

Abstract
A thiophile promoted synthesis of di-substituted 4H-[1,2,4]triazole-3-yl-amines as urea mimetics from the corresponding 1,3-disubstituted thioureas has been studied and the scope and limitations of this reaction are presented. The reaction proceeds through the formation of a carbodiimide, followed by a sequential addition-dehydration with acyl hydrazides. 1,3-Branched dialkylthioureas result in the formation of the corresponding ureas. The electronic and steric effect of the substitution on the phenyl rings of the 1,3-diarylthioureas play an important role in the formation of the intermediary carbodiimde and the direction of the subsequent ring closure of the N-acyl hydrazide adduct.
INTRODUCTION
Several compounds that belong to the class of 1,3-disubstituted ureas are being developed as drug candidates against a variety of diseases. For example, the Raf-1 inhibitor BAY 43–9006 is developed for cancer and inflammatory disorders1 and the cyclic urea Mozenavir is studied for HIV.2 1,3-Diarylurea is frequently encountered as a privileged structure for many biological targets.3 They constitute an essential part of pharmacophores for biological targets such as p38 and raf kinases,6,4 vascular endothelial growth receptor 2 (VRGFR-2),5 inosine monophosphate dehydrogenase (IMPDH),6 highly potent and selective human A2B adenosine receptor antagonists7 and cyclin-dependent kinase 2 (CDK2) inhibitors.8 The mode of interaction of different 1,3-diarylureas with their targets varies. For example, while some of these urea-based inhibitors bind to an allosteric site causing a significant rearrangement of the target protein structure9,10 others bind to the active site such as the ATP pocket with the urea function participating in a bi-dentate hydrogen bond.11
Bioisosteric replacements of the amide and thioamide functions included heterocyclic systems such as the [1,2,4]- and [1,3,4]oxadiazoles and [1,2,4]triazoles.12 Such replacements led to [1,2,4]triazoles which were active antimycobacterials13 and potent, selective 5-HT1D receptor agonists.14 Another interesting application of triazole was to employ it as a cis-amide bond surrogate.15 Replacement of the Ala-NMe-Tyr(OMe) amide bond in RA-VII, a plant-origin anti-tumor bicyclic hexapeptide, with [1,2,4]triazole yielded the pseudopeptide.16 As anticipated, the pseudopeptide reproduced the conformation of the minor cis-peptide isomer but was devoid of cytotoxic activity. Interestingly, replacement of urea function in β3-adrenergic receptor agonists with triazole resulted in improved oral bioavailability accompanied by retention of potency, selectivity, and in vivo efficacy.17 Replacement of the amide bond in Phe-Gly in substance P with [1,2,4]triazole peptide bond surrogate led to dramatic loss of affinity to the NK1 receptor.18 On the other hand, introduction of the same modification to dermorphin led to potent and selective pseudopeptides toward the μ receptor sub-type.18 We anticipate that replacement of the amide/thioamide bond in the urea/thiourea with heterocyclic bioiosteric moieties such as the triazoles may increase the structural diversity and generate novel and interesting bioactive compounds of therapeutic potential.
In an effort to increase structural diversity and modify the degrees of conformational freedom of the 1,3-disubstituted moieties in the urea/thiourea we introduced a bioisosterism used to mimic the amide/thioamide function and developed a synthetic transformation of 1,3-disubstituted thioureas to the corresponding alkyl-(4-aryl/alkyl-4H-[1,2,4]triazol-3-yl)-amine (Figure 1). Transformation of an amide to a thioamide using Lawesson’s reagent can be applied to convert the ureas to the corresponding thioureas,19 thus generating the starting material for the synthesis of [1,2,4]triazole. In addition, thioureas can be generated directly by the addition of amines to isothiocyanates.
Figure 1.

