engraftment. No reduction was seen in U55 mice treated with OA-5D5, suggesting that the efficacy of OA-5D5 was mediated by blocking HGF stimulation [3]. Furthermore, in the U87 group, it was noted that treated cells demonstrated less proliferation, cellularity, and a lower vascular density.
Based on the success of OA-5D5 with glioblastoma cells, treatment efficacy was investigated in pancreatic cancer cells, a disease which also highly expresses HGF and c-MET [38]. Two cell lines, BxPC-3 (c-MET+, HGF-) and KP4 (c-MET+, HGF+) were evaluated in murine xenograft and orthotopic models. In the case of BxPC-3, mice were implanted with osmotic pumps to provide human HGF, as mouse HGF does not bind to the c-MET receptor [39].
Administration of OA-5D5 in both xenograft models led to a reduction of cellular proliferation, reduced c-MET and downstream target (Gab-1, Akt, ERK) phosphorylation, and decreased tumor growth. A KP4 orthotopic model was also developed and drug administration abolished tumor growth and led to a significant survival advantage [39]. Mechanisms supporting these promising findings included reduced KP4 cellular proliferation and reduced c-MET phosphorylation [39].
6.2 Clinical Data
MetMAb has been studied in both Phase I and Phase II studies in solid tumor malignancies. The phase I study was an evaluation of the safety and efficacy of MetMAb in locally advanced and metastatic solid tumor malignancies refractory to previous therapies. Data was collected from patients presenting with a wide range of solid tumor malignancies, a majority of whom having received at least three prior systemic therapies. The study used a 3+3 dose escalation design testing 1, 4, 10, 20, and 30 mg/kg doses given on day 1 of three week cycles in 21 patients; an additional 13 patients were enrolled in the recommended phase 2 dose cohort (15 mg/kg every three weeks). All doses were generally well-tolerated, and a maximum tolerated dose was not reached. One patient with gastric cancer exhibited a complete response; this patient had a tumor profile consistent with “autocrine” biology, associated with high intra-tumoral c-MET and HGF by IHC, without evidence for Met mutation/amplification [37].
In phase II clinical testing, erlotinib was tested with or without MetMAb as second or third line treatment in patients with stage IIIb and IV NSCLC (OAM4558g). The rationale for combining a c-MET inhibitor with an EGFR inhibitor in refractory NSCLC comes, in part, from studies which found c-MET amplification and over-expression in EGFR mutant cell lines resistant to EGFR inhibition [40]. Furthermore, a study by Engelman et al. suggests that targeted c-MET inhibition may restore EGFR inhibitor sensitivity [41].
Over 120 patients were equally randomized to receive erlotinib (150mg PO daily) + MetMAb (15mg/kg IV q 3 w) or erlotinib (150mg PO daily) + placebo (IV q3 w). Tissue was used to determine MET expression status by IHC. “MET diagnostic positive” tumors were those in which >50% of tumor cells stained with an intensity of 2 or 3 on a immunohistochemistry scale of 0–3. Approximately 50% of patients were categorized as “MET diagnostic positive,” and initial results, reported at ESMO Congress in 2010 revealed that patients with “MET diagnostic positive” tumors treated with MetMAb plus erlotinib had a trend towards improved survival (HR=0.55, p=0.11), compared to those treated with erlotinib plus placebo. The converse was observed in patients with “MET diagnostic negative” tumors, i.e. worse overall survival was associated with MetMAb plus erlotinib treatment compared to with erlotinib plus placebo (HR = 3.02, p=0.02). The same pattern was also seen with progression free survival. In “MET diagnostic positive” patients, PFS was improved in the MetMAb arm compared to placebo (median PFS 12.4 weeks vs. 6.4 weeks respectively, (HR 0.56, p=0.05) [42]. The overall safety profile of the combination was very good; MetMAb did not exacerbate erlotinib related toxicity, and the most frequent MetMAb related toxicity was peripheral edema, occurring in ~20% of all MetMAb-treated patients.
7. Conclusions
c-MET receptor tyrosine kinase activation has been demonstrated in a number of malignancies [5, 7, 8, 18, 21]. Targeting this receptor with the monoclonal antibody MetMAb is promising as it demonstrates good specificity for the c-MET receptor and is generally well tolerated at indicated doses, both as a single agent and in combination with other agents. The combination of MetMAb and erlotinib holds promise in NSCLC and additional studies to determine whether MetMAb is beneficial with other agents in NSCLC, or in other malignancies, is warranted.
