

Abstract
A 5-μM docking hit has been optimized to an extraordinarily potent (55 pM) non-nucleoside inhibitor of HIV reverse transcriptase. Use of free energy perturbation (FEP) calculations to predict relative free energies of binding aided the optimizations by identifying optimal substitution patterns for phenyl rings and a linker. The most potent resultant catechol diethers feature terminal uracil and cyanovinylphenyl groups. A halogen bond with Pro95 likely contributes to the extreme potency of compound 42. In addition, several examples are provided illustrating failures of attempted grafting of a substructure from a very active compound onto a seemingly related scaffold to improve its activity.
INTRODUCTION
Though inhibition of multiple HIV proteins is therapeutically viable, HIV reverse transcriptase (RT) has been the key target.1 Nucleoside RT inhibitors (NRTIs) including AZT are incorporated into the product DNA causing premature strand termination, while the non-nucleoside RT inhibitors (NNRTIs) bind to an allosteric site ca. 10-Å away from the polymerase active site.2 Our efforts at discovery of new NNRTIs are intended to address continuing issues concerning the possible emergence of new viral strains, improved dosing, long-term tolerability, and safety.3 Numerous compounds in multiple series have been prepared that are both potent against the wild-type (WT) virus and that have auspicious computed pharmacological properties.4,5 Improvement in the performance of our compounds against clinically relevant viral variants is still desired. To address resistance from the outset, docking was done on multiple RT structures to seek consensus high-scoring hits. More than two million compounds from the ZINC library were screened with Glide using a conventional WT structure (1rt4), one with an alternative “down” conformation for Tyr181 (2be2), and a structure that incorporated the troublesome Tyr181Cys mutation (1jla).6 Though only nine compounds were purchased, three showed 5-12 μM activity against one or both viral strains in infected T-cell assays.
As described here, among the three actives, we have most pursued lead-optimization for compound 1, which showed 4.8 μM potency towards WT HIV-1.6 1 bears some structural similarity to the ligands, TNK-651 (2) and R221239 (3), from the 1jla and 2be2 crystal structures, respectively.7,8 Their roots can be traced back further to thymine analogs in the HEPT class including emivirine (MKC-442, 4), which progressed to phase III clinical trials.9 Various attributes of 1 are appealing including that it is a diphenylmethane derivative with a novel terminal uracil group, it likely has diminished metabolic liabilities compared to 3 and better computed aqueous solubility according to QikProp,10 and refinement of substituents in the phenyl rings can be expected to be productive. Thus, optimization of 1 was initiated using a computationally driven approach, primarily guided by results of free-energy perturbation (FEP) calculations for complexes of the inhibitors with HIV-RT.4

EXPERIMENTAL AND COMPUTATIONAL METHODS
Ultimately, the synthetic efforts focused on preparation of diphenylmethanes (Scheme 1) and catechol diethers (Schemes 2-4). The o-benzylphenols in Scheme 1 arose from Friedel–Crafts reactions of arylmethyl halides or alcohols with phenols.11,12 The catechol ether intermediates were prepared from substituted phenols and the aryl fluorides using SNAr reactions followed by treatment with boron tribromide or lithium chloride (Schemes 2 and 3). The final compounds were prepared in a two-step sequence via Mitsunobu reaction to install the bromoethoxy linker, followed by 2,4-bis(trimethylsiloxy)pyrimidine alkylation (5-15, 20-32).13
Scheme 1.

a. Synthesis of compounds 5-15
aReagents: (a) DIAD, bromoethanol, Ph3P, THF, rt, overnight; (b) 180 °C, 1.5 h.
Scheme 2.

a Synthesis of compounds 20-27, 36 and 44.
aReagents: (a) K2CO3, DMSO, 110 °C, 2-5 h; (b) BBr3, CH2Cl2, -78 °C, overnight; (c) DIAD, bromoethanol, Ph3P, THF, rt, overnight; (d) 2,4-bis((trimethylsilyl)oxy)pyrimidine, 180 °C, 1.5 h, or 3-benzoylpyrimidine-2,4(1H,3H)-dione, K2CO3, DMF, overnight, rt then MeOH, NH4OH, 2h, rt; (e) Fe, NH4Cl, EtOH, H2O, 75 °C; (f) NaNO2, conc HCl, KI, 80 °C, 2h; (g) Acrylonitrile, PdCl2(PPh3)2, Et3N, DMF, 140 ° C, 2 h.
Scheme 4.

a Synthesis of compounds 33-35.
aReagents: (a) DIAD, bromoethanol, Ph3P, THF, rt, overnight; (b) NaBH4, MeOH, 0 °C to rt, 3h; (c) NaH, DMF, rt, 2h.
Scheme 3.

a Synthesis of compounds 28 – 32 and 40-47
aReagents: (a) K2CO3, DMSO, 110 °C 2-5 h; (b) Fe, NH4Cl, EtOH, H2O, 75 °C; (c) NaNO2, conc HCl, CuCl, 80 °C, 1h; (d) CH3B(OH)2, Cu(OAc)2, pyr, dioxane, reflux; (f) Acrylonitrile, PdCl2(PPh3)2, Et3N, DMF, 140 °C, 2 h; (g) BBr3, CH2Cl2, -78 °C, overnight or LiCl, DMF 160 °C, overnight; (h) DIAD, bromoethanol, Ph3P, THF, rt, overnight; (i) 3-benzoylpyrimidine-2,4(1H,3H)-dione, K2CO3, DMF, rt, overnight then NH4OH, MeOH, rt.
However, when substituent ‘X’ was (E)-cyanovinyl (CV), methoxyethoxy (MOEO) or 3-hydroxypropan-1-oxy (HOPO), the last step did not yield the desired product. To circumvent this, the N-benzoyl uracil derivatives14 were made followed by alkylation with the alkyl bromide and cleavage of the benzoyl moiety to afford the target compounds in good yields (35-49).15 In particular, when 3-cyanovinyl- substituted catechol ethers were required, Heck coupling of aryl iodides with acrylonitrile using PdCl2(PPh3)2 as catalyst was effective. This reaction afforded separable mixtures of E:Z (70:30) stereoisomers in 50-70% yield.
