

Abstract
MicroRNAs (miRNAs) have been implicated in B cell lineage commitment, regulation of T cell differentiation, TCR signalling, regulation of IFN signalling, and numerous other immunological processes. However, their function in autoimmunity, and specifically in systemic lupus erythematosus (SLE), remains poorly understood. B6.Sle123 is a spontaneous genetic mouse model of SLE characterized by autoantibody production, lymphosplenomegaly, and glomerulonephritis. We identified several differentially regulated miRNAs in B and T lymphocytes of B6.Sle123 mice. We found that miR-21 expression in lupus B and T cells is up-regulated and that in vivo silencing of miR-21 using a tiny seed-targeting LNA reversed splenomegaly, one of the cardinal manifestations of autoimmunity in B6.Sle123 mice, and de-repressed PDCD4 expression in vivo and in vitro. In addition, treatment with anti-miR-21 altered CD4/CD8 T cell ratios and reduced Fas receptor-expressing lymphocyte populations. Our study shows that tiny LNAs can be used to efficiently antagonize endogenous miRNAs in peripheral lymphocytes in vivo and in primary lymphocytes cultured ex vivo and can alter the course of a spontaneous genetic disease in mice.
Keywords: autoimmunity, miR-21/PDCD4, SLE, splenomegaly
INTRODUCTION
Systemic lupus erythematosus (SLE, lupus) is an autoimmune disease in which a combination of genetic predisposition and possible environmental influences triggers an exaggerated immune reaction against self-antigens and loss of immune tolerance. Antibody production by B cells and aberrant antibody-independent B and T cell functions have placed B and T cells at the centre of development of new therapeutic strategies for treatment of SLE [reviewed in (Crispin et al, 2010; Sanz & Lee, 2010)]. The tri-congenic B6.Sle1.Sle2.Sle3 (B6.Sle123) mouse model bears three lupus susceptibility loci from the NZM2410 lupus-prone strain, backcrossed onto a C57BL/6 (B6) background (Morel et al, 2000). B6.Sle123 mice spontaneously develop an autoimmune syndrome that strongly resembles human lupus, characterized by autoantibodies against H2A/H2B/DNA nucleosomes, splenomegaly, lymphadenopathy, and immune complex-mediated glomerulonephritis. Anti-histone autoantibodies are detected early in the life of B6.Sle123 mice; however, splenomegaly and kidney disease are not typically present until the age of 4–6 months (Morel et al, 2000). As in human SLE, autoimmune manifestations in B6.Sle123 mice are associated with lymphocyte signalling defects [reviewed in (La Cava, 2009; Liu & Mohan, 2009)] and perturbation of cell proliferation and apoptosis (Mohan et al, 1997; Mohan et al, 1999).
MicroRNAs (miRNAs) are ∼22 nt non-coding RNAs that regulate gene expression post-transcriptionally by mediating translational repression or promoting degradation of their target mRNAs (Filipowicz et al, 2008; Nelson et al, 2003). Animal miRNAs have emerged as key players in diverse immunological processes, such as B cell lineage commitment, regulation of T cell differentiation, T cell receptor (TCR) signalling and regulation of interferon (IFN) signalling (Lu & Liston, 2009; Tang et al, 2009). However, their function in autoimmunity, and specifically in SLE, remains poorly understood. Induction of lymphoproliferative syndromes in mice by failed interaction of miR-101 with ICOS (Yu et al, 2007) or by transgenic expression of the miR-17-92 cluster (Xiao et al, 2008) highlights an important role for miRNAs in autoimmunity. Furthermore, aberrant miRNA expression in lymphocytes and peripheral blood mononuclear cells (PBMCs) from patients with SLE (Dai et al, 2007; Te et al, 2010) and miRNA regulation of signalling pathways involved in the induction or maintenance of lupus (Divekar et al, 2011; Tang et al, 2009; Zhao et al, 2011) add another layer of complexity to the molecular pathways that are disordered in lupus.
In this study, we identified miRNAs differentially regulated in peripheral lymphocytes in the mouse lupus model B6.Sle123. We identified miR-21 as a constitutively overexpressed miRNA in mouse SLE lymphocytes and showed that in vivo silencing of miR-21 reversed cardinal manifestations of autoimmunity, de-repressed PDCD4 expression and altered lymphocyte populations in B6.Sle123 mice.
RESULTS AND DISCUSSION
Differential miRNA expression in B6.Sle123 peripheral lymphocytes
We investigated whether miRNAs implicated in hematopoiesis (Xiao et al, 2007), in activation of innate immune responses (Taganov et al, 2006), and in apoptosis or cell proliferation (He et al, 2007; Rokhlin et al, 2008; Yamakuchi & Lowenstein, 2009) are differentially regulated in SLE and whether these miRNAs play a role in SLE pathogenesis or course of the disease. To this end, we isolated total RNA from fluorescence-activated cell sorting (FACS)-sorted splenic B and T lymphocytes from individual B6.Sle123 mice and compared their miRNA expression profiles to those of control age-matched B6 mice. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis showed that expression of several miRNAs is differentially regulated in B6.Sle123 splenic B cells, naïve (CD44LO CD62LHI) and memory (CD44HI CD62LLO) T cells (Table 1), particularly from older mice (12 months of age), which typically have advanced splenomegaly and kidney disease (Fairhurst et al, 2008; Morel et al, 2000). For up-regulated miRNAs, expression differences ranged from 1.8- to 13-fold. Specifically, in B6.Sle123 splenic B cells, miRs-21 and 222 were constitutively up-regulated relative to wild-type controls and their expression increased with disease severity. miR-181a was down-regulated in young and also in old mice with severe disease. Expression of microRNAs miR-34a, 146a, 221, 223, 142-5p and miR-155 was up-regulated in old B6.Sle123 mice with severe disease (p < 0.05, Table 1A). In naïve T cells, miR-21 was also constitutively up-regulated relative to wild-type controls and its expression increased with disease severity. miRs-221 and 155 were down-regulated in young B6.Sle123 mice but up-regulated in mice with severe disease. Expression of miR-34a, 146a and 223 increased significantly relative to wild-type controls in mice with severe disease, while expression of miR-222, 150 and 181a decreased (p < 0.05, Table 1B). In memory T cells, miR-21 and miR-146a were up-regulated relative to wild-type controls in young and older mice. miR-221 expression increased in mice with severe disease. miR-155 was down-regulated in young and old mice while miRs-34a, 221 and 223 were up-regulated and miR-181a was down-regulated in older mice with severe disease (p < 0.05, Table 1C). In summary, interestingly, miR-21 expression was up-regulated in lupus B cells as well as in naïve and memory T cells compared to B6 controls, regardless of the age of mice (Table 1 and Fig 1A). In contrast, miR-181a and miR-150 expression was down-regulated in most lymphocyte subsets examined in lupus mice compared to B6 controls (Table 1A–C).
