

Abstract
The National Blood Foundation (NBF) support was critical in my research career development. The NBF support came in the form of a start-up seed grant that I got from the American Association of Blood Banks (AABB), an organization that advances the practice and standards of transfusion medicine and cellular therapies and an organization in which I am a proud member. The NBF grant enabled me to keep up with my transfusion medicine practice while pursuing my passion to be a physician scientist. During its funding period, I was able to obtain critical preliminary bench data and to secure several NIH grants with over million dollar direct cost. In addition, the knowledge gained from the NBF-supported projects is currently being translated into medical practice in my lab by testing on cord blood expansion. I am looking forward to spending the upcoming years of my professional career bridging bedside observations on transfusion medicine with bench experiences and then utilizing that bench-derived knowledge in the practice of transfusion medicine.
Background
While traditional blood banking focuses on the manufacture and distribution of non-nucleated blood components such as red cells, platelets and plasma obtained from healthy donors, modern blood bankers are pioneers in cellular therapy wherein nucleated live cells are introduced into a tissue for treatment or regeneration. One well-known example of cellular therapy is the use of hematopoietic stem/progenitor cell transplantation for treating patients with hematopoietic malignancies, which requires an understanding of the biology of hematopoiesis.
Hematopoiesis consists of a cascade of tightly regulated multistage events, during which pluripotent, self-renewing stem cells give rise to all blood cell lineages1. Myelopoiesis is a subdivision of this process wherein myeloid progenitor cells that are derived from uncommitted pluripotent stem cells differentiate into mature and functioning myeloid cells 2–6. These developmental and maturational processes depend on tightly controlled lineage-specific gene expression, which is regulated by numerous transcription factors. The activity of these transcription factors is controlled, at least in part, by cytokines 7. The complex interactions between these factors lead to exquisite control of hematopoietic differentiation and proliferation. Disruption of this delicate developmental cascade can result in leukemia.
Normal hematopoiesis and leukemogenesis are two very closely related biological processes. I first started my research lab in 2003 after a clinical transfusion medicine fellowship with Dr. Edward Snyder and a research fellowship with Dr. Diane S Krause from Yale University. At that time, I proposed that conducting basic research on both normal and leukemic hematopoiesis would facilitate our understanding on the molecular basis of cell therapies and help us to optimize existing therapeutic strategies as well as design new ones. However, as a young investigator and a transfusion physician who bears clinical duties, it was very challenging for me to receive grant support to initiate my proposed research projects. I am very grateful to the National Blood Foundation (NBF), which awarded me a research grant in 2005. This grant allowed me to use comparative approaches to study the gene family of transcription factor HSAL (SALL), particularly the role of HSAL2 and HSAL4, in normal hematopoiesis and leukemogenesis.
The function of the HSAL (SALL) gene family in development
Members of the HSAL gene family, HSAL1 to HSAL4, were originally cloned based on DNA sequence homology to Drosophila gene spalt (sal) 8. Sal is a non-clustered region-specific homeobox gene and is essential for the development of the posterior head and anterior tail segments of the fly9,10. Furthermore, sal plays an important role in the embryonic development of the larval tracheal system and the adult wing. Sal-related genes have been isolated from C. elegans11, fish 12, frogs (xenopus) 13,14, mice 15 and humans 8. Each of these homologues is expressed during embryonic development as well as in specific adult tissues. In humans, HSAL1 is mutated in patients with Townes-Brockes Syndrome (TBS) which is associated with urogenital, limb, anal and cardiac malformations 16–18. Defects in hematopoiesis have not been reported to date in patients with TBS. Similar to HSAL1, HSAL2 is expressed in the developing neuroectoderm of the brain, inner ear and urogenital ridge-derived structures such as testes, ovaries and kidneys. HSAL3 is mapped to human chromosome 18q2319. It has been suggested that the HSAL3 gene product may be involved in the phenotype of patients with 18q deletion syndrome, which is characterized by developmental delay, hypotonia, growth retardation, midface hypoplasia, hearing loss and tapered fingers 19. The latest HSAL gene member, HSAL4, is mutated in human Duane-radial ray syndrome (DRRS) and Instituto Venezolano de Investigaciones Cientìficas (IVIC) syndrome 20–23. Both are autosomal-dominant developmental disorders involving radial-sided hand anomalies and congenital strabismus. IVIC is also characterized by leukocytosis and thrombocytopenia, suggesting that HSAL4 may be involved in normal hematopoiesis 24. Murine Hsal4 or Sall4 plays a crucial role in development as well. Sall4-null mice die at E6.5 25–28. We have shown that murine HSal4 is essential in the maintenance of pluripotency and self-renewal properties of ES cells by interacting with two other key regulators in ES cells, Nanog and Oct4 28.