Finally, compounds 33 and 34 were obtained via Mitsunobu reaction with the corresponding alcohols and catechol ethers (Scheme 4). For compound 35, the aliphatic linker was added via alkylation of 66 with 4-bromomethylpyridine. The identity of all assayed compounds was confirmed by 1H and 13C NMR and high-resolution mass spectrometry; purity was >95% as judged by high-performance liquid chromatography. Small molecule crystal structures were obtained by direct methods on data collected using a Rigaku Mercury2 CCD area detector with graphite monochromated Mo-Kα radiation.
For the biology, activities against the IIIB and variant strains of HIV-1 were measured using MT-2 human T-cells; EC50 values are obtained as the dose required to achieve 50% protection of the infected cells by the MTT colorimetric method. CC50 values for inhibition of MT-2 cell growth by 50% are obtained simultaneously.5,16,17 The antiviral and toxicity curves used triplicate samples at each concentration.
The principal computations were conjugate-gradient energy minimizations and Monte Carlo/FEP (MC/FEP) calculations, which yield relative free energies of binding. The calculations were performed with the MCPRO program18 and followed standard protocols.4,5 Coordinates of HIV-RT complexes were mostly constructed from the 2be2 crystal structure8 using the BOMB program.4 The model included the 175 amino acid residues nearest the ligand. Short conjugate-gradient minimizations were carried out on the initial structures for all complexes to relieve any unfavorable contacts. Coordinates for the free ligands were obtained by extraction from the complexes. The unbound ligands and complexes were solvated in TIP4P water spheres (“caps”) with a 25-Å radius; after removal of water molecules in too close contact with solute atoms, ca. 2000 and 1250 water molecules remained for the unbound and bound MC simulations. The FEP calculations utilized 11 windows of simple overlap sampling.19 Each window covered 10-15 million (M) configurations of equilibration and 10-30 M configurations of averaging at 25 °C. The ligand and side chains with any atom within ca. 10 Å of the ligand were fully flexible, while the protein backbone was kept fixed during the MC runs. The energetics were evaluated with the OPLS-AA force field for the protein,20 OPLS/CM1A for the ligands,21 and TIP4P for water.22
RESULTS AND DISCUSSION
A typical structure for the complexes is illustrated in Figure 1. As expected from the crystal structures for HEPT analogs and 3, the terminal phenyl ring resides in the π-box formed by Tyr181, Tyr188, Phe227, and Trp229, and the uracilylethoxy side chain projects into the channel lined by Pro225, Phe227, Pro236, and Tyr318. The computations further indicate that alternative conformations are possible for the side chain with the two illustrated in Figure 1 as the most probable. The gauche-anti-anti (gaa) conformer on the left is close to that found for the methoxymethyl fragment in the crystal structures for HEPT analogs,7 while the aag alternative allows formation of a hydrogen bond between the carbonyl group of Lys103 and the uracilyl NH. It should also be noted that the “down” orientation of the phenolic unit of Tyr181 follows from the 2be2 structure.8 It is possible that for some of the inhibitors considered here that the “up” orientation with Tyr181 and Tyr188 more parallel, as in the HEPT structures,7 is preferred.
Figure 1.

Computed structures of a catechol diether (31) bound to HIV-RT starting from the 2be2 crystal structure. Two possible conformations of the uracilylethoxy sidechain, gaa (left) and aag (right), are illustrated. Carbon atoms of the inhibitor are colored gold.
Diphenylmethanes
The MC/FEP calculations began with a chlorine scan for the terminal phenyl ring in 1. With numbering the ring such that the chlorines in Figure 1 are at the 2- and 5-positions, replacement of hydrogen by chlorine was predicted to be favorable (more negative free energy of binding, ΔGb) by 1.4, 1.9 and 3.8 kcal/mol at C2, C5 and C6 and unfavorable by 1.4 and 0.7 kcal/mol at C3 and C4. The computed uncertainties are ±0.2 kcal/mol. However, since double substitution was envisioned and the results might not be additive, MC/FEP calculations were executed for all 10 unique dichloro possibilities, as summarized in Table 1. Such exhaustive double substituent scans should find general utility in ligand design. The results continued to support the viability of substitutions at C2, C5, and C6 with the most favorable disubstitutions being C2/C5, C2/C6, and C4/C6, which correspond to the 2,5-, 2,6-, and 2,4-dichloro analogs. The 3,5-dichloro analog is also predicted to be better bound than the parent compound. A term that could be added for the 3,5- and 2,6-analogs is a factor of two symmetry benefit (-0.4 kcal/mol) over the unsymmetrical 2,5- or 2,4-analogs, since only one conformer is favorable in the latter cases (C2/C5 and C4/C6).
Table 1.
MC/FEP Results for the Change in Free Energy of Binding upon Introducing Two Chlorines in the Terminal Phenyl Ringa
 | ||
|---|---|---|
| H to Cl | ΔΔGb | σ |
| C2/C3 | 2.00 | 0.18 |
| C2/C4 | 2.43 | 0.20 |
| C2/C5 | -2.12 | 0.22 |
| C2/C6 | -3.69 | 0.25 |
| C3/C4 | 6.27 | 0.23 |
| C3/C5 | -1.13 | 0.19 |
| C3/C6 | 3.63 | 0.24 |
| C4/C5 | 1.79 | 0.23 |
| C4/C6 | -2.26 | 0.21 |
| C5/C6 | 2.52 | 0.20 |
ΔΔGb is the computed change in free energy of binding (kcal/mol) for introducing the two chlorines; ±σ is the computed uncertainty. C6 is proximal to Tyr181 in the binding site (see Figure 1).
Related experimental results are presented in Table 2. The methyl group in 1 was replaced by chlorine to give 5, which is more potent against the WT virus with an EC50 of 1.2 μM. For substitution in the terminal phenyl ring, 2-Cl substitution (6) was found to be more favorable than the 3-Cl alternative (7) consistent with the computed results for C2(C6) and C3(C5). The disubstituted compounds also mostly followed the computed trends with the most potent being the 2,6-analog (11) at 310 nM, which was followed by the 2,5- and 3,5-analogs, 10 and 9, at 380 nM and 1.3 μM. The 2,4-dichloro analog 8 was less active than anticipated and the range of activities was compressed from what might have been expected from the computed ΔΔGb values. The compression is normal and may result from comparing free energies of binding with results of cell-based assays and force-field inadequacies.4,5 Overall, a ca. 15-fold gain in potency was achieved in going from 1 to 10 or 11. It is notable that the FEP calculations pointed out the viability of 2,5- and 2,6-disubstitution, whereas the literature on more potent HEPT and emivirine analogs is dominated by 3,5-disubstituted cases.23 The same is true for the compounds in the 3-series.24 In the absence of the FEP results, even with display of the optimized structures, the preferences are not obvious. Most of the disubstituted possibilities look reasonable with the possible exception of X or Y = 4-Cl, which appears to yield a steric conflict with Trp229.