Table 1.
miRNA expression in B6.Sle123 B and T lymphocytes
| miRNA | 2 mo. (mild disease) | 6 mo. (moderate disease) | 12 mo. (severe disease) | |||
|---|---|---|---|---|---|---|
| RQ1 ± SEM | p-value | RQ1 ± SEM | p-value | RQ1 ± SEM | p-value | |
| A. Differential miRNA expression in B6.Sle123 splenic B Cells | ||||||
| miR-21 | 1.7 ± 0.15 | 0.001 | 2.2 ± 0.42 | 0.036 | 5.8 ± 0.64 | 0.016 |
| miR-34a | 0.9 ± 0.38 | 0.670 | 1.2 ± 1.33 | 0.821 | 13.2 ± 0.65 | 0.000 |
| miR-146a | 1.0 ± 0.51 | 0.996 | 0.6 ± 0.25 | 0.044 | 5.3 ± 0.75 | 0.032 |
| miR-221 | 1.2 ± 0.29 | 0.504 | 1.2 ± 0.28 | 0.398 | 13.3 ± 1.05 | 0.023 |
| miR-222 | 1.4 ± 0.22 | 0.040 | 2.1 ± 0.26 | 0.015 | 5.8 ± 0.96 | 0.037 |
| miR-223 | 0.9 ± 0.53 | 0.720 | 2.3 ± 1.61 | 0.507 | 11.4 ± 0.68 | 0.006 |
| miR-142-5p | 1.1 ± 0.21 | 0.374 | 0.4 ± 0.34 | 0.017 | 2.7 ± 0.45 | 0.032 |
| miR-155 | 1.3 ± 0.12 | 0.026 | 1.2 ± 0.39 | 0.504 | 2.8 ± 0.53 | 0.050 |
| miR-150 | 0.7 ± 0.45 | 0.314 | 0.5 ± 0.27 | 0.098 | 0.5 ± 0.18 | 0.006 |
| miR-181a | 0.5 ± 0.20 | 0.007 | 0.3 ± 0.78 | 0.069 | 0.4 ± 0.36 | 0.020 |
| B. Differential miRNA expression in B6.Sle123 splenic naïve T cells | ||||||
| miR-21 | 2.5 ± 0.37 | 0.012 | 3.6 ± 1.00 | 0.110 | 3.0 ± 0.58 | 0.032 |
| miR-34a | 1.5 ± 0.91 | 0.528 | 4.2 ± 1.05 | 0.094 | 6.2 ± 0.50 | 0.006 |
| miR-146a | 1.0 ± 1.20 | 0.955 | 4.9 ± 1.56 | 0.216 | 4.1 ± 0.31 | 0.002 |
| miR-221 | 0.3 ± 0.47 | 0.013 | 2.3 ± 1.40 | 0.433 | 4.0 ± 0.39 | 0.002 |
| miR-222 | 0.6 ± 0.49 | 0.187 | 1.2 ± 0.50 | 0.672 | 0.5 ± 0.20 | 0.006 |
| miR-223 | 1.2 ± 0.55 | 0.697 | 4.6 ± 0.58 | 0.019 | 11.1 ± 0.23 | 0.000 |
| miR-142-5p | 1.0 ± 0.33 | 0.906 | 1.3 ± 0.56 | 0.550 | 2.6 ± 0.46 | 0.027 |
| miR-155 | 0.5 ± 0.12 | 0.000 | 2.1 ± 1.40 | 0.475 | 2.1 ± 0.17 | 0.000 |
| miR-150 | 1.0 ± 0.84 | 0.943 | 0.7 ± 0.69 | 0.553 | 0.6 ± 0.22 | 0.008 |
| miR-181a | 1.4 ± 0.77 | 0.562 | 1.0 ± 0.97 | 0.980 | 0.2 ± 0.66 | 0.015 |
| C. Differential miRNA expression in B6.Sle123 splenic memory T cells | ||||||
| miR-21 | 2.3 ± 0.26 | 0.00371 | 2.3 ± 0.32 | 0.00935 | 2.9 ± 0.31 | 0.00734 |
| miR-34a | 1.2 ± 0.46 | 0.61619 | 2.8 ± 0.98 | 0.17000 | 6.1 ± 1.0 | 0.03999 |
| miR-146a | 2.0 ± 0.27 | 0.00815 | 1.9 ± 0.23 | 0.00692 | 1.8 ± 0.31 | 0.03301 |
| miR-221 | 0.9 ± 0.29 | 0.78955 | 2.2 ± 0.33 | 0.02462 | 3.5 ± 0.59 | 0.01522 |
| miR-222 | 3.0 ± 0.66 | 0.04896 | 2.5 ± 0.39 | 0.02568 | 4.7 ± 0.91 | 0.03822 |
| miR-223 | 1.1 ± 0.44 | 0.80584 | 1.7 ± 0.14 | 0.00425 | 5.3 ± 0.89 | 0.02602 |
| miR-142-5p | 1.2 ± 0.22 | 0.35640 | 1.3 ± 0.39 | 0.38912 | 0.9 ± 0.52 | 0.72454 |
| miR-155 | 0.1 ± 0.86 | 0.02879 | 1.3 ± 0.30 | 0.91145 | 0.1 ± 0.31 | 0.00023 |
| miR-150 | 1.3 ± 0.36 | 0.34351 | 1.1 ± 0.50 | 0.80519 | 1.0 ± 0.68 | 0.92495 |
| miR-181a | 0.8 ± 0.33 | 0.49138 | 0.9 ± 0.98 | 0.93433 | 0.2 ± 0.79 | 0.01509 |
A. CD19+ splenic B cells from individual B6.Sle123 and age- and gender-matched B6 mice at 2, 6 and 12 months of age were FACS purified. Twenty nanograms total B cell RNA were reverse-transcribed with TaqMan miRNA-specific reverse-transcription primers followed by quantification using Taqman miRNA-specific qRT-PCR assays. B6.Sle123 miRNA RQ values represent the average of triplicate measurements from at least three independent experiments in individual B6.Sle123 mice relative to the expression of the same miRNA in individual B6 B cells. Prior to obtaining RQ values, the Ct values for each miRNA in each sample, were normalized against sno429 expression (ΔCt). The mean B6.Sle123 miRNA RQ was calculated by evaluating 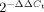 where ΔΔCt = ΔCt Sle123 ave − ΔCt B6 ave.
where ΔΔCt = ΔCt Sle123 ave − ΔCt B6 ave.
B. As in A except total splenic CD44Low CD62LHigh naïve T cells were FACS purified from individual mice.
C. As in A except total splenic CD44High CD62LLow memory T cells were FACS purified from individual mice.
Figure 1. miR-21 expression in B6.Sle123 is regulated transcriptionally and correlates with age and disease severity.