[1,2,4]Triazoles as amide mimics and 4H-[1,2,4]Triazol-3-yl amine as urea mimics
Katritzky and coworkers reported on a solid-phase method to prepare [1,2,4]triazoles from acyl hydrazide resins and amidines in the presence of molecular sieves.20 Larsen and DiPaolo converted N-resin-bound thioamides to amidrazones which were then acylated by acyl halides and cyclized in the presence of acetic acid at room temperature to the triazoles of interest.21 An efficient one-pot, three component synthesis of substituted [1,2,4]triazoles was reported by Stocks and coworkers.22 Reaction of an amine with N′-acetyl-N,N-dimethylhydrazonoformamide and acyl hydrazides led to moderate yields when aromatic amines were employed.
We chose to employ a method used to transform the thioamide function in a thionopeptide into a [1,2,4]triazole as a cis-amide bond surrogate by treating the thionopeptide with formic hydrazide and mercury (II) acetate as a thiophile.15 This reaction proceeds through the formation of acyl hydrazide adduct which is cyclized to the corresponding [1,2,4]triazole under acidic conditions.
However, the problem in extending this methodology for the thiourea lies in the regioselectivity of the cyclization (Figure 1). Unlike the unidirectional cyclization of intermediary acylamidrazone (II) generated from the thioamides (I), which yields a single [1,2,4]triazole (III), the intermediary acylureidrazone (V) generated from the thiourea (IV) can in principle cyclize in either direction yielding two isomeric [1,2,4]triazoles (VIa and VIb) (Figure 1). Herein we report a general method used to generate substituted [1,2,4]triazol-3-ylamines as amide bond mimetics of corresponding ureas. Additionally, we observe interesting steric and electronic effects of the 1,3-substitutents on the thiourea that play an important role on the product distribution and regioselectivity of the cyclization.
RESULTS AND DISCUSSION
The 1,3-disubstituted thioureas were either commercially available or were generated in good yields by the reaction of the amine with the corresponding isothiocyanate in either dichloromethane or acetonitrile. The objective was to select reagents and optimize various reaction conditions to develop a general method that can be used to generate regioselective [1,2,4]triazol-3-yl amines as amide bond mimics of 1,3-disubstituted urea compounds. Mercury salts23 and Mukaiyama reagent24 have been used as thiophiles in the guanylation of amines by thioureas. Our initial objective was to identify a suitable thiophile to carryout this reaction in one-pot without isolation of intermediates. We chose to compare a variety of mercury (II) salts and the Mukaiyama reagent as our thiophile (Table 1) and monitored the formation of phenyl-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (2) by LCMS in a model reaction using the 1,3-diphenylthiourea 1. Based on the product yield it was clear that Hg(OAc)2 was the optimal thiophile for this model reaction hence we decided to use Hg(OAc)2 for all subsequent reactions as the thiophile. We then turned our attention to investigate the time course for this reaction employing equimolar amounts of 1 and formylhydrazide. In about 15 minutes from the start of the reaction ~65% of the 1,3-diphenylthiourea is converted to the 1,3-diphenylcarbodiimide which was isolated and characterized (Figure 2). Therefore, we followed the course of this reaction by monitoring the disappearance of the carbodiimide and the formation 2 by LCMS. We observed that in about 2h the reaction results in ~70% of 2 and there is not much improvement in the product yield for the next 22h. Hence, we decided to use 2h as optimal time of incubation for further studies. By using 2.5 equivalents of the formylhydrazide in the above reaction we were able to increase the yield of the isolated product to 91%.
Table 1.
Reaction of 1,3-diphenylthiourea 1 and formylhydrazide to generate phenyl-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine 2 mediated by a thiophile
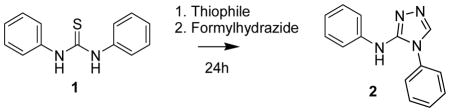 | ||
|---|---|---|
| Thiophile | % Yield | |
| a | Hg(OAc)2 | 75 |
| b | Hg(OCOCF3)2 | 10 |
| c | HgCl2 | 6 |
| d | HgBr2 | 11 |
| e | HgI2 | 6 |
| f | HgSO4 | 22 |
| g | Mukaiyama reagent | 25 |
Figure 2.