Table 2.
Anti-HIV-1 Activity (EC50) and Cytotoxicity (CC50), μM, of Diphenylmethane Derivatives
 | ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | X | Y | R2 | EC50 | CC50 |
| 1 | 4-Me | H | H | H | 4.8 | 72 |
| 5 | 4-Cl | H | H | H | 1.2 | 23 |
| 6 | 4-Cl | 2-Cl | H | H | 0.62 | 28 |
| 7 | 4-Cl | 3-Cl | H | H | 1.5 | 11 |
| 8 | 4-Cl | 2-Cl | 4-Cl | H | 2.9 | 88 |
| 9 | 4-Cl | 3-Cl | 5-Cl | H | 1.3 | 10 |
| 10 | 4-Cl | 2-Cl | 5-Cl | H | 0.38 | 15 |
| 11 | 4-Cl | 2-Cl | 6-Cl | H | 0.31 | 49 |
| 12 | 4-Cl | H | H | Me | 2.4 | 22 |
| 13 | 4-Cl | H | H | F | 2.5 | 27 |
| 14 | 4-Cl | H | H | Cl | 2.2 | 21 |
| 15 | 5-Cl | 2-Cl | H | H | 0.41 | 12 |
Several additional diphenylmethane derivatives were prepared. The results for 12 - 14 in comparison with 5 demonstrate that substitution at the 5-position in the uracil ring is not beneficial, while the results for 15 vs. 6 show that a 5-Cl substituent in the central ring is somewhat preferred to the 4-Cl isomer.
Catechol Diethers
The next consideration was the position of the oxygen in the linkers, especially given the alternatives suggested by 2 and 3. Thus, MC/FEP calculations were executed for perturbing structure 16 with X = Y = Z = CH2 to the three compounds in which X, Y, or Z are individually oxygen. The resultant ΔΔGb values were -5.95, 0.64, and -2.07 kcal/mol, respectively, with uncertainties of ±0.3 kcal/mol. Thus, the phenoxy substructure as in 3 was predicted to be much favored. This likely reflects conformational preferences.25 It is easier for a diphenyl ether fragment than a diphenylmethane one to achieve the near perpendicular arrangement, illustrated in Figure 1, which is most complimentary to the positioning of Tyr188 and Trp229. In fact, when a gas-phase conformational search is performed for the parent o-methoxy derivatives of diphenylmethane and diphenyl ether with the OPLS-AA force field, conformers 17 and 18 are the global minima. For 17, there is only this minimum, while there are 5 unique minima for 18. Conformer 19 is the second minimum, only 0.37 kcal/mol higher in energy than 18. Thus, catechol diethers like 18 are well pre-organized to bind to HIV-RT in the desired manner (Figure 1).

Given these results, a complete dichlorine FEP scan was performed for the model inhibitor in Table 3. The results favor 2,3-, 2,5-, and 3,5-substitution with the 2,5-pattern, illustrated in Figure 1, the most favored. A mono-chlorine scan for the catechol ring was also performed for the corresponding 3,5-dichlorophenyl analog. This yielded ΔΔGb values of -1.2, -2.6, -3.1, and -2.8 kcal/mol (±0.2 kcal/mol) for introducing a chlorine at the 3’, 4’, 5’, and 6’ positions. Consequently, the synthetic focus turned to catechol diethers with the favored substitution patterns in both rings. However, since SNAr chemistry was envisioned for the synthesis of the core (Schemes 2 and 3), an activating group was needed in one of the rings; cyano groups were chosen as they are the most similar sterically to chlorine and model-building indicated that they should be viable.
Table 3.
MC/FEP Results for the Change in Free Energy of Binding (kcal/mol) upon Introducing Two Chlorines in a Catechol Diether
 | ||
|---|---|---|
| H to Cl | ΔΔGb | σ |
| C2/C3 | -3.87 | 0.14 |
| C2/C4 | 1.88 | 0.20 |
| C2/C5 | -5.56 | 0.18 |
| C2/C6 | 1.44 | 0.21 |
| C3/C4 | 2.82 | 0.18 |
| C3/C5 | -3.28 | 0.15 |
| C3/C6 | 6.26 | 0.24 |
| C4/C5 | 2.16 | 0.17 |
| C4/C6 | 6.42 | 0.30 |
| C5/C6 | 4.48 | 0.19 |
As recorded in Table 4, headway was rapidly made. The results for 20 – 25 show that the 2,5-, 3,5-, and 2,6-substitution patterns for the phenoxy ring all provide active compounds and a chlorine is indeed preferred at the 5’ position in the catechol ring over the 4’-alternative. 25 is a potent NNRTI with an EC50 of 14 nM, and it has a large safety margin, since no cytotoxicity was observed to the limit of the tested concentration range, 100 μM. The results for 26 and 27 then reconfirm that small substituents at C5 in the uracil ring have little effect. However, potency is dramatically lost by replacement of the 5’-Cl by cyano in 28 and 29. Modeling indicates that the 5’-nitrile nitrogen in the complexes is ca. 3.1 Å from the carbonyl oxygen of Lys101. In addition, if a 5-cyano group in the phenoxy ring is positioned over the catechol ring, there would be dipole-dipole repulsion between the two cyano groups. The situation is strikingly relieved by replacing the phenoxy cyano group with chlorine in progressing to 32, which brings the anti-HIV activity back to 20 nM. The good potency, 43 nM, for the 2,5-dichloro isomer 31 (Figure 1) is also consistent with the expectations from the FEP results, and the benefit of the second chlorine is apparent in comparison to the results for 30. The difference in performance of the 3-chloro,5-cyanophenoxy substituent depending on the substitution of the catechol ring is notable (22, 25, 28). The spectroscopic characterization of the structures for some of the compounds was also confirmed by crystallography, e.g., for 20 in Figure 2. It is noted that the conformation of 20 in the crystal lattice is identical to that for the aag conformer of 31 in Figure 1.