The role of HSAL2 in ovary cancer
Sal proteins belong to a group of C2H2 zinc finger transcription factors characterized by multiple finger domains distributed over the entire protein 8. Structural characteristics of human HSAL include a single C2H2 zinc finger near the N-terminus and a cluster of three C2H2 zinc fingers in the middle portion of the protein considered essential for DNA binding. HSAL2, like other Sal family proteins, has glutamate, proline and alanine rich sequences, suggesting potential transcriptional modulator functions. The HSAL2 gene is mapped to chromosome 14q12.1–13, a region that associates with loss of heterozygosity in 49% ovarian cancer 29 and 25% of bladder cancers. These findings, along with our studies that demonstrated loss of HSAL2 expression in some solid tumors30, suggest that HSAL2 may function as a tumor suppressor. Initial experiments in the Benjamin lab at the Harvard Medical School showed that by using a yeast two-hybrid system, mouse Sall2 (Hsal2) was able to bind to the large T antigen of polyoma virus, a DNA virus that induces a broad variety of neoplasms in its natural host 31. Disruption of this interaction results in resistance to tumor induction by large T antigen31. Re-expression of the HSAL2 gene by transfection inhibits the growth and DNA synthesis of the ovarian cancer cell line SKVO3 and inhibits tumor formation in nude mice, associated with induction of p21Cip1/Waf1 32. These observations further indicate that HSAL2 may have roles in suppressing tumorigenesis.
Loss of p53 and murine HSAL2 (Sall2) synergistically promotes lymphomagenesis
We have previously characterized HSAL2 expression in normal human tissues and some solid tumors 33. During my NBF funding period using the same approach we observed that HSAL2 expression was lost or reduced in some human leukemic cell lines, such as HL-60, and primary leukemic samples. As shown in Figure 1, in contrast to normal bone marrow, six cases of primary acute myeloid leukemia had decreased HSAL2 expression, suggesting HSAL2 may be involved in leukemogenesis.
Figure 1. Decreased HSAL2 transcripts in human acute myeloid leukemias.
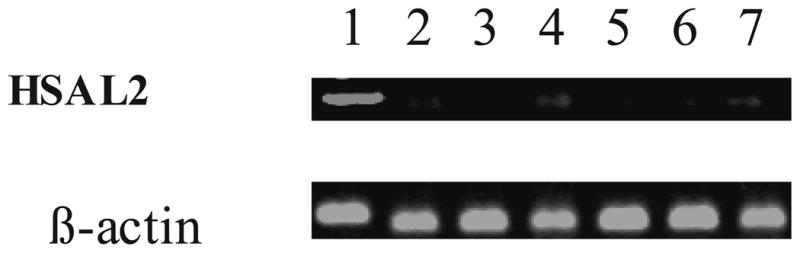
Primers used for RT-PCR are the same as those in our previous publication33. Products of HSAL2 mRNA were investigated in various human leukemias and normal BM after 35 cycles of amplification. Lane 1 is normal bone marrow, Lane 2, 4&7 are AML M2, Lane 3&6 are AML M4, and Lane 5 is AML M1. A comparable amount of β-actin cDNA, as determined by RT-PCR, after 23 cycles of amplification was observed.