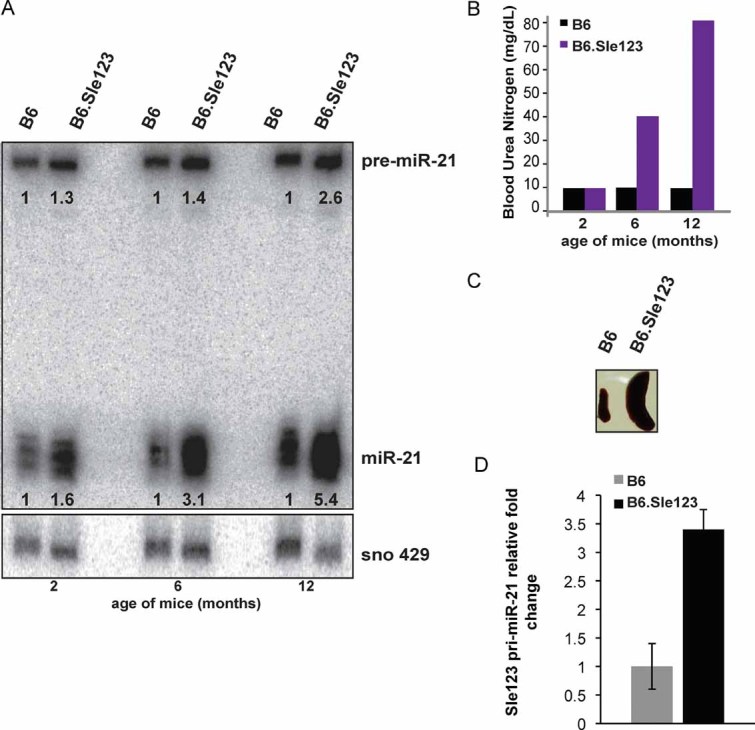
- pre-miR-21 and mature miR-21 expression in B6.Sle123 splenic B cells. Twenty micrograms of total splenic B cell RNA from individual age-matched 2, 6 and 12 months old B6.Sle123 and B6 mice were analysed under denaturing conditions, transferred and hybridized with LNA probe complementary to miR-21. For mass-normalization, the same membrane was stripped and re-probed with mouse snoRNA-429 radiolabelled DNA probe. The data were normalized to snoRNA-429 expression and relative fold-expression values calculated by taking the B6.Sle123/B6 ratio. A shorter exposure of the same membrane was used for the detection of the miR-21.
- B6.Sle123 disease severity correlates with age: BUN of 2, 6 and 12 months old female B6 and B6.Sle123 mice used as a measure of disease severity.
- Comparison of spleens extracted from 6 months old B6 and B6.Sle123 mice showing correlation of BUN and splenomegaly.
- qRT-PCR amplification of primary miR-21 (pri-miR-21) using splenic B cell RNA from individual, age-matched 9 months old B6.Sle123 and B6 mice. The average of three individual experiments is shown. Error bars represent SEM.
The Sle2 locus in B6.Sle123 mice confers expansion of the peritoneal and splenic B1-a B cell compartment (Mohan et al, 1997), even though the B2 subset remains the most abundant. To evaluate the relative contribution of each cell subset to individual miRNA abundance in the total pool of splenic CD19+ B cells from older SLE mice, we quantified the expression of miR-21, miR-146a and miR-155 in B1-a and B2 subsets. We detected the same trend for miR-21 and miR-146a expression in both lupus B cell subgroups whereas miR-155 was up-regulated only in B2 cells (Table 2). When lupus T cell subsets were examined in older mice, a 10- and 24-fold up-regulation of miR-21 was detected in CD4+ and CD8+ T cells, respectively, as well as a significant up-regulation of miR-146a and miR-155 compared to controls (Table 2).
Table 2.
miRNA expression in B6.Sle123 lymphocyte subsets
| miRNA | RQ1 ± SEM (p-value) | |||
|---|---|---|---|---|
| CD5− B2 B cells | CD5+ B1a B cells | CD4+ T cells | CD8+ T cells | |
| miR-21 | 5.2 ± 0.85 (0.048) | 2.7 ± 1.30 (0.329) | 10.2 ± 0.76 (0.011) | 24.4 ± 0.97 (0.009) (0.0090) |
| miR-146a | 1.9 ± 1.09 (0.456) | 1.9 ± 0.83 (0.319) | 5.4 ± 0.48 (0.007) | 19.4 ± 0.93 (0.010) |
| miR-155 | 2.1 ± 0.35 (0.038) | 0.94 ± 1.6 (0.954) | 2.9 ± 0.03 (0.0001) | 7.5 ± 0.69 (0.014) |
As in Table 1A expect CD5− B2 and CD5+ B1a B cells and CD4+ and CD8+ T cells were FACS purified from individual ∼12 months old B6.Sle123 and age- and gender matched B6 mice. Standard error and p-values were calculated using an un-paired, two-tailed Student's t-test.
Transcriptional activation of miR-21 in B6.Sle123
We found miR-21 expression to be consistently up-regulated in all SLE cell subsets that we investigated. Notably, the fold change of miR-21 expression in B6.Sle123 B lymphocytes, compared to that from age- and gender-matched B6 mice, positively correlated with the age of mice and thus with severity of their disease (Fig 1A–C). To further dissect the role of miR-21 in mouse SLE, we asked whether transcription of miR-21 gene is activated in lupus by comparing the expression of pri-miR-21 in CD19+ B cells isolated from 9 months old B6.Sle123 and age/gender-matched B6. qRT-PCR analysis revealed up-regulation of pri-miR-21 transcript in B cells from lupus mice compared to controls (Fig 1D). This indicates that, at least in part, miR-21 up-regulation in B6.Sle123 B lymphocytes results from transcriptional activation of miR-21 gene.
In vivo inhibition of miR-21 de-represses PDCD4 expression in B6.Sle123 T cells
Since miR-21 has been shown to regulate apoptosis and cell proliferation pathways (Chan et al, 2005; Cheng et al, 2005; Lu et al, 2008; Sayed et al, 2010; Yang et al, 2004) and was overexpressed in all subsets of lupus lymphocytes, we decided to test whether in vivo inhibition of miR-21 in B6.Sle123 mice could affect the course of their disease. The use of LNA-modified anti-miR oligonucleotides for miRNA silencing in vivo has been previously demonstrated in rodents and non-human primates (Elmen et al, 2008; Worm et al, 2009). Furthermore, recent studies have described an approach that enables efficient antagonism of miR-21 function in vivo using a short LNA-modified seed-targeting anti-miR-21 oligonucleotide (Obad et al, 2011; Patrick et al, 2010). Thus, we designed a short-term in vivo study in which three B6.Sle123 mice were injected intravenously at a dose of 25 mg/kg of saline-formulated anti-miR-21 compound on three consecutive days and sacrificed within 24 h after the last dose. In control experiments, three age- and gender-matched B6.Sle123 mice were treated with a LNA scramble compound. Interestingly, our short-term study showed that silencing of miR-21 in vivo results in approximately 20% de-repression of PDCD4 in naïve CD4+ T cells from anti-miR-21 treated mice, as compared to the PDCD4 expression in naïve CD4+ T cells from mice treated with control compound (Fig 2B). PDCD4 is an inhibitor of translation initiation (Zakowicz et al, 2005) and a tumour suppressor (Cmarik et al, 1999; Jansen et al, 2005). Expression of PDCD4 is post-transcriptionally regulated by miR-21 (Asangani et al, 2008; Frankel et al, 2008). The function of PDCD4 in lymphocytes is not yet understood but it is likely involved in cell proliferation and/or apoptosis (Lankat-Buttgereit & Goke, 2009).