Table 4.
Anti-HIV-1 Activity (EC50) and Cytotoxicity (CC50), μM, of Catechol Diether Derivatives
 | ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | Xa | Y | R2 | EC50 | CC50 |
| 20 | 4-Cl | 3-CN | 5-CN | H | 0.14 | 80 |
| 21 | 4-Cl | 2-Cl | 6-CN | H | 0.12 | 50 |
| 22 | 4-Cl | 3-Cl | 5-CN | H | 0.090 | 33 |
| 23 | 5-Cl | 2-Cl | 5-CN | H | 0.100 | 60 |
| 24 | 5-Cl | 3-CN | 5-CN | H | 0.046 | >100 |
| 25 | 5-Cl | 3-Cl | 5-CN | H | 0.014 | >100 |
| 26 | 5-Cl | 3-Cl | 5-CN | F | 0.013 | 17 |
| 27 | 5-Cl | 3-Cl | 5-CN | Cl | 0.017 | 31 |
| 28 | 5-CN | 3-Cl | 5-CN | H | 6.0 | >100 |
| 29 | 5-CN | 3-Cl | 5-CN | Cl | 5.3 | 94 |
| 30 | 5-CN | 2-Cl | H | H | 0.27 | >100 |
| 31 | 5-CN | 2-Cl | 5-Cl | H | 0.043 | 71 |
| 32 | 5-CN | 3-Cl | 5-Cl | H | 0.020 | >100 |
| 40 | 5-CN | 3-CV | H | H | 0.015 | >100 |
| 41 | 5-Cl | 3-CV | H | H | 0.005 | 19 |
| 42 | 5-Cl | 3-CV | 5-Cl | H | 0.000055 | 10 |
| 43 | 5-Cl | 3-CV | 5-F | H | 0.0032 | 48 |
| 44 | 5-F | 3-CV | 5-F | H | 0.00032 | 45 |
| 45 | 5-NHMe | 3-CV | 5-Cl | H | 0.020 | 30 |
| 46 | 3-F,5-CN | 2-Cl | H | H | 0.16 | >100 |
| 47 | 3-Cl,5-Cl | 2-Cl | H | H | 0.83 | 13 |
| 48 | 5-CN | 3-MOEO | H | H | 0.54 | >100 |
| 49 | 5-CN | 3-HOPO | H | H | 1.8 | >100 |
CV = (E)-cyanovinyl; MOEO = methoxyethoxy; HOPO = 3-hydroxypropan-1-oxy.
Figure 2.

Small molecule crystal structure of 20. Details are in the Supplementary Information.
At this point, consideration was given to possible replacement of the uracil group and/or variation of the linking chain. Though multiple options were tried, none emerged as competitive with the uracilylethoxy substituent. For example, 33, the 4-pyridinyl analog of 31, was synthesized and found to worsen the EC50 value 18-fold to 0.76 μM. The isomer 34 with the methylenoxymethyl linker was significantly less potent still (12 μM), and shortening the linker to OCH2 in 35 also did not improve the activity (1.8 μM). The 3-pyridinyl-, 2,4-pyrimidinyl-, 5-pyrazolyl-, 5-oxazolyl-, and 4-pyridinyl-N-oxide-analogs of 34 also showed no activity below μM-levels.

Thus, the transfer of parts of a potent inhibitor to a less potent one, e.g., the methyleneoxymethyl linker of 2 to 33 or 35, is an undependable strategy.26 Conformational differences are affected by the rest of the structures. For the present catechol derivatives, in conformational searches using OPLS-AA for 2-ethoxyanisole, the aa and ga conformers for the ethoxy group are the lowest in energy; they correspond to the conformers in Figure 1. For 2-methoxymethylanisole, the corresponding ga conformer is not an energy minimum; it collapses to aa. Finally, the importance of the uracilylmethyl group was clearly demonstrated through the preparation of 36, whose EC50 of 9.9 μM can be compared to the 14 nM for 25. The benefits of additional van der Waals interactions and the hydrogen bond between Lys103 and the uracilyl NH are likely contributors. Thus, deletion of the uracil, which might be suggested by considering emivirine (4), is detrimental.
By this juncture, some results on the activity of the more potent compounds towards variant strains of HIV-1 containing the Tyr181Cys mutation and the challenging Lys103Asn/Tyr181Cys double mutation in HIV-RT had been obtained (Table 5). Optimism might be justified from the general topological similarities of 3 and the present catechol diethers and the fact that 3 is reported to show 1-10 nM activity towards WT HIV-1 and many variants including Y181C and K103N/Y181C.8 However, the catechol diethers through compound 32 are not as potent against WT HIV-1 as 3 (2 nM), and the performance against the Y181C variant and double mutant was also found to be diminished (Table 5). The best compound at this point was 27, which has EC50 values of 17, 240, and 570 nM towards WT, Y181C, and the K103N/Y181C variants. The 14- and 34-fold ratios between the variant and WT activities are better than typical,24 so it seemed that a prerequisite for further improvement was to drive down the WT EC50 by another factor of 10 or more.
Table 5.
Anti-HIV Activity (EC50) and Average Cytotoxicity (CC50), μM, of Catechol Diether Derivatives and Reference NNRTIs for WT and Viral Variants.
| Compd | EC50 |
CC50 | ||
|---|---|---|---|---|
| WT | Y181C | K103N/Y181C | ||
| 20 | 0.14 | 21 | 2.2 | 77 |
| 22 | 0.090 | 6.7 | 1.2 | 32 |
| 25 | 0.014 | 0.52 | 1.7 | >100 |
| 26 | 0.013 | 0.62 | 1.7 | 13 |
| 27 | 0.017 | 0.24 | 0.57 | 21 |
| 31 | 0.043 | 0.92 | 2.8 | 59 |
| 32 | 0.020 | 0.80 | >43 | >61 |
| 40 | 0.015 | 0.98 | 8.0 | >90 |
| 41 | 0.005 | 0.37 | 1.6 | 22 |
| 42 | 0.000055 | 0.049 | 0.22 | 10 |
| 43 | 0.0032 | 0.15 | 0.90 | 48 |
| 44 | 0.00032 | 0.016 | 0.085 | 45 |
| 45 | 0.020 | 2.8 | ND | 30 |
| nevirapine | 0.11 | >100 | >100 | >100 |
| nevirapinea | 0.081 | >10 | >10 | |
| efavirenz | 0.002 | 0.010 | 0.030 | 15 |
| efavirenza | 0.0014 | 0.002 | 0.037 | |
| etravirine | 0.001 | 0.008 | 0.005 | 11 |
| etravirinea | 0.001 | 0.007 | 0.004 | |
| rilpivirine | 0.00067 | 0.00065 | 0.002 | >1 |
| rilpivirinea | 0.0004 | 0.0013 | 0.001 | 8 |
Literature data; see ref. 28.