8. Expert Opinion
MetMAb is a novel therapy which targets the tyrosine kinase receptor, c-MET. It was developed as a one armed antibody, and through this innovation, it is able to selectively bind to and inhibit the c-MET receptor, rather than agonize or partially antagonize the receptor as observed with other bivalent antibodies. Early research has supported the belief that MetMAb holds great promise in the treatment of malignancies, particularly among those exhibiting c-MET activation.
The key to the success of MetMAb therapy will be to clearly identify patients exhibiting c-MET activation and to target them with single agent or combination therapies. The current trial data is promising and if the beneficial effects of MetMAb translate during larger trials, clinical care strategies may be modified. One area where the use of MetMAb may be of benefit is with non-small cell lung cancer. Though erlotinib therapy is already considered a mainstay of treatment in advanced refractory non-small cell lung cancer, outcomes may be improved with the addition of a c-MET inhibitor, in order to lessen the impact of EGFR inhibitor resistance. If the phase III trial testing the combination of erlotinib with MetMAb is positive and it is FDA approved, this targeted combination inhibitor therapy may be used more broadly as second line therapy in NSCLC. This combination may improve outcomes without resulting in greater morbidity.
The use of MetMAb is not limited to lung cancer. A phase II investigation in the potential use of MetMAb in triple negative breast cancer in combination with paclitaxel and bevacizumab is currently underway. Furthermore, MetMAb has the potential to be of benefit in other solid tumor malignancies with high MET expression, such as pancreatic cancer and glioblastoma multiforme. Large trials are helpful not only in identifying which patients benefit the most, but also determining which patients experience additional harm from MetMAb therapy. Though the agent has been well tolerated thus far, larger studies will provide additional information on patient selection criteria.
It is anticipated by the authors that MetMAb will be accepted as part of the treatment of malignancies demonstrating c-MET overexpression, as in lung and breast cancers. As c-MET overexpression occurs in many other malignancies, it will be important to develop a reliable biomarker assay that will quickly and accurately identify patients demonstrating overexpression. It has already been demonstrated that “MET diagnostic negative” patients may have poorer outcomes with MetMAb treatment. At this time it is unclear why these patients did not benefit; therefore, it will be essential to correctly categorize patients by their MET status. Further efforts should be used to validate IHC as a sufficient litmus test for classifying patients. Other testing could include searching for Met gene amplification, mutation, or MET phosphorylation status. By treating appropriate candidates, MetMAb has the potential to improve progression free survival and possibly overall survival, in c-MET active patients.
Article Highlights.
An overview of the structure and function of c-MET and HGF
An overview of the HGF-MET dyad in physiologic and pathologic processes
A description of select targeted HGF and c-MET inhibitors
A review of the most up to date preclinical and clinical data surrounding the MET inhibitor MetMAb (OA-5D5)
An expert summary regarding the future of MetMAb in clinical practice with a focus on lung malignancy
Drug Summary Box.
| Drug name | MetMAb |
| Phase | Phase III |
| Indications (to date) | Non-small cell lung cancer Breast cancer Solid tumor malignancies |
| Pharmacology description | MET tyrosine kinase inhibitor Hepatocyte growth factor receptor antagonist |
| Route of administration | Parenteral/intravenous |
| Pivotal trial(s) |
NCT01068977 NCT00854308 NCT01186991 |
Footnotes
Declaration of Interest
Ravi Salgia is supported by following grants from the NIH/NCI: 5R01CA100750-08, 5R01CA125541-05, and 5P01HL058064-16. He is receives support from the Respiratory Health Association of Metropolitan Chicago. Premal Patel is an employee of Genentech. Amy Peterson was an employee of Genentech at the time of the writing of this manuscript.
Contributor Information
Mosmi Surati, Email: msurati@uchicago.edu, Medical student at the University of Chicago Pritzker School of Medicine; 924 E. 57th St., Chicago, IL 60637; (p) 847-293-4470.
Premal Patel, Email: patel.premal@gene.com, Associate Medical Director, Exploratory Clinical Development; Genentech; 1 DNA Way, MS 44-2B South San Francisco, CA 94080 (p) 650-225-679; (f) 650-467-3165.