Time course for the conversion of 1,3-diphenylthiourea (1) to phenyl-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (2).
Since the [1,2,4]triazole formation is an addition-dehydration reaction, we hypothesized that the substituents on the thiourea nitrogens will play an important role in the rate of addition and the regioselectivity of ring closure. In order to study the substituent effect on the formation of the [1,2,4]triazole a series of symmetrical 1,3-disubstituted thioureas were generated. A one-pot reaction was carried out with the 1,3-disubstituted thiourea, Hg(OAc)2 and an acylhydrazine (1:1.05:0.95 equivalents) mixed together at room temperature and incubated for 2h. It is interesting to note that the major product from the reaction in the case of the 1,3-dialkyl substitutions viz., tert-butyl, cyclohexyl and n-propyl thioureas (3d, 3e and 3f Table 2) was the corresponding 1,3-dialkylurea. This is because the acetate, released from the thiophile Hg(OAc)2 in the course of de-sulfurization, reacts with the intermediary 1,3-dialkylcarbodiimide faster than the addition of acylhydrazide to yield the 2-acetyl-1,3-dialkyl-isourea (Scheme 1). By using the Mukaiyama reagent (N-methyl 2-chloro-pyridinium iodide) as the thiophile and 1,3-dicylcohexylthiourea we were able to get ~ 15% of the [1,2,4]triazole, longer incubation times however did not improve the yields. The de-sulfurization of the substituted 1,3-diphenylthioureas to form the corresponding carbodiimides was visually monitored based on the formation of the HgS black precipitate. HgS formation was the fastest in the thioureas in which phenyl rings substituted with electron withdrawing groups and the slowest in those where phenyl rings were substituted with electron donating groups. The data in Table 2 also suggests that 1,3-diphenylthioureas substituted with electron withdrawing group on the aromatic rings generate lower yields of [1,2,4]triazole compared to the electron rich ring systems (cf 3a, 3b and 3c Table 2). In the 4-Cl and 3,5-di-CF3 disubstituted phenyl thioureas a significant amount of a high molecular weight product composed of two units of carbodiimide and one unit of fromylhydrazide was observed by LCMS. This product is generated by the addition of a second carbodiimide to the intermediary N-acyl hydrazide (V) prior to the ring closure. This pattern could be explained if the carbodiimides generated with electron withdrawing groups on the phenyl rings are much more reactive towards the formylhydrazide compared to the one with electron donating group on the phenyl rings, in addition these are slower to cyclize (cf 3b and 3c with 3a Table 2). The stereoelectronic effects of the acyl group on the acylhydrazine affect the yields of the [1,2,4]triazole (3g–i Table 2). A similar pattern is observed in the [1,2,4]triazole yields (4g, 4j, and 4k Table 2) generated using acetylhydrazide with varying substitutions. The acid catalyzed dehydration to generate the corresponding [1,2,4]triazole 4h (Table 2) could be pushed to ~ 75% completion with a stronger acid (p-TsOH) and higher temperature (reflux).
Table 2.
Substituent effect on the formation of [1,2,4]triazoles from symmetrical thioureas
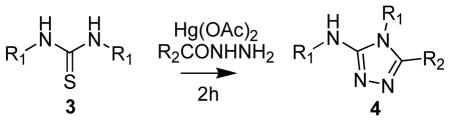 | |||
|---|---|---|---|
| R1 | R2 | % Yield | |
| a | 4-OMe-Phenyl | H | 89 |
| b | 4-Cl-Phenyl | H | 74 |
| c | 3,5-di-CF3-Phenyl | H | 36 |
| d | tert-Butyl | H | Urea |
| e | Cyclohexyl | H | Urea |
| f | n-Propyl | H | Urea |
| g | Phenyl | Me | 68 |
| h | Phenyl | Phenyl | 12 |
| i | Phenyl | H | 73 |
| j | 4-OMe-Phenyl | Me | 77 |
| k | 3,5-di-CF3-Phenyl | Me | 60 |
Scheme 1.