Cyanovinyl analogs
To this end, introduction of a cyanovinyl group in the terminal phenyl ring was considered. This had proved profitable in another series, which included 37,27 and it also finds precedent in R165481 (38)8 and rilpivirine (39).28 All three of these cyanovinyl containing NNRTIs have activities below 5 nM for both WT HIV-1 and the Y181C-containing variant. However, in view of the caveats above concerning the methylenoxymethyl linker and uracil, modeling was carried out, which did indicate that a m-cyanovinyl group could be incorporated in the current series (Figure 3). It should make favorable van der Waals contact with Trp229 and be positioned between Tyr188 and Phe227, as is observed in the crystal structures for the complexes of 38 and 39.8,28 Since rilpivirine is a marketed drug, potential liabilities of the cyanovinyl group as a weak Michael acceptor seem acceptable.
Figure 3.

Computed structure of 42 (JLJ0494) bound to HIV-RT illustrating the positioning of the cyanovinyl group between Tyr188 and Phe227. Carbon atoms of 42 are colored gold.

Thus, continuing from 32 in Table 4, the 3-cyanovinyl analog 40 was prepared and showed improved activity at 15 nM. Switching to chlorine instead of cyano at the 5-position in the catechol ring brought an expected further enhancement, specifically, to 5 nM for 41. Then, for reintroduction of a chlorine in the phenoxy ring, the 4-, 5-, and 6-Cl isomers were considered with the cyanovinyl group at the 3-position. Energy minimizations for the complexes of the three alternatives indicated a preference for substitution at C5 or C6 based on the protein-ligand interaction energies of -70.0, -73.9, and -73.1 kcal/mol. The interaction energy for the reference compound 41 is -71.4 kcal/mol. The 4-chlorine leads to a steric clash with Trp229, while the 5-chlorine projects into a pocket formed by Pro95, Pro97, Leu100, and Tyr181, and the 6-chlorine nuzzles between Leu100 and Tyr181. However, for the C5 and C6 options, evaluation of the conformational energetics suggests more ligand strain for the C6 isomer owing to the placement of the 6-Cl over the catechol ring. In view of the interaction energy improvement for addition of the 5-chlorine and the anticipated benefits of burial of more hydrophobic surface area, a significant activity boost was expected.
Thus, synthesis of the 3-cyanovinyl, 5-Cl analog 42 was carried out and the activity results were gratifying. It is an extraordinarily potent NNRTI. The initial WT assay yielded an EC50 of 24 pM. To our knowledge, this is the lowest EC50 for an NNRTI that has been reported. Several pyrimidinone “SDABO” analogs have been reported to have EC50 values for WT HIV-1 (NL4-3) in the 70 – 440 pM range,29 and 39 is another very potent NNRTI.28 42 was re-assayed side-by-side with 39 yielding EC50s of 55 and 670 pM, which are the values reported in Table 5. A 5- or 6-substituent in the phenoxy ring is also expected to help fill the space vacated upon mutation of Tyr181 to cysteine. This notion and the benefits of the improved activity for the cyanovinyl-containing inhibitors yielded EC50 values for 42 of 49 and 220 nM for the variant HIV-1 strains containing the Y181C and K103N/Y181C mutations (Table 5).
Close analogs of 42 with replacement of the 5-Cl with fluorine (43) or both chlorines with fluorines (44) were then prepared. 43 is significantly less active at 3.2 nM for the WT virus, while 44 is still very potent at 320 pM. Furthermore, 44 shows improved potency towards the viral variants, 16 nM for Y181C and 85 nM for K103N/Y181C (Table 5). The activity patterns for NNRTIs towards viral variants are often not straightforward to interpret. For example, the most potent S-DABO in one report have EC50 values of 100-400 pM towards WT HIV-1, but are no better than 870 nM towards a K103N-bearing variant.29a However, it is generally thought that smaller, flexible inhibitors have an advantage in being able to better adjust to the structural changes that accompany point mutations.28b This can help rationalize the improved performance of the difluoro analogue 44 over 42 towards the mutant strains and the still better results with 39. In particular, greater contact with Lys103 is expected for the present compounds (Figure 3) than for rilpivirine. Thus, it is not surprising that mutations of Lys103 may be more detrimental for 42– 44.
Table 5 includes data for four approved drugs with results from both our measurements using infected MT2 cells and those of Janssen et al. using MT4 cells;28 the directly comparable results are almost all within a factor of 2. In comparison to the approved drugs in Table 5, 42 is by far the most potent towards the wild-type virus. 42 and 44 also show good potency towards the two mutant strains, though not at the low-nanomolar levels of rilpivirine (39). The relatively low cytotoxicity, CC50, towards human T-cells of many of the present compounds in comparison to the most potent drugs is also notable.