Figure 2. Silencing of miR-21 by tiny LNA-anti-miR in vivo and in vitro derepresses PDCD4 in B6.Sle123 T cells.
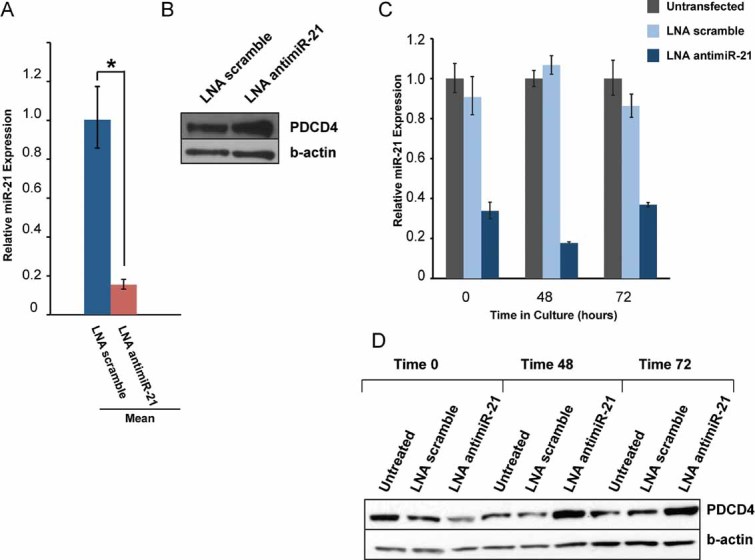
- Three groups of two B6.Sle123 mice were treated in vivo with seed-targeting LNA antimiR-21 or LNA scramble control compound daily for 3 days. miR-21 qRT-PCR quantification using total RNA from CD4+ T cells FACS purified from LNA antimiR-21- and LNA scramble compound-treated mice. Mean RQs of three independent experiments. *p = 8.1 × 10−6.
- Western blot analysis of PDCD4 expression in CD4+ T cells derived from in vivo LNA antimiR-21 treated mice and controls. Results are representative of three independent experiments.
- Unassisted uptake of LNA antimiR-21 by primary B6.Sle123 CD4+ T cells maintained in culture. Cells were purified, cultured in the presence of tiny seed-targeting LNA antimiR-21 or control compound as described in Materials and Methods section. At selected time points, cells were harvested and miR-21 expression was assessed by real-time PCR.
- PDCD4 expression in CD4+ T cells treated in vitro with seed-targeting LNA antimiR-21 or control compound.
In vitro inhibition of miR-21 de-represses PDCD4 expression in B6.Sle123 CD4+ T cells
We studied the expression of miR-21 and PDCD4 in primary CD4+ T cells purified from 3 months old B6.Sle123 mice cultured in presence of seed targeting LNA antimiR-21 or controls. Primary CD4+ T cells were purified and then mixed with culture media containing LNA anti-miR-21, control compound or no LNA. Cells were subsequently plated and harvested either immediately (time 0) or at the indicated time points (Fig 2D). Interestingly, miR-21 inhibition was observed at time 0, indicating efficient uptake of LNA compounds in the time required to mix, plate and harvest the cells. This unassisted uptake of LNA anti-miR-21 by primary CD4+ T cells cultured ex vivo resulted in a functional inhibition of miR-21 as shown by de-repression of PDCD4 (Fig 2C and D). As expected, although efficient LNA uptake by the primary T cells occurs within a short time period, de-repression of PDCD4 is only observed at subsequent time points (48 and 72 h). Our results indicate that seed-targeting LNA anti-miR-21 can silence endogenous miR-21 efficiently and in a very short period of time in primary B6.Sle123 T lymphocytes cultured ex vivo.
In vivo inhibition of miR-21 ameliorates splenomegaly in B6.Sle123 mice
To study the effect of in vivo miR-21 silencing in lupus, we treated 9-week-old female SLE mice by injecting them with anti-miR-21. Control mice were injected with LNA scramble or saline. Twelve 9-weeks-old female B6.Sle123 mice were randomly assigned to four groups. Mice in each group received injections of either LNA anti-miR-21 at a dose of 25 mg/kg, LNA scramble control compound at the same dose, or saline vehicle. All mice received intraperitoneal (IP) injections on three consecutive days and subsequent maintenance IP injections of 25 mg/kg every 3 weeks over a period of 3 months. The last injection was given intravenously. All mice were sacrificed within 24 h after the last dose. Spleens were extracted, weighed and subsequently, splenic lymphocytes were sorted. Northern blot analysis of total RNA isolated from liver or splenic B cells indicated that miR-21 was sequestered in a slower-migrating heteroduplex with the anti-miR-21 in the liver and in B cells (Fig 3A). Notably, silencing of miR-21 in vivo resulted in a significant reduction of splenomegaly in the SLE mice treated with anti-miR-21 compared to the LNA scramble- and vehicle control-treated mice in all four groups (Fig 3B and C). All mice survived the treatment with no observable change in the behaviour to suggest toxicity or intolerance to the treatment. Furthermore, no gross pathologic abnormalities were observed in the spleens (Fig 3B), livers, kidneys or other organs examined macroscopically during the necropsy at the completion of the study.
Figure 3. Silencing of miR-21 in vivo by tiny LNA-anti-miR ameliorates splenomegaly in B6.Sle123.