Formation of 1,3-dilakylurea through the 1,3-dialkyl O-acetylisourea intermediate formed from a 1,3-dialkylthiourea
In order to address the substituent effects on the regioselectivity of ring closure generating the [1,2,4]triazoles, we studied thioureas in which both nitrogens were differentially substituted. The corresponding [1,2,4]triazoles were generated using the optimized procedure {1,3-disubstituted thiourea, Hg(OAc)2 and an acylhydrazine (1:1.05:0.95 equivalents) mixed together and incubated at room temperature}, the results of which are summarized in Table 3. In the 1-cyclohexyl-3-aryl thiourea (5a–5c Table 3) series the electronics on the phenyl ring was modified by substituting it with either an electron donating group (5b, Table 3) or electron withdrawing group (5c, Table 3). By LCMS we observed a significant amount of the carbodiimide and the intermediate that results from the addition of formyl hydrazide to the carbodiimide. These reactions were allowed to proceed for 48h at room temperature. The 1-cyclohexyl-3-phenylthiourea (5a, Table 3) yields the urea as a major product, while the aryl substituted 1-cyclohexy-3-arylthioureas (5b and 5c, Table 3) yields the desired products in good to moderate yields. These observations do not follow an obvious trend and at present we can only speculate that the kinetics of the intermediate reactions might play an important role in the product ([1,2,4]triazole) formation. The reaction tolerates electron withdrawing groups (5e and 5f, Table 3), electron donating groups (5d and 5h, Table 3) and a combination of the two (5g Table 3) resulting in good yields of the corresponding [1,2,4]triazole. A strong electron withdrawing group on the phenyl ring such as m,m′-di-CF3 (f, Table 3) yields a regioselective product, however, with p-OMe and p-Cl substituents (d and e, Table 3) we obtain a mixture of regioisomers. In the case of a reaction wherein we get a mixture of the two regioisomers the ratio and regioselectivity was confirmed by synthesizing exclusively a single regioisomer using a benzyl protection-deprotection strategy. In entry 5d of Table 3 the [1,2,4]triazoles obtained were a 1:5 mixture of regioisomers 6d and 7d. The individual regioisomers were synthesized starting from the corresponding 1-benzyl-1,3-diphenylthioureas summarized in Scheme 2. The assignments of 6d and 7d were made based on chemical shifts in the NMR spectra and retention times on the LC.
Table 3.
Substituent effect on the direction of ring closure to yield the [1,2,4]triazoles generated from unsymmetrical 1,3-disubstituted thioureas (General Procedure II)
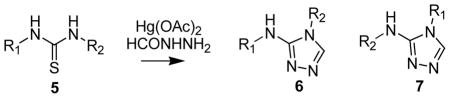 | |||
|---|---|---|---|
| R1 | R2 | % Yield | |
| a | Cyclohexyl | Phenyl | Urea |
| b | Cyclohexyl | 4-OMe-Phenyl | 75(6b) |
| c | 3,5-di-CF3-Phenyl | Cyclohexyl | 44(6c) |
| d | Phenyl | 4-OMe-Phenyl | 76 (1:5) |
| e | Phenyl | 4-Cl-Phenyl | 70 (2:3) |
| f | 3,5-di-CF3-Phenyl | Phenyl | 84(6f) |
| g | 3,5-di-CF3-Phenyl | 4-OMe-Phenyl | 77 |
| h | 3,5-di-CF3-Phenyl | p-Tolyl | 55 |
| i | 3,5-di-CF3-Phenyl | o-Tolyl | 50 |
| j | 3,5-di-CF3-Phenyl | 2-i-Propyl-phenyl | 45 |
| k | 3,5-di-CF3-Phenyl | 2-t-Butyl-phenyl | 8 |
Reaction times a – c = 48h and d – k = 2h
Scheme 2.