The computed structure for the complex of 42 (Figure 3) indicates that 42 fills the space extremely well in the channels running from Tyr181 past Trp229 and towards Phe227 and Pro236. The hydrogen bond with the carbonyl group of Lys103 remains notable. Furthermore, there may also be a boost for a halogen bond between the 5-Cl and the carbonyl oxygen of Pro95.30 This is illustrated in Figure 4, though standard force fields including OPLS do not account for the benefits of halogen bonding.31 The simple point-charge model with a partial negative charge on chlorine is inadequate to describe correctly the electrostatic field along the C-Cl bond axis.30 Nevertheless, the present, computed O-Cl distance of 3.43 Å is near the ideal value of ca. 3.3 Å for such interactions.30,31 It may be noted that when the complex is reoptimized with the only change being replacement of the cyanovinyl group in 42 by chloro or cyano, the O-Cl distance increases to 3.62 and 3.72 Å, respectively. The shift towards Pro95 for 42 improves the contact between the first methine unit of the vinyl group and Trp229. Thus, the optimal halogen bonding likely requires both the 3-chloro and 5-cyanovinyl groups. The significant decrease in activity in going from 42 to 43 is also consistent with loss of the halogen bond upon replacement of chlorine with fluorine. Notably, a recent report on inhibition of cathepsin L also found large binding enhancements upon introduction of a halogen bond between a halophenyl group of the inhibitors and a backbone carbonyl group.32 In comparison to the unsubstituted case, introduction of a halogen bond with chlorine, bromine, and iodine enhanced the binding by factors as large as 24, 49, and 74, respectively.32
Figure 4.
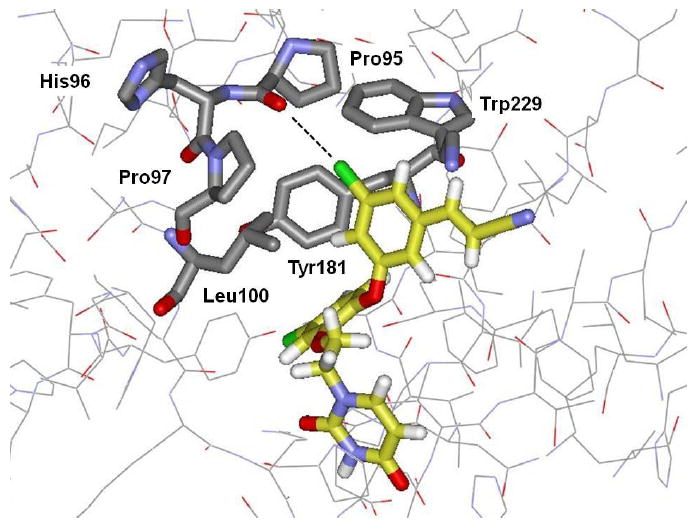
Computed structure of 42 bound to HIV-RT illustrating the possible halogen bond between the oxygen of Pro95 and the 5-chlorine in the terminal phenyl ring of the inhibitor. The O-Cl distance is 3.4 Å.
A few additional compounds were synthesized. (1) The analog of 42 with a methylamino group at C5 in the catechol ring, 45, is much less active (20 nM). Though it might benefit from a favorable electrostatic interaction between the amino group and the carbonyl of Lys101, the dehydration penalty would be greater than for 42. (2) Returning to 2 and 3, one might be motivated to include a substituent at the 3-position in the catechol ring, though the FEP results noted above indicated that substitution at that site with chlorine would be the least beneficial of the four possibilities. Addition of the 3-fluoro group in 46 (0.16 μM) was found to provide slight improvement over the results for 30 (0.27 μM), while the 3-chlorine in 47 (0.83 μM) was detrimental, since the reference compound would be expected to be more potent than 30. In this case, the reason why the mapping from the precedents fails is likely associated with the fact that 2 and 3 both form a hydrogen bond with their NH groups and the carbonyl of Lys101. As the 5-Cl or 5-CN in 46 or 47 aligns with the NH, the present compounds are pushed away and rotated from Lys101, which changes the positioning of the catechol ring versus the central rings in 2 and 3. (3) As the region behind Tyr188, Phe227, and Trp229 in Figure 3 opens into a solvent filled channel heading towards the polymerase active site, replacement of the (E)-cyanovinyl group with longer chains that could extend into this region was explored. In comparison to 40, 48 with a methoxyethoxy substituent was found to retain significant activity (0.54 μM), while the more hydrophilic hydroxypropoxy alternative in 49 diminishes the activity to 1.8 μM.
Other diarylethers
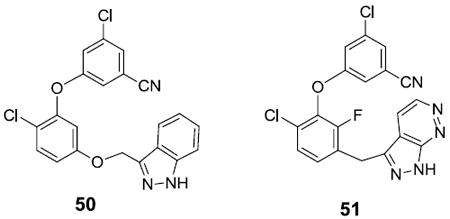
While this work was in progress, reports of other NNRTIs with diarylether substructures appeared from Merck and Roche.33,34 The illustrated 50 and 51 appear to be the most promising compounds from these efforts. They are clearly far more similar to each other than to 42 - 44. Notably, the central benzene ring in 50 and 51 is 1,3-disubstituted with the aryl appendages, while here it is 1,2-disubstituted. The lengths of the linkers to the heteroaryl group in the compounds are also different, the heterocycle is a monocyle in 42 - 44, the cyanovinyl group is not present in 50 and 51, and they have a chlorine adjacent to the phenoxy oxygen, which was not found to be desirable in the present series (e.g., 47). 50 is reported to have anti-HIV activities in cell assays of 4.7, 13.8, and 141 nM for the WT, Y181C, and K103N/Y181C variants,33 while the corresponding values for 51 are 1, 1, and 4 nM.34a Thus, these compounds are less potent towards wild-type HIV-1 than 42 or 44, while 51 has the best results for the mutant strains.
CONCLUSION
In summary, the present closely-coupled experimental and computational efforts began with virtual screening, which led to the intriguing core structure 1 starting from the crystal structures of 2 and 3. Though the anti-HIV activity of 1 was modest, 5 μM, with the aid of the computational analyses especially MC/FEP results, it was possible to evolve 1 into the 55-pM anti-HIV agent 42 (Scheme 5). Key advances included recognition of optimal substitution patterns for the terminal phenyl ring, and the benefits of progressing to the catechol diether core, placement of a substituent at the 5-position in the catechol ring, and introduction of a cyanovinyl group in the terminal phenyl ring. Along the way examples were provided where false assumptions could be made about expected gains from precedents such as transferabilty of the methylenoxymethyl linker from 2, deletion of the terminal heterocycle as in 4, or addition of a halogen adjacent to the phenoxy ring as in 3. When a change as large as replacement of a central pyridinone ring by a benzene ring is made, optimization must start afresh without assumptions of fragment transferability from active precedents. Further evolution of 42 - 44 is on-going including crystallographic investigations and preparation of analogues to tune physical properties and activity towards variant strains of HIV-1.
Scheme 5.