The Benjamin’s group has provided strong evidence to show that HSAL2 acted as an ovarian cancer suppressor gene in vitro 31,34. However, to convincingly demonstrate an HSAL2-mediated cancer phenotype, we should be able to observe enhanced tumor susceptibility in Hsal2-deficient mice. To achieve this, we have obtained Hsal2 knockout mice from Dr. Ryuichi Nishinakamura from the University of Tokyo, Japan, and have been able to use these founder mice to characterize HSAL2 as a tumor suppressor gene in our laboratory. Genotyping of these mice confirmed the absence of the Hsal2 gene. The Hsal2 null mice (Hsal2−/−) appeared grossly normal, as demonstrated by Nishinakamura et al. 35; however, when we crossed these mice with tumor susceptible mice (p53−/−), the resulting mice, Hsal2−/− or Hsal2 −/+/p53−/− mice, exhibited significantly accelerated tumorigenesis and mortality rate compared to Hsal2+/+/p53−/− mice (Table 1 and Figure 2). The Hsal2−/− or Hsal2−/+/p53−/− mice also showed more advanced tumor progression with thymus T-cell lymphoma that metastasized to the liver, lung, kidney, bone marrow, peripheral blood and central nervous system, while in most Hsal2+/+/p53−/− mice, the lymphoma is limited to the thymus and adjacent organs such as lung. No myeloid leukemia was observed, which might be due to the fact that in the p53 mouse model, mice are more prone to develop and die from thymic tumors.
Table 1.
Mean Survival
| Genotype | Mice | Mean survival age (days) |
|---|---|---|
| Hsal2+/+/p53−/− | 10 | 139 |
| Hsal2+/−or−/−/p53−/− | 11 | 102 |
| Hsal2+/+/p53+/+ | 40 | >300 |
|
| ||
| Hsal2+/−or−/−/p53+/+ | 28 | >300 |
Note: None of the Hsal2−/−or+/−/p53+/+ animals developed tumors or needed to be sacrificed for ill health during the period of observation which lasted for 10 month. Hsal2+/−or−/−/p53−/− mice showed accelerated tumorigenesis and shorter survival age compared to that of Hsal2+/+/p53−/− mice (P=0.002)
Figure 2. Enhanced tumor susceptibility and shortened survival in Hsal2 -deficient mice.

Survival curves of Hsal2−/− or +/−/p53−/− and Hsal2+/+/p53−/−. All mice died from aggressive T cell lymphoblastic lymphomas.
The role of HSAL2 in normal hematopoiesis
Additionally, detailed phenotypic analysis of Hsal2-null mice by our group showed a decreased committed myeloid population in Hsal2−/− mouse bone marrow. Manual differentials were performed on 500 cells from four to six random fields. The obtained data correlated well with fluorescent activated cell sorting (FACS) analysis of the same cells using panels of antibodies against progenitors, myeloid, lymphoid and erythroid populations. Analysis using the above-mentioned two approaches on these Hsal2−/− mice in my lab showed a statistically significant decrease in the myeloid population in the bone marrow (Hsal2−/− vs WT mean 40.6+/−7.2% vs 51.5+/−4.6%, n=40, p<0.01). Because the absolute cellularity of the bone marrow was the same in Hsal 2−/− and WT mice, the decreased percentage in myeloid cells indicates a decrease in the absolute numbers of myeloid cells in the bone marrow.
To determine if the decrease in bone marrow myeloid population was due to a defect in myeloid progenitors, we performed CFU experiments using M3434 medium (Stem Cell Tech., Vancouver, Canada) with whole bone marrow cells from Hsal2−/− mice and age-matched WT controls.