N-Benzyl protection-deprotection strategy to access regioselective [1,2,4]triazoles
Based on 1H and 13C NMR and LCMS all the other unsymmetrical 1,3-disubstituted thioureas in Table 3 resulted in an overwhelming excess of single regioisomer of [1,2,4]triazole (>95:5). The regiochemistry of the compounds was assigned based on the diagnostic NOESY peak for example, Figure 3 shows the NOESY spectrum of compound 6g displaying cross peaks between Hb and Hc and lack of cross peaks between Hc and Hd (see supplementary material for TOCSY and NOESY data for 6c, 6f and 6g).
Figure 3.

NOESY spectrum showing the diagnostic peak confirming the direction of the regioselective ring closure to yield [1,2,4]triazole 6g.
From the symmetrical 1,3-disubstituted urea series we know that N-3,5-bis-trifluoromethylphenyl substituted thioureas undergo rapid formation of the carbodiimide intermediate thereby making the addition-dehydration the rate limiting steps. Also, N-1,3-bis-substituted thioureas that have the 3,5-ditrifluoromethylphenyl substituent yield regioselective products (see 5f–5k Table 3). To investigate the steric effects of substituents on the rate of the addition-dehydration reaction we synthesized a series of mixed 1,3-disubstituted thioureas which maintain 3,5-bis-trifluoromethylphenyl on one nitrogen and a phenyl substituted with varying steric bulk on the other (5h – 5k, Table 3). The yields of the reactions with N-p-tolyl and N-o-tolyl substituted thioureas (5h and 5i, Table 3) are almost equivalent. However, the 1-p-tolyl-3-(3,5-bis-CF3-phenyl)thiourea (5h) yielded a high molecular weight compound that corresponds to the addition of two carbodiimides to one formyl hydrazide as the minor product, while the 1-o-tolyl-3-(3,5-di-CF3-phenyl)thiourea (5i) gave the intermediate generated by the addition of formyl hydrazide to carbodiimide as the minor product. The reactions were monitored by LCMS and ratios of the major to minor products were 5:1 for h (N-p-tolyl substitution) and 6:1 for i (N-o-tolyl substitution). Based on the product distribution the steric hinderance in N-o-tolyl-3-(3,5-di-CF3-phenyl)thiourea (5i) prevents the addition of second carbodiimde. Similar to 5i the steric hindered ring closure is also compromised in the N-i-propyl- (5j) and remarkably inhibited in the N-t-butyl-3-(3,5-di-CF3-phenyl)thiourea (5k) as shown by the lower yields of the [1,2,4]triazoles 45% and 8%, respectively, obtained under similar reaction conditions. Extending the reactions overnight these ortho substituted phenyl thiourea reactions proceed to give isolated yields of ~70–75% of the corresponding [1,2,4]triazoles.
It is clear from the studies of the series of 1,3-disubstituted thioureas that the reaction can be fine tuned to yield regioselective 1,3-disubstituted [1,2,4]triazoles by modifying the substituents on the nitrogens of the thiourea. At least, for the compounds described the rate limiting step in this reaction is the addition-dehydration. The ring closure preferentially occurs on the nitrogen that has the electron donating substituents. In the case where the difference between the two N-substituents is not significant leading to the formation of a mixture of regioisomeric [1,2,4]triazoles a protection-deprotection strategy employing a benzyl group as a transient protecting group can be considered to generate the regioisomeric [1,2,4]triazole of choice. In summary, we describe a new synthesis of useful 1,3-disubstituted triazoles as amide bond mimetics of urea compounds. Furthermore the central [1,2,4]triazol-3-yl amine scaffold under optimized conditions can be used to generate a library with four points of diversity, which is currently underway in our laboratories and will be reported in due course.