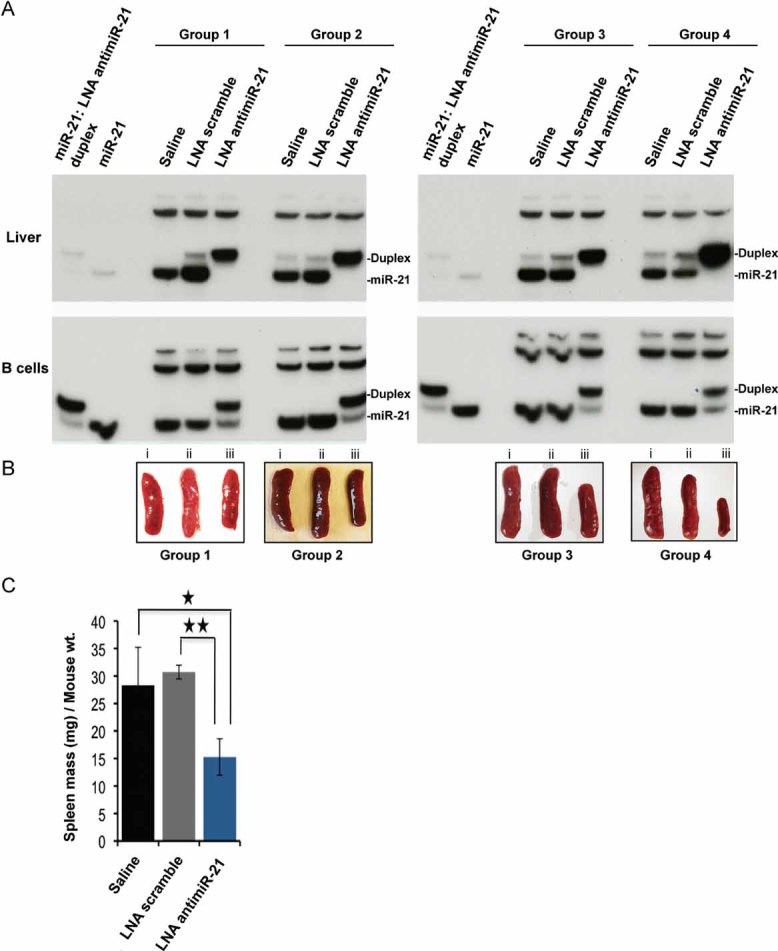
- In vivo sequestration of miR-21 by seed-targeting LNA antimiR-21 in a slower migrating heteroduplex. Four groups of mice were treated with either saline (n = 4), or LNA-scramble control compound (n = 4) or tiny LNA antimiR-21 (n = 4) over a period of 12 weeks. Native Northern blot analysis of total RNA extracted from liver tissue (top panels) or from purified splenic B220+ B cells (lower panels) was performed using a radiolabelled DNA probe complementary to miR-21. The first and second sample in each panel corresponds to in vitro annealed miR-21/LNA anti-miR-21 duplex and synthetic miR-21, respectively, used as controls.
- Effect of in vivo miR-21 silencing in autoimmune splenomegaly. Individual spleens harvested from the four groups of mice treated in the study are shown. (i, ii, iii indicate treatment with saline, LNA scramble and LNA antimiR-21, respectively).
- Mean spleen masses plotted as spleen-mass (mg)/mouse body mass (g) ratios. Error bars represent the SEM of the four independent experiments. p-values were calculated using an un-paired, two-tailed t-test. *p = 0.0149, **p = 0.000127.
In addition to the observed reduction in splenomegaly, in vivo miR-21 silencing resulted in a significant decrease in the splenic CD4+ to CD8+ T cell ratio and a reduction in the number of Fas receptor-expressing splenic B cells (Fig 4 and Table 3). Interestingly, elevated CD4+ to CD8+ ratios are a characteristic phenotypic alteration in Sle3-bearing T lymphocytes (Mohan et al, 1999). Thus, our results suggest that miR-21 inhibition skews the CD4+ to CD8+ ratio towards that of the background non-autoimmune strain (Mohan et al, 1999). In the LNA scramble-treated group, we observed increased CD4+ to CD8+ ratios due to an expansion of the CD4+ cells. Anti-chromatin antibody titers and blood urea nitrogen (BUN) levels were not affected by miR-21 silencing in this study (data not shown).
Figure 4. Silencing of miR-21 in vivo results in altered CD4/CD8 T cell ratios and reduced populations of Fas receptor-expressing B cells.

Four groups of B6.Sle123 mice were treated in vivo with LNA antimiR-21 or controls for 12 weeks, as described in the text. Splenocytes from B6.Sle123 mice injected with saline (left panels), LNA scramble control (middle panels) or LNA antimiR-21 (right panels) were stained with appropriate fluorophore-conjugated antibodies and subjected to flow cytometry.
- Top panels: CD4+ and CD8+ populations from the four treatment groups plotted as the percentage of viable lymphocytes. Horizontal bar indicates the mean value. Top Right: Chart of the mean CD4/CD8 ratios in the four treatment groups. *p = 0.0135, **p = 0.0025. Bottom panels: representative flow cytometry plots from one treatment group.
- Top panels: FasR+ IgD− populations from the four treatment groups plotted as the percentage of CD19+ B220+ lymphocytes. Horizontal bar indicates the mean value. Bottom panels: representative flow cytometry plots from one treatment group.
Table 3.
In vivo miR-21 silencing skews T and B lymphocyte subsets in B6.Sle123a
| Treatment group | |||
|---|---|---|---|
| Saline | LNA scramble | LNA antimiR-21 | |
| Total splenocytes (×106)b | 392 ± 74.5 | 335 ± 82.2 | 373 ± 106.5 |
| T cells | |||
| Events | 250 × 103 | 250 × 103 | 250 × 103 |
| Viable (×103)c | 199.3 ± 15.1 | 201.6 ± 9.5 | 207.8 ± 3.9 |
| CD4+ (×103)d | 36.2 ± 7.5 | 35.6 ± 4.4 | 39.8 ± 2.5 |
| CD8+ (×103)d | 7.8 ± 2.1 | 5.2 ± 0.4 | 17.0 ± 5.9*† |
| CD4+/CD8+ ratio | 4.7 ± 0.6 | 6.9 ± 1.3 | 2.6 ± 1.1 |
| B cells | |||
| Events | 1 × 106 | 1 × 106 | 1 × 106 |
| Viable (×103)c | 617.9 ± 77.5 | 565.2 ± 56.1 | 607.6 ± 93.0 |
| CD19+ B220+ (×103)c | 265.0 ± 88.2 | 127.3 ± 88.8 | 268.8 ± 50.6 |
| FasR+ IgD− (×103)e | 38.8 ± 19.4 | 25.4 ± 14.7 | 20.9 ± 7.1 |
Values represent the mean ± SD (n = 4 mice in each treatment group).
Spleens were harvested and single-cell suspensions prepared as described in Materials and Methods section. Viable cells were identified on the basis of trypan blue staining.
Viable lymphocyte populations were identified post-collection.
Gated on the viable lymphocyte population.
Gated on the CD19+ B220+ population.
p = <0.01 compared to LNA scramble.
p = 0.01 compared to Saline.
Our work demonstrates aberrant miRNA expression in B6.Sle123 lymphocytes at different stages of mouse SLE. Although we focused only on a small set of miRNAs, it is likely that other miRNAs not tested in this study are also differentially regulated. The up-regulation of miR-21 was of particular interest since it is detected in old and young mice and thus precedes severe SLE manifestations. Importantly, up-regulation of miR-21 in B and T lymphocytes preceding SLE manifestations was reported recently in the MRL/lpr, another genetic mouse model of lupus. Furthermore, miR-21 is also upregulated in human lupus CD4+ T and B cells and PBMCs (Pan et al, 2010; Te et al, 2010). Combined with our studies, this recent evidence implies an important role for miR-21 in the disordered immunoregulation in SLE.
Our study shows that seed-targeting tiny LNAs can be used for in vivo inhibition of miR-21 in rapidly proliferating cells, such as splenic lymphocytes. Efficient, unassisted uptake of tiny LNAs in cultured cell lines has been recently reported (Obad et al, 2011); our study is the first to show unassisted uptake of tiny seed-targeting LNAs by primary T lymphocytes.