Supplementary Material
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551) for support. Receipt of reagents through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is also greatly appreciated. Gratitude is also expressed to Dr. Julian Tirado-Rives for computational assistance and to Dr. Christopher D. Incarvito for the small molecule X-ray crystallography.
ABBREVIATIONS USED
- HIV
human immunodeficiency virus
- HIV-RT
HIV reverse transcriptase
- NRTI
nucleoside inhibitor of HIV-RT
- NNRTI
non-nucleoside inhibitor of HIV-RT
- OPLS
optimized potentials for liquid simulations
- OPLS-AA
OPLS all-atom
- CM1A
charge model 1A
- DIAD
diisopropyl azodicarboxylate
Footnotes
Supporting Information. Synthetic details, NMR and HRMS spectral data for the new compounds in Tables 2 and 4, WT assay curves for 42, and X-ray data for 20. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Code CCDC ID: The cif file has been included in the Supporting Information for deposit by the journal.
References
- 1.(a) Flexner C. HIV drug development: the next 25 years. Nature Rev Drug Disc. 2007;6:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]; (b) De Clerq E. The design of drugs for HIV and HCV. Nature Rev Drug Disc. 2007;6:1001–1018. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]; (c) Jochmans D. Novel HIV-1 reverse transcriptase inhibitors. Virus Res. 2008;134:171–185. doi: 10.1016/j.virusres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal Structure at 3.5 Å Resolution of HIV-1 Reverse Transcriptase Complexed with an Inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403.. For a review, see: Prajapati DG, Ramajayam R, Yadav MR, Giridhar R. The search for potent, small molecule NNRTIs: A review. Bioorg Med Chem. 2009;17:5744–5762. doi: 10.1016/j.bmc.2009.06.060.
- 3.(a) Adams J, Patel N, Mankaryous N, Tadros M, Miller CD. HIV/AIDS: Nonnucleoside Reverse Transcriptase Inhibitor Resistance and the Role of the Second-Generation Agents. Ann Pharmacotherapy. 2010;44:157–165. doi: 10.1345/aph.1M359. [DOI] [PubMed] [Google Scholar]; (b) Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The Challenge of Finding a Cure for HIV Infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 4.For a review, see: Jorgensen WL. Efficient Drug Lead Discovery and Optimization. Acc Chem Res. 2009;42:724–733. doi: 10.1021/ar800236t.
- 5.(a) Zeevaart JG, Wang L, Thakur VV, Leung CS, Tirado-Rives J, Bailey CM, Domaoal RA, Anderson KS, Jorgensen WL. Optimization of Azoles as Anti-HIV Agents Guided by Free-Energy Calculations. J Am Chem Soc. 2008;130:9492–9499. doi: 10.1021/ja8019214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leung CS, Zeevaart JG, Domaoal RA, Bollini M, Thakur VV, Spasov K, Anderson KS, Jorgensen WL. Eastern extension of azoles as non-nucleoside inhibitors of HIV-1 reverse transcriptase; cyano group alternatives. Bioorg Med Chem Lett. 2010;20:2485–2488. doi: 10.1016/j.bmcl.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nichols SE, Domaoal RA, Thakur VV, Bailey CM, Wang L, Tirado-Rives J, Anderson KS, Jorgensen WL. Discovery of Wild-type and Y181C Mutant Non-nucleoside HIV-1 Reverse Transcriptase Inhibitors Using Virtual Screening with Multiple Protein Structures. J Chem Inf Model. 2009;49:1272–1279. doi: 10.1021/ci900068k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Hopkins AL, Ren J, Esnouf RM, Willcox BE, Jones EY, Ross C, Miyasaka T, walker RT, Tanaka H, Stammers DK, Stuart DI. Complexes of HIV-1 Reverse Transcriptase with Inhibitors of the HEPT Series Reveal Conformational Changes Relevant to the Design of Potent Non-Nucleoside Inhibitors. J Med Chem. 1996;39:1589–1600. doi: 10.1021/jm960056x. [DOI] [PubMed] [Google Scholar]; (b) Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S, Stuart DI, Stammers DK. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J Mol Biol. 2001;312:795–805. doi: 10.1006/jmbi.2001.4988. [DOI] [PubMed] [Google Scholar]
- 8.Himmel DM, Das K, Clark AD, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. Crystal structures for HIV-1 reverse transcriptase in complexes with three pyridinone derivatives: a new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J Med Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 9.Baba M, Tanaka H, Miyasaka T, Yuasa S, Ubasawa M, De Clercq E. Hept derivatives: 6-Benzyl-1-ethoxymethyl-5-isopropyluracil (MKC-442) Nucleosides Nucleotides. 1995;14:575–583. [Google Scholar]
- 10.Jorgensen WL. QikProp, v 3.0. Schrödinger LLC; New York: 2006. [Google Scholar]
- 11.McKay AF, Baker HA, Gaudry R, Garmaise DL, Ranz RJ. Bacteriostats. VII. Substituted Benzylphenols. J Med Chem. 1963;6:816–817. doi: 10.1021/jm00342a049. [DOI] [PubMed] [Google Scholar]
- 12.Langer R, Buysch HJ. Verfahren zur Monobenzylierung von p-substituierten Phenolen. 1993 EP0538704. [Google Scholar]
- 13.Novikov MS, Ozerov AA. The Silyl Method for the Synthesis of 1[-2(Phenoxy)ethyl]uracils. Chem Het Comp. 2005;41:905–908. [Google Scholar]
- 14.Frieden M, Giraud M, Reese CB, Song Q. Synthesis of 1-[cis-3-(hydroxymethyl)cyclobutyl]-uracil, -thymine and -cytosine. J Chem Soc Perkin Trans. 1998;1:2827–2832. [Google Scholar]
- 15.Reese CB, Stewart JCM. Methoxyacetyl as a protecting group in ribonucleoside chemistry. Tetrahedron Lett. 1968;40:4273–4276. doi: 10.1016/s0040-4039(00)76405-7. [DOI] [PubMed] [Google Scholar]
- 16.Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Antiviral activity of 2’,3’-dideoxy-β-L-5-fluorocytidine (β-L-EddC) and 2’,3’-dideoxy-β-L-cytidine (β-L-ddC) against hepatitis B virus and human immunodeficiency virus type 1 in vitro. Biochem Pharmacol. 1994;47:171–174. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 17.Ray AS, Yang Z, Chu CK, Anderson KS. Novel use of a guanosine prodrug approach to convert 2’,3’-didehydro-2’,3’-dideoxyguanosine into a viable antiviral agent. Antimicrob Agents Chemother. 2002;46:887–891. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Tirado-Rives J. Molecular Modeling of Organic and Biomolecular Systems Using BOSS and MCPRO. J Comput Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 19.For a review, see: Jorgensen WL, Thomas LT. Perspective on Free-Energy Perturbation Calculations for Chemical Equilibria. J Chem Theory Comput. 2008;4:869–876. doi: 10.1021/ct800011m.