EXPERIMENTAL SECTION
Unless otherwise specified, all reagents were purchased from commercial sources and were used without further purification. Flash chromatography was carried out on silica gel (200–400 mesh). Melting points were recorded on a electrothermal melting point apparatus and are uncorrected. Mass spectra were obtained from a single quad instrument with APCI source. 1D- and 2D-NMR spectra were colleted using standard pulse sequences.
N-Phenyl-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (2)
(following general procedure II) 72%, colorless solid: mp 214–216 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 8.50 (bs, 1H), 8.47 (s, 1H), 7.57-7.53 (m, 2H), 7.51-7.46 (m, 3H), 7.40 (d, 2H, J = 8 Hz), 7.20 (t, 2H, J = 8 Hz), 6.83 (t, 1H, J = 7.2 Hz); 13CNMR (DMSO-d6, 100 MHz) δ 150.6, 142.6, 142.1, 134.0, 130.5, 129.4, 129.3, 126.0, 120.9, 117.3; MS+(APCI) 236.9 [M + H]+; Anal. Calcd for C14H12N4: C, 71.17; H, 5.12; N, 23.71. Found: C, 70.95; H, 4.89; N, 23.77.
1,3-Di-(4-methoxy-phenyl)-thiourea (3a)
98%, colorless solid: mp 201–203 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.42 (s, 2H), 7.29 (d, J = 8.8 Hz, 4H), 6.87 (d, J = 8.8 Hz, 4H), 3.72 (s, 6H); 13CNMR (DMSO-d6, 100 MHz) δ 181.1, 157.2, 132.9, 126.8, 114.3, 55.9; MS+(APCI) 288.84 [M + H]+
1,3-Bis-(3,5-bis-trifluoromethyl-phenyl)-thiourea (3c)
71%, colorless solid: mp 180–181 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.65 (s, 2H), 8.19 (s, 4H), 7.85 (s, 2H); MS+(APCI) 499.99 [M + H]+
(4-Methoxy-phenyl)-[4-(4-methoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-amine (4a)
89%, colorless solid: mp 183–184 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 8.29 (s, 1H), 8.13 (s, 1H), 7.43-7.39 (m, 4H), 7.09 (dd, J = 6.8, 2.0 Hz, 2H), 6.79 (dd, J = 6.8, 2.0 Hz, 2H), 3.80 (s, 3H), 3.67 (s, 3H); 13CNMR (DMSO-d6, 100 MHz) δ 159.4,153.4, 150.9, 141.1, 134.9, 127.3, 125.9, 118.4, 114.9, 113.8, 55.5, 55.2; MS+(ESI) 296.30 [M + H]+
(4-Chloro-phenyl)-[4-(4-chloro-phenyl)-4H-[1,2,4]triazol-3-yl]-amine (4b)
74%, colorless solid: mp 226–228 ºC; 1HNMR (DMSO-d6, 500 MHz) δ 8.74 (s, 1H), 8.48 (s, 1H), 7.61 (dd, J = 40.5, 9.0 Hz, 2H), 7.41 (dd, J = 125.0, 9.0 Hz, 2H); 13CNMR (DMSO-d6, 125 MHz) δ 149.7, 141.2, 140.5, 133.5, 132.0, 129.8, 128.4, 127.6, 123.9, 118.3; MS+(ESI) 305.5 [M + H]+
(3,5-Bis-trifluoromethyl-phenyl)-[4-(3,5-bis-trifluoromethyl-phenyl)-4H-[1,2,4]triazol-3-yl]-amine (4c)
36%; 1HNMR (DMSO-d6, 400 MHz) δ 9.49 (bs, 1H), 8.65 (s, 1H), 8.44 (s, 2H), 8.35 (s, 1H), 8.18 (s, 2H), 7.53 (s, 1H); 13CNMR (DMSO-d6, 100 MHz) δ 149.9,143.6, 142.2, 135.3, 132.5, 132.2, 131.6, 131.3, 128.8, 125.4, 124.8, 123.9, 122.7, 122.1, 117.3, 113.7; MS+(APCI) 508.93 [M + H]+
(5-Methyl-4-phenyl-4H-[1,2,4]triazol-3-yl)-phenyl-amine (4g)
68%, colorless solid: mp 236–238 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 8.14 (s, 1H), 7.58-7.50 (m, 3H), 7.44-7.36 (m, 4H), 7.18-7.13 (m, 2H), 6.81-6.77 (m, 1H), 2.10 (s, 3H); 13CNMR (DMSO-d6, 100 MHz) δ 151.3, 148.3, 142.7, 133.8, 130.6, 130.0, 129.2, 128.3, 120.6, 117.1, 11.9; MS+(APCI) 251.01 [M + H]+; Anal. Calcd for C15H14N4: C, 71.98; H, 5.64; N, 22.38. Found: C, 71.78; H, 5.47; N, 22.41.