Despite a similar total number of colonies observed in both groups, there were more and larger CFU-GMs in Hsal2-null CFU. In addition, these CFU-GMs in Hsal2-null CFU were mainly comprised of macrophages and immature myeloid cells rather than a mixed population of neutrophils and macrophages. The defect in myeloid maturation was supported by morphological examination of the cellular components of the CFUs. After 9–10 days, in WT CFU, all the myeloid cells were matured (band and segment neutrophils). In contrast, after 9–10 days, Hsal2-null CFUs (Figure 3) were composed of 50% mature myeloid cells and 50% immature myeloid cells (blasts, promyelocytes). These studies indicate that the decrease in the myeloid population in Hsal2−/− mice may be due to an impairment of or a delay in myeloid maturation.
Figure 3. Myeloid differentiation impairment of Hsal2 −/− bone marrow cells in vitro.
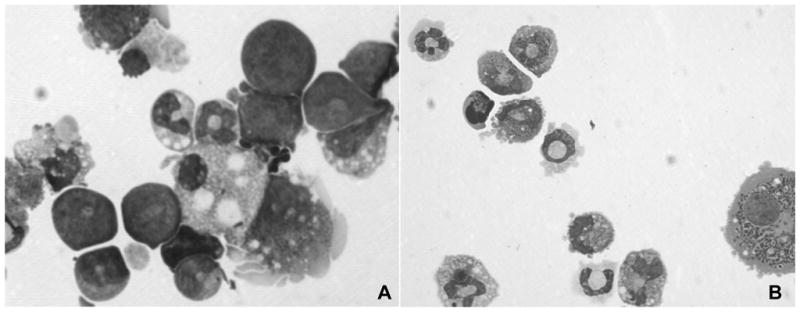
In day 9 CFU, a significant number of immature myeloid cells (50% blasts, indicated by double arrows) were still present in Hsal2 −/− bone marrow (A), while no immature myeloid cells remained in WT bone marrow (B). (Single arrow indicates mature neutrophiles).
To rule out the possibility that the hematopoietic defects associated with interruption of Hsal2 were caused, at least in part, by defects in the environment or bone marrow niche, we performed bone marrow transplantation (repopulation) assays. Whole bone marrows (1×106 cells/recipient) from age-matched WT and Hsal2−/− mice were transplanted into C57BL6 recipient mice after lethal irradiation (1000 cGy) (n=30). After 4 weeks, we studied the engraftment of the recipient bone marrows. Interestingly, the Hsal2−/− bone marrow recipients showed impaired myeloid engraftment with decreased myeloid population and myeloid differentiation arrest in comparison to WT bone marrow recipients (Figure 4A and B). This finding was also supported by FACS analysis. Recipients of Hsal2−/− bone marrow lacked the mature neutrophil population that co-expresses Gr-1 and Mac-1 (Figure 4C and D). However, long term post-transplantation follow-up at 4 and 8 months showed no difference in myeloid population between the WT and Hsal2−/− recipients. These results suggest a defect in myeloid maturation of Hsal2−/− mice is partially intrinsic to the hematopoietic cells, probably at the committed myeloid progenitor level. There may be additional environmental factor(s) that contribute to the myeloid maturation defects. No other hematologic defect was found in these Hsal2-null mice.
Figure 4. FACS analysis of recipient bone marrows from Hsal2 −/− and WT donors.

Forward and side scatter (FSC&SSC) shows that a recipient of WT donor (A) had a much larger myeloid population (single arrow) than a recipient of a Hsal2 −/− donor (B). Panels C&D show Gr-1 vs Mac-1 expression for cells gated on the indicated (single arrow) myeloid population in panels A&B, respectively. This myeloid population co-expressed Gr-1 and Mac-1 (double arrows) in the recipient of WT donor (C), which was lacking in the recipient of Hsal2−/− donor (D).
P21 has been shown to be a HSAL2 target gene. To understand the mechanism of HSAL2 involvement in hematopoiesis, we test if HSAL2 plays a role in cell cycle regulation by examining p21 expression in thymus and bone marrow. As shown in Figure 5, p21 was down-regulated in both organs in Hsal2 KO mice. This finding is consistent with previous reports on HSAL2 involvement in cell cycle regulation and may account for the hematologic defects that we observed.