Amy Peterson, Email: peterson.amy@gene.com, Associate Group Director, Exploratory Clinical Development, Genentech; 1 DNA Way, MS 44-2B, South San Francisco, CA 94080 (p) 650-467-4752; (f) 650-742-8618.
Ravi Salgia, Email: rsalgia@medicine.bsd.uchicago.edu, Professor of Medicine, Pathology, and Dermatology, Section of Hematology/Oncology, Department of Internal Medicine, University of Chicago Pritzker School of Medicine; 5841 S. Maryland Ave.; Chicago, IL 60637; (p) 773-702-6149; (f) 773-702-3002.
Annotated Bibliography
- 1.Schilsky RL. Personalized medicine in oncology: The future is now. Nature Reviews Drug Discovery. 2010;9:363–6. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 2.Liotta LA, Kohn EC, Petricoin EF. Clinical proteomics: Personalized molecular medicine. JAMA. 2001;286(18):2211–4. doi: 10.1001/jama.286.18.2211. [DOI] [PubMed] [Google Scholar]
- 3.Maertens T, Nils-Ole S, Eckerich C, Fillbrandt R, Merchant M, Schwall R, Westphal M, Lamszus K. A novel one-armed anti-c-met antibody inhibits glioblastoma growth in vivo. Clinical Cancer Research. 2006;12:6144. doi: 10.1158/1078-0432.CCR-05-1418. [DOI] [PubMed] [Google Scholar]
- 4.Altekruse SF, Kosary CL, Krapcho M, Neyman N, Aminou R, Waldron W, Ruhl J, Howlader N, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Cronin K, Chen HS, Feuer EJ, Stinchcomb DG, Edwards BK. SEER Cancer Statistics Review, 1975–2007. National Cancer Institute; Bethesda, MD: [Internet] [Google Scholar]
- 5.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, Hensen M, Schaefer E, Naoki K, Lader A, Richards W, Sugarbaker D, Husain AN, Christensen JG, Salgia R. Functional expression and mutations of c-met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Research. 2005;65:1479. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 6.Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 7▪ ▪.Maulik G, Kijima T, Ma PC, Ghosh SK, Lin J, Shapiro GI, Schaefer E, Tibaldi E, Johnson BE, Salgia R. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clinical Cancer Research. 2002;8:620. Provides excellent review of HGF/Met signaling pathway in disease and in normal state and of potential c-MET inhibitors. [PubMed] [Google Scholar]
- 8.Sattler M, Hasina R, Reddy M, Gangadhar T, Salgia R. The role of the c-met pathway in lung cancer and the potential for targeted therapy. Ther Adv Med Oncol. 2011;3(4):171–84. doi: 10.1177/1758834011408636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y. The human hepatocyte growth factor receptor gene: Complete structural organizaiton and promoter characterization. Gene. 1998;215:159–169. doi: 10.1016/s0378-1119(98)00264-9. [DOI] [PubMed] [Google Scholar]
- 10▪.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Research. 2003;63:6272. Important discovery of c-MET mutation and the significance of this mutation. [PubMed] [Google Scholar]
- 11.Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. 2009;4(4):444. doi: 10.1097/JTO.0b013e31819d6f91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang YW, Vande Woude GF. HGF/SF-met signaling in the control of branching morphogenesis and invasion. J Cell Biochem. 2003;88:408–17. doi: 10.1002/jcb.10358. [DOI] [PubMed] [Google Scholar]
- 13.Comoglio PM. Pathway specificity for met signaling. Nature Cell Biol. 2001;3:E161–2. doi: 10.1038/35083116. [DOI] [PubMed] [Google Scholar]
- 14▪ ▪.Nakamura T, Nawa K, Ichihara A, Kaise N, Nishino T. Purification and subunit structure of hepatocyte growth factor from rat platelets. FEBS Lett. 1987;224(2):311–6. doi: 10.1016/0014-5793(87)80475-1. Seminal paper describing structure of HGF. [DOI] [PubMed] [Google Scholar]