Furthermore, our in vivo and in vitro studies showed that miR-21 regulates PDCD4 in SLE T lymphocytes. PDCD4 is directly targeted by miR-21 (Asangani et al, 2008; Frankel et al, 2008; Reis et al, 2010) and regulates pathways involved in apoptosis, cell cycle and differentiation [reviewed in (Lankat-Buttgereit & Goke, 2009)]. In pancreatic adenoma, miR-21 silencing induces PDCD4, promotes cell proliferation, and inhibits apoptotic cell death (Bhatti et al, 2011). PDCD4 promotes activation of the transcription factor NF-κB, suppresses IL10 and regulates TLR4 signalling (Sheedy et al, 2010). PDCD4-deficient mice develop B cell lineage lymphomas and their lymphocytes produce increased IL-10, IL-4 and IFN gamma, cytokines with important functions in T cell differentiation (Hilliard et al, 2006). The function of PDCD4 in SLE is not yet understood, however, it is likely that PDCD4 controls apoptotic or cell proliferation pathways contributing to the immunologic phenotype of B6.Sle123. It is also likely that PDCD4, via miR-21, regulates cell-signalling pathways participating in the orchestration of the cross talk between hyperactive B and T cells in lupus.
To investigate the function of miR-21 in mouse SLE, we designed a long term in vivo miR-21 knockdown study and demonstrated significant amelioration in splenomegaly, induced specifically by treatment with the anti-miR-21 but not the LNA control compound.
Mice treated in our study were young and miR-21 inhibition likely interfered with mechanisms inducing splenomegaly in lupus mice, altering the natural course of the disease. The severity of kidney disease by the age of 5 months varies in B6.Sle123. This could be the reason that in this model, miR-21 inhibition did not result in statistically significant BUN change between LNA antimiR-21-treated mice and controls. Inhibition of splenomegaly suggests that miR-21 controls cellular pathways important for cardinal disease manifestations in B6.Sle123 and is consistent with recent reports showing that miR-21 silencing reverses lymphosplenomegaly in mice with pro-B cell lymphoma (Medina et al, 2010). Two studies published while our manuscript was under review have shed further light on the role of miR-21/PDCD4 in immune activation and tolerance (Iliopoulos et al, 2011) and demonstrated correlation of miR-21/PDCD4 expression with human SLE disease activity/remission (Stagakis et al, 2011). Furthermore, aberrant T cell responses in active SLE and T cell-driven B cell functions in human lupus were connected to the function of miR-21 (Stagakis et al, 2011). Together with our functional results, this new evidence supports a central role for miR-21/PDCD4-controlled pathways in mouse and human lupus. Further elucidation of these pathways with studies in mouse models and human cells will shed light on the molecular complexity of this prototype autoimmune disease.
In summary, our study shows that tiny seed-targeting LNAs can be used to efficiently antagonize endogenous miRNAs in peripheral lymphocytes in vivo and in vitro and that pharmacological inhibition of a miRNA using such compounds can alter the course of a spontaneous genetic disease in mice. Our work, together with a recent study showing efficient silencing of miRNA families by seed-targeting LNAs (Obad et al, 2011), opens new directions for investigations of tiny LNAs as novel therapeutic tools. In addition, our results in a mouse model of lupus corroborated by recent studies in human SLE, indicate that miR-21 plays a central role in regulating autoimmune responses in lupus and that miR-21 inhibition can favourably alter the course of a systemic autoimmune process, opening new directions for explorations of miRNA therapeutics in SLE.
The paper explained
PROBLEM
Systemic lupus erythematosus (SLE) is a potentially fatal systemic autoimmune disease. The pathogenesis of SLE is poorly understood, however, aberrant functions of the immune system place B and T lymphocytes at the centre of investigations for novel therapeutic targets. miR-21 is implicated in cancer, however, its function in autoimmunity and specifically in SLE is poorly understood.
RESULTS
This study identifies a set of miRNAs differentially expressed in peripheral lymphocytes in a mouse model of SLE. miR-21 is up-regulated in all lymphocyte subsets examined. In vivo and in vitro silencing of miR-21 using small, seed targeting LNA inhibitors de-represses PDCD4, a protein involved in cell proliferation and apoptosis and ameliorates cardinal SLE manifestations in mouse lupus.
IMPACT
This work provides evidence for the involvement of miR-21 in SLE. Furthermore, this study is the first to show that pharmacological in vivo inhibition of miR-21 using seed-targeting LNAs can alter the course of a spontaneous, genetic disease in mice. Our work opens new directions for the use of small LNA inhibitors as therapeutic tools in autoimmune diseases.
MATERIALS AND METHODS
Mice
Breeder pairs of B6.Sle1Sle2Sle3 (B6.Sle123) (Morel et al, 2000) mice were a kind gift from Dr. Laurence Morel (University of Florida, Gainesville). B6.Sle123 or C57BL/6 (B6) (Jackson Laboratory, Bar Harbor, ME, USA) mice were bred and maintained in pathogen-free, Institutional Animal Care and Use Committee (IAUCAC)-approved animal facility at the University of Pennsylvania. Care and experimental procedures were approved by the IACUC at the University of Pennsylvania. BUN was measured in freshly drawn cardiac blood (Azostix reagent strips, Siemens Healthcare Diagnostics Inc.). BUN of 5–15 mg/dL was considered consistent with no detectable renal disease, 15–26 mg/dL consistent with mild disease, 30–40 mg/dL consistent with moderate disease and 50–80 mg/dL consistent with severe disease.
Antibodies
Rat anti-mouse CD19 (1D3), CD44 (IM7), CD44 (30-F11), CD62L (MEL-14), CD19 (1D3) and hamster anti-mouse CD3ε (145-2C11), CD69 (H1.2F3), CD11c (HL3), were purchased from BD Pharmingen. Rat anti-mouse CD45R (B220; RA3-6B2), CD5 (53-7.3), CD4 (L3T4), CD4 (GK1.5), CD4 (GK1.5), CD8 (53-6.7), IgD (11-26c), hamster anti-mouse CD3ε (145-2C11), and mouse anti-mouse CD95 (APO-1/FasR; 15A7) were from eBioscience.
Splenocyte preparation and lymphocyte purification
All procedures were performed on ice in FACS buffer (1X DPBS (Ca2+/Mg2+ free); 0.5% BSA; 2 mM EDTA). Single-cell suspensions were prepared by crushing spleens between frosted glass slides. Cells were blocked with rat anti-mouse CD16/32 (2.4G2) monoclonal antibody (mAb) for 15 min at room temperature (RT) and stained for 30 min on ice with appropriate fluorophor-conjugated antibodies. Dead cells were excluded from FACS by staining with DAPI. FACS sorting was performed at 4°C on a FACSAria cytometre (BD Biosciences) to a purity >95%. Alternatively, B cells were purified using autoMACS magnetic cell separation columns. Following purification, cells were either pelleted and snap-frozen or processed immediately.