Figure 5. The expression of P21 is altered in Hsal2−/− mice.

Western blot showing P21 downregulation in Hsal2-null mice (KO) as compared to that of the wild type controls (WT) in cells obtained from thymus and bone marrow (N=4). β-actin was used as a control.
The story of HSAL4 (SALL4)
Towards the second half of my NBF grant supporting period in 2005, my lab started to work on another HSAL gene family member, HSAL4 (or SALL4). In contrast to HSAL2, HSAL4 is an oncogene. We have cloned SALL4 and its isoforms (SALL4A and SALL4B). We used immunohistochemistry to demonstrate that SALL4 was constitutively expressed in primary acute myeloid leukemia cells (French American British, FAB: M1 to M5, N=81). Moreover, the expression of SALL4 RNA was quantitatively evaluated in bone marrow cells derived from acute myeloid leukemia (AML) and compared to that of the non-neoplastic hematopoietic cells from a purified CD34 progenitor pool, normal bone marrow and peripheral blood by qRT-PCR. SALL4 expression was present in the purified normal CD34+ cells, AML blasts, but absent in mature myeloid cells. We then tested the leukemogenic potential of constitutive overexpression of SALL4 in a murine model. All transgenic mice overexpressing SALL4B developed hematopoietic disorders, including myelodysplastic (MDS)-like symptoms and an AML transformation that disseminated to peripheral tissues including the spleen, liver, kidney and lymph nodes. The myelodysplastic features in these transgenic mice were pathologically similar to human MDSs and manifested as ineffective myelopoiesis. Dysplastic features such as Pseudo-Pelger-Huet-like atypical white cells and increased immature cells were detectable in the transgenic mouse peripheral blood. Both SALL4A and SALL4B were able to bind to β-catenin in vitro and synergistically enhanced the Wnt/β-catenin signaling pathway in a reporter gene assay. Our data suggests that the constitutive expression of SALL4 is causal to the leukemic phenotype. Our model should provide a useful platform to analyze the interaction of SALL4 with Wnt/β-catenin pathway in leukemia stem cells 36.
Going forward
Our later work on HSAL4 loss-of-function studies have demonstrated that HSAL4 is a key regulator of cell survival and apoptosis in leukemic cells37. The important role of SALL4 in normal hematopoietic stem cells and leukemic stem or initiating cells (LICs) is supported by its interactions with several key players implicated in self-renewal of HSCs and LICs - Wnt/β-catenin, Bmi137,38, and Pten (phosphatase and tensin homolog deletion on chromosome 10)39. In addition, HSAL4 is the only HSAL gene that can regulate other HSAL gene family members 40. It is also one of the few genes that play an important role in maintaining stem cell properties in embryonic stem cell, HSC and LIC.
Many questions still remain to be investigated. We are currently working on elucidating the mechanism of regulation of HSAL gene family. Despite our early promising in vitro data on Wilms’ tumor suppressor (WT1) gene in regulating HSAL2 33, the expression of these two genes in human leukemia samples is not correlated. Identification of upstream regulators of HSAL genes will be important in controlling the expression of these genes and therefore important in controlling their function(s).
We are also in the process of using the knowledge gained by the support of NBF to elucidate the roles of downstream targets of HSAL gene family, such as P21, Bmi-1, Wnt/β-catenin, and Pten in targeting leukemia as well as in expanding the hematopoietic stem/progenitor cells in cord blood. A small molecule screen is under way in order to identify the regulators of HSAL gene function using HSAL-expressing cell systems.
In summary, the NBF support was critical in my research career development. This grant helped me make strides in research while keeping up with my medical practice. With the funding it provided, I obtained data critical for securing subsequent NIH grants. Additionally, the results of the research I did while being supported by the NBF grant are now being used in projects that will hopefully translate to medical practice on expanding cord blood. I am excited to continue crossing the bridge between medical practice and bench research in my future career.