- 15.Basilico C, Arnesano A, Galluzzo M, Comoglio PM, Michieli P. A high affinity hepatocyte growth factor-binding site in the immunolobulin-like region of met. J Biol Chem. 2008;283(30):21267–77. doi: 10.1074/jbc.M800727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF B-chain in complex with the sema domain of the met receptor. EMBO. 2004;23:2325–35. doi: 10.1038/sj.emboj.7600243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17▪ ▪.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376(31):768–71. doi: 10.1038/376768a0. Seminal paper demonstrating function of c-met. [DOI] [PubMed]
- 18.Peruzzi B, Bottaro DP. Targeting the c-met signaling pathway in cancer. Clinical Cancer Research. 2006;12:3657. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- 19.Jeffers M, Rong S, Vande Woude GF. Hepatocyte growth factor/scatter factor-met signaling in tumorigenicity and invasion/metastasis. J Mol Med. 1996;74:505. doi: 10.1007/BF00204976. [DOI] [PubMed] [Google Scholar]
- 20▪ ▪.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nature Reviews Drug. 2008;7:504. doi: 10.1038/nrd2530. Review of c-MET inhibitors to date. [DOI] [PubMed] [Google Scholar]
- 21.Scott A, Salgia R. Biomarkers in lung cancer: From early detection to novel therapeutics and decision making. Biomarker Med. 2008;2(6):577. doi: 10.2217/17520363.2.6.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abounader R, Raganathan S, Lal B, Fielding K, Dietz H, Burger P, Laterra J. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst. 1999;91(18):1548–56. doi: 10.1093/jnci/91.18.1548. [DOI] [PubMed] [Google Scholar]
- 23.Stella MC, Comoglio PM. HGF: A multifunctional growth factor controlling cell scattering. Int J Biochem Cell Biol. 1999;31:1357–62. doi: 10.1016/s1357-2725(99)00089-8. [DOI] [PubMed] [Google Scholar]
- 24.Rosen EM, Lamszus K, Laterra J, Polverini PJ, Rubin JS, Goldberg ID. HGF/SF in angiogenesis. Ciba Found Symp. 1997;212:215–29. doi: 10.1002/9780470515457.ch14. [DOI] [PubMed] [Google Scholar]
- 25.Dong G, Chen Z, Li Z, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth Factor/Scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911–18. [PubMed] [Google Scholar]
- 26.Burgess T, Coxon A, Meyer S, Sun J, Rex K, Tsuruda T, Chen Q, Ho S, Li L, Kaufman S, Zhang K, Feng X, Jia X, Green L, Radinsky R. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-met dependent human tumors. Cancer Research. 2006;66:1721. doi: 10.1158/0008-5472.CAN-05-3329. [DOI] [PubMed] [Google Scholar]
- 27.Rosen PJ, Sweeny CJ, Park DJ. A phase ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin Cancer Res. 2010;16:2677–87. doi: 10.1158/1078-0432.CCR-09-2862. [DOI] [PubMed] [Google Scholar]
- 28.Wen PY, Schiff D, Cloughesy TF, Raizer JJ, Laterra J, Smitt M, Wolf M, Oliner KS, Anderson A, Zhu M, Loh E, Reardon DA. A phase II study evaluating the efficacy and safety of AMG102 (rilotumumab) in patients with recurrent glioblastoma. Neuro-Oncology. 2011;13(4):437–446. doi: 10.1093/neuonc/noq198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KJ, Whang L, Su Y, Gillespie Y, Salhotra A, Lal B, Laterra J. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clinical Cancer Research. 2006;12:1292. doi: 10.1158/1078-0432.CCR-05-1793. [DOI] [PubMed] [Google Scholar]
- 30.Stabile LP, Rothstein ME, Keohavong P, Jin J, Yin J, Land SR, Dacic S, Luong TM, Kim KJ, Dulak AM, Siegfried JM. Therapeutic targeting of human hepatocyte growth factor with a neutralizing monoclonal antibody reduces lung tumorigenesis. Molecular Cancer Therapeutics. 2008;7:1913. doi: 10.1158/1535-7163.MCT-07-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31▪.Prat M, Crepaldi T, Pennacchietti S, Bussolino F, Comoglio PM. Agonistic monoclonal antibodies against the met receptor dissect the biological responses to HGF. J Cell Sci. 1998;111:237–47. doi: 10.1242/jcs.111.2.237. Describes the drug development and balancing the need for c-MET antagonism in the presence of partial agonistic effects. [DOI] [PubMed] [Google Scholar]