- 20.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 21.Jorgensen WL, Tirado-Rives J. Potential energy functions for atomic-level simulations of water, and organic and biomolecular systems. Proc Natl Acad Sci U S A. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 23.Tanaka H, Takashima H, Ubasawa M, Sekiya K, Inouye N, Baba M, Shigeta S, Walker RT, De Clerq E, Miyasaka T. Synthesis and Antiviral Activity of 6-Benzyl Analogs of 1-[(2-Hydroxyethoxy)methyl]-5-(phenylthio)thymine (HEPT) as Potent and Selective Anti-HIV-1 Agents. J Med Chem. 1995;38:2860–2865. doi: 10.1021/jm00015a008. [DOI] [PubMed] [Google Scholar]
- 24.Benjahad A, Guillemont J, Andries K, Nguyen CH, Grierson DS. 3-Iodo-4-phenoxypyridinones (IOPY’s), a new family of highly potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2003;13:4309–4312. doi: 10.1016/j.bmcl.2003.09.045. [DOI] [PubMed] [Google Scholar]
- 25.Brameld KA, Kuhn B, Reuter DC, Stahl M. Small Molecule Conformational Preferences Derived from Crystal Structure Data. A Medicinal Chemistry Focused Analysis. J Chem Inf Model. 2008;48:1–24. doi: 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- 26.Pierce AC, Rao G, Bemis GW. BREED: Generating Novel Inhibitors through Hybridization of Known Ligands. Application to CDK2, P38, and HIV Protease. J Med Chem. 2004;47:2768–2775. doi: 10.1021/jm030543u. [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen WL, Bollini M, Thakur VV, Domaoal RA, Spasov K, Anderson KS. Efficient Discovery of Potent Anti-HIV Agents Targeting the Tyr181Cys Variant of HIV Reverse Transcriptase. J Am Chem Soc. 2011;133:15686–15696. doi: 10.1021/ja2058583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune M-P, Pauwels R, Das K, Clark AD, Jr, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48:1901–1919. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]; (b) Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc Nat Acad Sci USA. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Manetti F, Este JA, Clotet-Codina I, Armand-Ugon M, Maga G, Crespan E, Cancio R, Mugnaini C, Bernardini C, Togninelli A, Carmi C, Alongi M, Petricci E, Massa S, Corelli F, Botta M. Parallel Solution-Phase and Microwave-Assisted Synthesis of New S-DABO Derivatives Endowed with Subnanomolar Anti-HIV-1 Activity. J Med Chem. 2005;48:8000–8008. doi: 10.1021/jm050744t. [DOI] [PubMed] [Google Scholar]; (b) Radi M, Maga G, Alongi M, Angeli L, Samuele A, Zanoli S, Bellucci L, Tafi A, Casaluce G, Giorgi G, Armand-Ugon M, Gonzalez E, Este JA, Baltzinger M, Bec G, Dumas P, Ennifar E, Botta M. Discovery of Chiral Cyclopropyl Dihydro-Alkylthio-Benzyl-Oxopyrimidine (S-DABO) Derivatives as Potent HIV-1 Reverse Transcriptase Inhibitors with High Activity Against Clinically Relevant Mutants. J Med Chem. 2009;52:840–851. doi: 10.1021/jm801330n. [DOI] [PubMed] [Google Scholar]
- 30.For a review, see: Politzer P, Murray JS, Clark T. Halogen bonding: an electrostatically-driven highly directional noncovalent interaction. Phys Chem Chem Phys. 2010;12:7748–7757. doi: 10.1039/c004189k.
- 31.Ibrahim MAA. Molecular Modeling Study of Halogen Bonding in Drug Discovery. J Comput Chem. 2011;32:2564–2574. doi: 10.1002/jcc.21836. [DOI] [PubMed] [Google Scholar]
- 32.Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher J-M, Hartmann G, Banner DW, Haap W, Diederich F. Angew Chem Int Ed. 2011;50:314–318. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- 33.Tucker TJ, Saggar S, Sisko JT, Tynebor RM, Williams TM, Felock PJ, Flynn JA, Lai M-T, Liang Y, McGaughey G, Liu M, Miller M, Moyer G, Munshi V, Perlow-Poehnelt R, Prasad S, Sanchez R, Torrent M, Vacca JP, Wan B-L, Yan Y. The design and synthesis of diaryl ether second generation HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) with enhanced potency versus key clinical mutations. Bioorg Med Chem Lett. 2008;18:2959–2966. doi: 10.1016/j.bmcl.2008.03.064. [DOI] [PubMed] [Google Scholar]
- 34.(a) Sweeney ZK, Harris SF, Arora N, Javanbakht H, Li Y, Fretland J, Davidson JP, Billedeau JR, Gleason SK, Hirschfeld D, Kennedy-Smith JJ, Mirzadegan T, Roetz R, Smith M, Sperry S, Suh JM, Wu J, Tsing S, Villasenor AG, Paul A, Su G, Heilek G, Hang JQ, Zhou AS, Jernelius JA, Zhang F-J, Klumpp K. Design of annulated pyrazoles as inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2008;51:7449–7458. doi: 10.1021/jm800527x. [DOI] [PubMed] [Google Scholar]; (b) Sweeney ZK, Acharya S, Briggs A, Dunn JP, Elworthy TR, Fretland J, Giannetti AM, Heilek G, Li Y, Kaiser AC, Martin M, Saito YD, Smith M, Suh JM, Swallow S, Wu J, Hang JQ, Zhou AS, Klumpp K. Discovery and optimization of pyridazinone non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2008;18:4352–4354. doi: 10.1016/j.bmcl.2008.06.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.