Acknowledgments
I thank Nikki R Kong, Kol Jia Yong and Claudia Tenen in assisting the preparation of the manuscript. In addition to NBF seed grant, this work was also supported in part through NIH grants RO1HL092437, RO1HL092437-A1S1 and PO1HL095489 and PO1DK080665 (to L.C).
Footnotes
Conflict of Interest: The author declares that she has no conflict of interest relevant to the manuscript submitted to Transfusion.
References
- 1.Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell. 1997;88:287–298. doi: 10.1016/s0092-8674(00)81867-x. [DOI] [PubMed] [Google Scholar]
- 2.Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol. 1996;16:4717–4725. doi: 10.1128/mcb.16.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ford AM, Bennett CA, Healy LE, Towatari M, Greaves MF, Enver T. Regulation of the myeloperoxidase enhancer binding proteins Pu1, C-EBP alpha, -beta, and -delta during granulocyte-lineage specification. Proc Natl Acad Sci U S A. 1996;93:10838–10843. doi: 10.1073/pnas.93.20.10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman AD. Transcriptional regulation of myelopoiesis. Int J Hematol. 2002;75:466–472. doi: 10.1007/BF02982108. [DOI] [PubMed] [Google Scholar]
- 5.Metcalf D. Stem cells, pre-progenitor cells and lineage-committed cells: are our dogmas correct? Ann N Y Acad Sci. 1999;872:289–303. doi: 10.1111/j.1749-6632.1999.tb08473.x. discussion 303–284. [DOI] [PubMed] [Google Scholar]
- 6.Metcalf D, Nicola NA. The Hemopoietic Colony-Stimulating Factors. Cambridge University Press; 1995. [Google Scholar]
- 7.Ward AC, Loeb DM, Soede-Bobok AA, Touw IP, Friedman AD. Regulation of granulopoiesis by transcription factors and cytokine signals. Leukemia. 2000;14:973–990. doi: 10.1038/sj.leu.2401808. [DOI] [PubMed] [Google Scholar]
- 8.Kohlhase J, Schuh R, Dowe G, Kuhnlein R, Jackle H, Schroeder B, Schulz-Schaeffer W, Kretzchmar H, Kohler A, Muller U, Raab-Vetter M, Burkhardt E, Engel W, Stick R. Isolation, characterization and organ-specific expression of two novel human zinc finger genes related to the Drosophila gene spalt. Genomics. 1996;38:291–298. doi: 10.1006/geno.1996.0631. [DOI] [PubMed] [Google Scholar]
- 9.Kuhnlein RP, Frommer G, Friedrich M, Gonsalez-Gaitan M, Weber A, Wagner-Bernholz UF, Gehring WJ, Jackle H, Schuh R. Spalt encodes an evolutionarily conservative zinc finger protein of novel structure which provides homeotic gene function in the head and tail region of Drosophila embryo. EMBO J. 1994;13:168–179. doi: 10.1002/j.1460-2075.1994.tb06246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurgens G. Head and tail development of the Drosophila embryo involves spalt, a novel homeotic gene. EMBO J. 1988;7:189–196. doi: 10.1002/j.1460-2075.1988.tb02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basson M, Horvitz HR. The Caenorhabditis elegans gene sem-4 controls neuronal and mesodermal cell development and encodes a zinc finger protein. Genes Dev. 1996;10:1953–1965. doi: 10.1101/gad.10.15.1953. [DOI] [PubMed] [Google Scholar]
- 12.Koster R, Stick R, Loosli F, Wittbrodt J. Medaka spalt acts as a target gene of hedgehog signaling. Development. 1997;124:3147–3156. doi: 10.1242/dev.124.16.3147. [DOI] [PubMed] [Google Scholar]
- 13.Onuma Y, Nishinakamura R, Takahashi S, Yokota T, Asashima M. Molecular cloning of a novel Xenopus spalt gene (Xsal-3) Biochem Biophys Res Commun. 1999;264:151–156. doi: 10.1006/bbrc.1999.1479. [DOI] [PubMed] [Google Scholar]