- 32.Petrelli A, Circosta P, Granziero L, Mazzone M, Pisacane A, Fenoglio S, Comoglio PM, Giordano S. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. PNAS. 2006;103(13):5090–5. doi: 10.1073/pnas.0508156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacchiana G, Chirlaco C, Stella MC, Petronzelli F, De Santis R, Galluzo M, Carminati P, Comoglio PM, Michieli P, Vigna E. Monovalency unleashes the full therapeutic potential of the DN-30 anti-met antibody. J Biol Chem. 2010;285(46):36149–57. doi: 10.1074/jbc.M110.134031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merchant M, Zheng Z, Romero M, Huang A, Adams C, Moffat B, Zhang M, Chen Y, Wang L, LaFleur M, Tien J, Ross S, Yan M, Mallet W, Reslan HB, Ross J, Carano R, Yang R, Jin R, Colbern G, Dennis M, Yansura D, Schwall R. One-armed 5D5 (OA5D5) is a potent humanized HGF-blocking anti-c-Met monovalent antibody that inhibits HGF-dependent activity in vitro and demonstrates anti-tumor efficacy in vivo. 98th AACR Annual Meeting; 2007 April 14–17, 2007; Los Angeles, CA. 2007. [Google Scholar]
- 35.Xiang H, Reyes A, Merchant M, Bender B, Jumbe N, Young J, Gelzeichter T, Vaidyanathan A, Peterson A, Damico L. Supporting MetMab entry into the clinic with nonclinical pharmacokinetic and pharmacodynamic information. 20th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; 2008 Oct 21–24, 2008; Geneva, Switzerland. 2008. [Google Scholar]
- 36.Bender B, Xiang H, Reyes AE, Damico LA, Merchant M, Peterson A, Forrest W, Jumbe NL. Translational pharmacokinetic, pharmacodynamic modeling and simulation analysis of MetMab. 20th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; 2008 Oct 21–24, 2008; Geneva, Switzerland. 2008. [Google Scholar]
- 37.Moss RA, Bothos JG, Patel PH, Peterson AC, Eppler S, Bai S, Nijem I, Desnoyers L, Kaur S, Zha J, Yu W, Simpson J, Ratain MJ, Tan AR, Stein MN, Nehnert JM, Salgia R. Final results from the Phase I study of MetMab, a monovalent antagonist antibody to the receptor Met, dosed as a single agent and in combination with bevacizumab in patients with advanced solid tumor malignancies. AACR 102nd meeting; 2011 April 5, 2011; Orlando, Florida. 2011. [Google Scholar]
- 38.Ebert M, Yokoyama M, Friess H, Buchler MW, Korc M. Coexpression of the c-met proto-oncogene and hepatocyte growth factor in human pancreatic cancer. Cancer Res. 1994;54:5775–78. [PubMed] [Google Scholar]
- 39.Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, Carano RAD, Kasman I, Mai E, Young J, Zha J, Zhang Z, Ross S, Schwall R, Colbern G, Merchant M. MetMab, the one-armed 5D5 anti-c-met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Research. 2008;68:4360. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 40.Zucali PA, Ruiz MG, Giovannetti E, Destro A, Varella-Garcia M, Floor K, Geresoli GL, Rodriguez JA, Garassino I, Comoglio P, Roncalli M, Santoro A, Giaccone G. Role of cMET expression in non-small-cell lung patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol. 2008;19:1605–12. doi: 10.1093/annonc/mdn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41▪ ▪.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. Excellent translational paper of primary data which elaborates mechanism for treatment failure. [DOI] [PubMed] [Google Scholar]
- 42▪.Spigel DR, Ervin TJ, Ramlau R, Daniel DB, Goldschmidt JH, Krzakowski M, Godber B, Yu W, Zha J, Yauch RL, Patel PH, Peterson AC. Randomized, phase 2, multicenter, double-blind, placebo-controlled study evaluating MetMab, an antibody to Met receptor, in combination with erlotinib, in patients with advanced non-small-cell lung cancer. 35th ESMO Conference; 2010 October 8–12, 2010; Milan, Italy. 2010. Good primary data suggesting possible new advance in treatment of NSCLC. [Google Scholar]