Flow cytometry
All flow cytometry experiments were performed on a Becton Dickinson FACSCalibur at the University of Pennsylvania and the acquired data analysed with the FloJo software package (Tree Star Inc.).
RNA purification and quantitative real-time PCR
Total RNA was extracted using either Trizol reagent (Invitrogen) or the mirVana miRNA Isolation Kit (Ambion). For miRNA-specific reverse transcription (RT), 20 ng purified total RNA was reverse-transcribed (TaqMan miRNA-specific RT PCR primers, Applied Biosystems). qRT-PCR was performed in triplicates using 1:10 dilution of the RT product (Applied Biosystems 7500 real-time PCR system, according to the manufacturer's instructions). snoRNA-429 expression was used as control. Relative quantification (RQ) of B6.Sle123 miRNA expression was assessed by calculating the 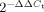 where
where 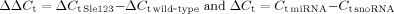 .
.
For qRT-PCR amplification of the primary miR-21 (pri-miR-21), primers (F: 5′-TGA CAT CGC ATG GCT GTA-3′ and R: 5′-GAT GCT GGG TAA TGT TTG AAT G-3′) spanning a 209 nt sequence containing the mouse pre-miR-21, were designed by identifying a mouse genomic sequence, with 91% similarity to the human primary miR-21. Splenic B cells (CD19+) were isolated from 3 nine months old B6.Sle123 and three age- and gender-matched C57BL/6 and B6.Sle123 mice using anti-CD19 MACS microbeads (Miltenyi Biotec, Germany). Total RNA was extracted with Trizol (Invitrogen) and double treated with TURBO DNase (Ambion, Inc., Austin, Texas, USA) to remove genomic DNA. Six hundred nanograms of total RNA was reverse-transcribed (Random primers, Applied Biosystems and Superscript III Reverse Transcriptase, Invitrogen). qRT-PCR was performed in triplicates using  of the RT product for pri-miR-21 and
of the RT product for pri-miR-21 and  for β-actin amplification (β-actin primers from Applied Biosystems Mm 00607939_S1). No-RT (-RT) RNA was used as negative control to exclude amplification due to genomic DNA contamination. RQ of B6.Sle123 pri-miR-21 expression was assessed by calculating the
for β-actin amplification (β-actin primers from Applied Biosystems Mm 00607939_S1). No-RT (-RT) RNA was used as negative control to exclude amplification due to genomic DNA contamination. RQ of B6.Sle123 pri-miR-21 expression was assessed by calculating the 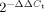 , where ΔΔCt = ΔCtB6.Sle123 − ΔCtC57BL/6 and ΔCt = Ctpri-miR-21 − Ctβ-actin.
, where ΔΔCt = ΔCtB6.Sle123 − ΔCtC57BL/6 and ΔCt = Ctpri-miR-21 − Ctβ-actin.
Northern blot analysis
Total RNA samples were resolved on 15% urea-polyacrylamide gels (20% polyacrylamide for native Northern blots), transferred to nitrocellulose membranes, hybridized with radiolabelled DNA or LNA probes complementary to miR-21. snoRNA-429 expression was used for normalization. Detection and analysis: Storm 860 (Molecular Dynamics) and ImageQuant (GE).
Protein isolation and Western blot analysis
Purified cell populations were pelleted, washed with PBS then re-suspended RIPA buffer (Pierce Biotechnology) supplemented with protease and phosphatase inhibitors (Roche Diagnostics). Proteins were resolved on NuPAGE 4-12% bis–tris polyacrylamide gels (Invitrogen Corp., Carlsbad, CA) and transferred to nitrocellulose membranes (Invitrogen). Primary antibodies: PDCD4 (Cell Signaling) and β-actin (Sigma). Proteins were visualized using the ECL Western Blotting Detection System (GE Healthcare). Protein bands were quantified using the ImageQuant software.
In vivo experiments
The 8-mer seed-targeting antimiR-21 (5′-GATAAGCT-3′) and 8-mer LNA scramble control (5′-TCATACTA-3′) oligonucleotides were synthesized as unconjugated, fully LNA-substituted oligonucleotides with a complete PS backbone as described (Patrick et al, 2010) and formulated in physiological saline (0.9% NaCl). For short-term experiments, six 8-weeks-old female B6.Sle123 mice were randomly assigned to three groups. The mice in each group received three intravenous (retro-orbital) injections of either LNA antimiR-21 or LNA scramble compounds (25 mg LNA/kg mouse body weight) on consecutive days. The mice were sacrificed within 24 h after the last dose. For long-term experiments, twelve 9-weeks-old female B6.Sle123 mice were randomly assigned to four groups and mice in each group received IP injections of either LNA antimiR-21, LNA scramble control compound, or saline. The mice were weighed immediately prior to dosing and received a dose of 25 mg LNA/kg mouse body wt. All mice received three initial IP injections on consecutive days and subsequent booster injections of 25 mg/kg every 3 weeks over a period of 3 months. The last booster injection was via intravenous delivery. All mice were sacrificed within 24 h after the last dose.
Statistical analysis
Reported B6.Sle123 mean miRNA expression differences represent triplicate measurements from at least three independent experiments in individual age- and gender-matched B6 and B6.Sle123 mice. Standard errors and p-values were calculated with an un-paired, two-tailed Student's t-test.
Ex vivo T cell culture and miR-21 inhibition
Spleens from 3 months old B6.Sle123 mice were harvested and single-cell suspensions prepared as described. Splenocytes were pooled and CD4+ T cells purified by incubation with anti-CD4 MACS microbeads (L4T4) followed by separation on MACS LS columns (Miltenyi Biotec, Auburn, CA) according to the Manufacturer's instructions. The purity was >90% CD4+, as determined by flow cytometry. Cells were resuspended (106 cells/ml) in RPMI 1640 media (supplemented with 10% FBS, 25 mM HEPES, 100 U/ml penicillin/streptomycin, 2 mM l-glutamine, 1% non-essential amino acids, 50 µM 2-mercaptoethanol and 100 U/ml IL-2) containing either PBS, 50 µM LNA scramble oligonucleotide or 50 µM LNA antimiR-21 and plated in 96-well round-bottom plates (1 × 106 cells/well) and maintained at 37°C + 5% CO2. At selected time points, cells were harvested and miR-21 expression was assessed by qRT-PCR.
Acknowledgments
We would like to thank Dr. Laurence Morel for permission to use the B6.Sle123 model for our studies. Grant support: National Institutes of Health/National Institute of Allergy and Infectious Diseases [K08AI063030 to MK; R01-AI063626 to RE], Lupus Research Institute [to MK and RE] and Alliance for Lupus Research [to MK, RC and RE]. National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases [T32-AR007442-22 to BG; R01-AR-34156 to RE]. Danish National Advanced Technology Foundation [009-2008-2 to SK]. Dr. Eisenberg is also supported by the American Autoimmune Related Disease Association.
Conflict of interest statement: SO, AP, and SK are employees of Santaris Pharma, a clinical stage biopharmaceutical company that develops RNA-based therapeutics. The other authors declare that they have no conflict of interest.