- 14.Hollemann T, Schuh R, Pieler T, Stick R. Xenopus Xsal-1, a vertebrate homolog of the region specific homeotic gene spalt of Drosophila. Mech Dev. 1996;55:19–32. doi: 10.1016/0925-4773(95)00485-8. [DOI] [PubMed] [Google Scholar]
- 15.Ott T, Kaestner KH, Monaghan AP, Schutz G. The mouse homolog of the region specific homeotic gene spalt of Drosophila is expressed in the developing nervous system and in mesoderm-derived structures. Mech Dev. 1996;56:117–128. doi: 10.1016/0925-4773(96)00516-3. [DOI] [PubMed] [Google Scholar]
- 16.de Vries-Van der Weerd MA, Willems PJ, Mandema HM, ten Kate LP. A new family with the Townes-Brocks syndrome. Clin Genet. 1988;34:195–200. doi: 10.1111/j.1399-0004.1988.tb02862.x. [DOI] [PubMed] [Google Scholar]
- 17.Marlin S, Toublanc JE, Petit C. Two cases of Townes-Brocks syndrome with previously undescribed anomalies. Clin Dysmorphol. 1998;7:295–298. doi: 10.1097/00019605-199810000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Kohlhase J, Taschner P, Burfeind P, Pasche B, Newman B, Blanck C, Breuning M, ten Kate L, Maaswinkel-Mooy P, Mitulla B, Seidel J, Kirkpatrick S, Pauli R, Wargowski D, Devriendt K, Proesmans W, Gabrielli O, Coppa G, Wesby-van Sway E, Trembath R, Schnizel A, Reardon W, Seemanova E, Engel W. Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am J Hum Genet. 1999;64:435–445. doi: 10.1086/302238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohlhase J, Hausmann S, Stojmenovic G, Dixkens C, Bink K, Schulz-Schaeffer W, Altmann M, Engel W. SALL3, a new member of the human spalt-like gene family, maps to 18q23. Genomics. 1999;62:216–222. doi: 10.1006/geno.1999.6005. [DOI] [PubMed] [Google Scholar]
- 20.Al-Baradie R, Yamada K, St Hilaire C, Chan WM, Andrews C, McIntosh N, Nakano M, Martonyi EJ, Raymond WR, Okumura S, Okihiro MM, Engle EC. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet. 2002;71:1195–1199. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borozdin W, Wright MJ, Hennekam RC, Hannibal MC, Crow YJ, Neumann TE, Kohlhase J. Novel mutations in the gene SALL4 provide further evidence for acro-renal-ocular and Okihiro syndromes being allelic entities, and extend the phenotypic spectrum. J Med Genet. 2004;41:e102. doi: 10.1136/jmg.2004.019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohlhase J, Schubert L, Liebers M, Rauch A, Becker K, Mohammed SN, Newbury-Ecob R, Reardon W. Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J Med Genet. 2003;40:473–478. doi: 10.1136/jmg.40.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohlhase J, Heinrich M, Schubert L, Liebers M, Kispert A, Laccone F, Turnpenny P, Winter RM, Reardon W. Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet. 2002;11:2979–2987. doi: 10.1093/hmg/11.23.2979. [DOI] [PubMed] [Google Scholar]
- 24.Paradisi I, Arias S. IVIC syndrome Is caused by a c.2607delA mutation in the SALL4 locus. Am J Med Genet A. 2007;143:326–332. doi: 10.1002/ajmg.a.31603. [DOI] [PubMed] [Google Scholar]
- 25.Elling U, Klasen C, Eisenberger T, Anlag K, Treier M. Murine inner cell mass-derived lineages depend on Sall4 function. Proc Natl Acad Sci U S A. 2006;103:16319–16324. doi: 10.1073/pnas.0607884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakaki-Yumoto M, Kobayashi C, Sato A, Fujimura S, Matsumoto Y, Takasato M, Kodama T, Aburatani H, Asashima M, Yoshida N, Nishinakamura R. The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development. 2006;133:3005–3013. doi: 10.1242/dev.02457. [DOI] [PubMed] [Google Scholar]