Author contributions
MK and BGG designed the studies and prepared the manuscript; BGG performed cell purification, miRNA expression, in vivo silencing, protein expression studies and statistical analysis; RC and RAE assisted with the design and analysis of cell sorting and flow cytometry studies; AP assisted with the statistical analysis; PYT performed immunoassays; YTL and OBE cloned and analysed pri-miR-21 expression; MK, BGG and SK designed the in vivo silencing studies and analysed the data; SO and BGG validated the LNA compounds; MK supervised the studies and performed data analysis.
References
- Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- Bhatti I, Lee A, James V, Hall RI, Lund JN, Tufarelli C, Lobo DN, Larvin M. Knockdown of microRNA-21 inhibits proliferation and increases cell death by targeting programmed cell death 4 (PDCD4) in pancreatic ductal adenocarcinoma. J Gastrointest Surg. 2011;15:199–208. doi: 10.1007/s11605-010-1381-x. [DOI] [PubMed] [Google Scholar]
- Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, Matsuhashi S, Colburn NH. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci USA. 1999;96:14037–14042. doi: 10.1073/pnas.96.24.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:317–325. doi: 10.1038/nrrheum.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, Xu ZM, Yin YB. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007;16:939–946. doi: 10.1177/0961203307084158. [DOI] [PubMed] [Google Scholar]
- Divekar AA, Dubey S, Gangalum PR, Singh RR. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. J Immunol. 2011;186:924–930. doi: 10.4049/jimmunol.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, Hedtjarn M, Hansen JB, Hansen HF, Straarup EM, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhurst AM, Mathian A, Connolly JE, Wang A, Gray HF, George TA, Boudreaux CD, Zhou XJ, Li QZ, Koutouzov S, et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol. 2008;38:1948–1960. doi: 10.1002/eji.200837925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight. Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard A, Hilliard B, Zheng SJ, Sun H, Miwa T, Song W, Goke R, Chen YH. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol. 2006;177:8095–8102. doi: 10.4049/jimmunol.177.11.8095. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Kavousanaki M, Ioannou M, Boumpas D, Verginis P. The negative costimulatory molecule PD-1 modulates the balance between immunity and tolerance via miR-21. Eur J Immunol. 2011;41:1754–1763. doi: 10.1002/eji.201040646. [DOI] [PubMed] [Google Scholar]
- Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 2005;65:6034–6041. doi: 10.1158/0008-5472.CAN-04-2119. [DOI] [PubMed] [Google Scholar]
- La Cava A. Lupus and T cells. Lupus. 2009;18:196–201. doi: 10.1177/0961203308098191. [DOI] [PubMed] [Google Scholar]
- Lankat-Buttgereit B, Goke R. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell. 2009;101:309–317. doi: 10.1042/BC20080191. [DOI] [PubMed] [Google Scholar]
- Liu K, Mohan C. Altered B-cell signaling in lupus. Autoimmun Rev. 2009;8:214–218. doi: 10.1016/j.autrev.2008.07.048. [DOI] [PubMed] [Google Scholar]
- Lu LF, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291–298. doi: 10.1111/j.1365-2567.2009.03092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Liu M, Stribinskis V, Klinge CM, Ramos KS, Colburn NH, Li Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene. 2008;27:4373–4379. doi: 10.1038/onc.2008.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- Mohan C, Morel L, Yang P, Wakeland EK. Genetic dissection of systemic lupus erythematosus pathogenesis: Sle2 on murine chromosome 4 leads to B cell hyperactivity. J Immunol. 1997;159:454–465. [PubMed] [Google Scholar]
- Mohan C, Yu Y, Morel L, Yang P, Wakeland EK. Genetic dissection of Sle pathogenesis: Sle3 on murine chromosome 7 impacts T cell activation, differentiation, and cell death. J Immunol. 1999;162:6492–6502. [PubMed] [Google Scholar]
- Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P, Kiriakidou M, Sharma A, Maniataki E, Mourelatos Z. The microRNA world: small is mighty. Trends Biochem Sci. 2003;28:534–540. doi: 10.1016/j.tibs.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Obad S, Dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, Li J, Zhou H, Tang Y, Shen N. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184:6773–6781. doi: 10.4049/jimmunol.0904060. [DOI] [PubMed] [Google Scholar]
- Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E, Olson EN. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest. 2010;120:3912–3916. doi: 10.1172/JCI43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis PP, Tomenson M, Cervigne NK, Machado J, Jurisica I, Pintilie M, Sukhai MA, Perez-Ordonez B, Grenman R, Gilbert RW, et al. Programmed cell death 4 loss increases tumor cell invasion and is regulated by miR-21 in oral squamous cell carcinoma. Mol Cancer. 2010;9:238. doi: 10.1186/1476-4598-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokhlin OW, Scheinker VS, Taghiyev AF, Bumcrot D, Glover RA, Cohen MB. MicroRNA-34 mediates AR-dependent p53-induced apoptosis in prostate cancer. Cancer Biol Ther. 2008;7:1288–1296. doi: 10.4161/cbt.7.8.6284. [DOI] [PubMed] [Google Scholar]
- Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–337. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, Abdellatif M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem. 2010;285:20281–20290. doi: 10.1074/jbc.M110.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q, Johnson DS, Chen Y, O'Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11:141–147. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, Iliopoulos D, Boumpas DT. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. 2011;70:1496–1506. doi: 10.1136/ard.2010.139857. [DOI] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, Huang X, Zhou H, de Vries N, Tak PP, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–1075. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- Te JL, Dozmorov IM, Guthridge JM, Nguyen KL, Cavett JW, Kelly JA, Bruner GR, Harley JB, Ojwang JO. Identification of unique microRNA signature associated with lupus nephritis. PLoS One. 2010;5:e10344. doi: 10.1371/journal.pone.0010344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worm J, Stenvang J, Petri A, Frederiksen KS, Obad S, Elmen J, Hedtjarn M, Straarup EM, Hansen JB, Kauppinen S. Silencing of microRNA-155 in mice during acute inflammatory response leads to derepression of c/ebp Beta and down-regulation of G-CSF. Nucleic Acids Res. 2009;37:5784–5792. doi: 10.1093/nar/gkp577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8:712–715. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 2004;24:3894–3906. doi: 10.1128/MCB.24.9.3894-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, Hutloff A, Giles KM, Leedman PJ, Lam KP, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature. 2007;450:299–303. doi: 10.1038/nature06253. [DOI] [PubMed] [Google Scholar]
- Zakowicz H, Yang HS, Stark C, Wlodawer A, Laronde-Leblanc N, Colburn NH. Mutational analysis of the DEAD-box RNA helicase eIF4AII characterizes its interaction with transformation suppressor Pdcd4 and eIF4GI. RNA. 2005;11:261–274. doi: 10.1261/rna.7191905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, Yin H, Lu Q. MicroRNA-126 regulates DNA methylation in CD4(+) T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011;63:1376–1386. doi: 10.1002/art.30196. [DOI] [PubMed] [Google Scholar]