- 27.Warren M, Wang W, Spiden S, Chen-Murchie D, Tannahill D, Steel KP, Bradley A. A Sall4 mutant mouse model useful for studying the role of Sall4 in early embryonic development and organogenesis. Genesis. 2007;45:51–58. doi: 10.1002/dvg.20264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Tam WL, Tong GQ, Wu Q, Chan HY, Soh BS, Lou Y, Yang J, Ma Y, Chai L, Ng HH, Lufkin T, Robson P, Lim B. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol. 2006;8:1114–1123. doi: 10.1038/ncb1481. [DOI] [PubMed] [Google Scholar]
- 29.Bandera CA, Takahashi H, Behbakht K, Liu PC, LiVolsi VA, Benjamin I, Morgan MA, King SA, Rubin SC, Boyd J. Deletion mapping of two potential chromosome 14 tumor suppressor gene loci in ovarian carcinoma. Cancer Res. 1997;57:513–515. [PubMed] [Google Scholar]
- 30.Ma Y, Li D, Chai L, Luciani AM, Ford D, Morgan J, Maizel AL. Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J Biol Chem. 2001;276:48223–48230. doi: 10.1074/jbc.M106468200. [DOI] [PubMed] [Google Scholar]
- 31.Li D, Dower K, Ma Y, Tian Y, Benjamin TL. A tumor host range selection procedure identifies p150(sal2) as a target of polyoma virus large T antigen. Proc Natl Acad Sci U S A. 2001;98:14619–14624. doi: 10.1073/pnas.251447198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Tian Y, Ma Y, Benjamin T. p150Sal2 Is a p53-Independent Regulator of p21WAF1/CIP. Mol Cell Biol. 2004;24:3885–3893. doi: 10.1128/MCB.24.9.3885-3893.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma Y, Li D, Chai L, Luciani AM, Ford D, Morgan J, Maizel AL. Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J Biol Chem. 2001;276:48223–48230. doi: 10.1074/jbc.M106468200. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Tian Y, Ma Y, Benjamin T. p150(Sal2) is a p53-independent regulator of p21(WAF1/CIP) Mol Cell Biol. 2004;24:3885–3893. doi: 10.1128/MCB.24.9.3885-3893.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato A, Matsumoto Y, Koide U, Kataoka Y, Yoshida N, Yokota T, Asashima M, Nishinakamura R. Zinc finger protein sall2 is not essential for embryonic and kidney development. Mol Cell Biol. 2003;23:62–69. doi: 10.1128/MCB.23.1.62-69.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Y, Cui W, Yang J, Qu J, Di C, Amin HM, Lai R, Ritz J, Krause DS, Chai L. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood. 2006;108:2726–2735. doi: 10.1182/blood-2006-02-001594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Chai L, Gao C, Fowles TC, Alipio Z, Dang H, Xu D, Fink LM, Ward DC, Ma Y. SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood. 2008;112:805–813. doi: 10.1182/blood-2007-11-126326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Chai L, Liu F, Fink LM, Lin P, Silberstein LE, Amin HM, Ward DC, Ma Y. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci U S A. 2007;104:10494–10499. doi: 10.1073/pnas.0704001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, Jeong H, Kong N, Yang Y, Carroll J, Luo HR, Silberstein LE, Yupoma, Chai L. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS ONE. 2009;4:e5577. doi: 10.1371/journal.pone.0005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Gao C, Chai L, Ma Y. A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PLoS One. 2010;5:e10766. doi: 10.1371/journal.pone.0010766. [DOI] [PMC free article] [PubMed] [Google Scholar]