(4,5-Diphenyl-4H-[1,2,4]triazol-3-yl)-phenyl-amine (4h)
12%, colorless solid: mp 201–202 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 8.36 (s, 1H), 7.54-7.49 (m, 5H), 7.44-7.42 (m, 2H), 7.35-7.33 (m, 1H), 7.33-7.31 (m, 4H), 7.23 (t, J = 7.2 Hz, 2H), 6.89 (t, J = 7.2Hz, 1H); 13CNMR (DMSO-d6, 100 MHz) δ 152.5, 150.6, 141.8, 133.8, 130.7, 130.6, 130.1, 129.4, 129.2, 129.1, 128.5, 127.7, 121.7, 118.3; MS+(APCI) 313.12 [M + H]+
N-Phenyl-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (4i)
(following General Procedure II) 73%. Compound 4i is identical to 2, which is fully characterized.
(4,5-Diphenyl-4H-[1,2,4]triazol-3-yl)-phenyl-amine (4k)
60%, colorless solid; 1HNMR (DMSO-d6, 500 MHz) δ 9.56 (s, 1H), 9.06 (s, 1H), 8.34 (s, 2H), 8.27 (s, 1H), 8.07 (s, 2H), 7.38 (s, 1H), 1.71 (s, 3H); 13CNMR (DMSO-d6, 125 MHz) δ 167.9, 149.6, 147.8, 142.9, 134.6, 132.1, 131.8, 130.8, 130.6, 129.8, 124.5, 123.9, 122.3, 121.7, 116.4, 112.8, 10.9; MS+(APCI) 523.13 [M + H]+
1-Cyclohexyl-3-phenyl-thiourea (5a)23
80%, colorless solid: mp 144–145 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.29 (bs, 1H), 9.31 (s, 1H), 7.61 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 7.2 Hz, 2H), 7.05 (t, J = 7.6 Hz, 1H), 4.07 (s, 1H), 1.89-1.87 (m, 2H), 1.68-1.64 (m, 2H), 1.56-1.52 (m, 1H), 1.32-1.11 (m, 5H); MS+(APCI) 235.23 [M + H]+
1-Cyclohexyl-3-(4-methoxy-phenyl)-thiourea (5b)23
84%, colorless solid: mp 138–139 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.12 (bs, 1H), 7.36 (s, 1H), 7.24 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.2 Hz, 2H), 4.04 (s, 1H), 3.72 (s, 3H), 1.86-1.82 (m, 2H), 1.70-1.66 (m, 2H), 1.54-1.52 (m, 1H), 1.30-1.09 (m, 5H); MS+(APCI) 264.89 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-cyclohexyl-thiourea (5c)25
86%, colorless solid: mp 164–165 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.84 (s, 1H), 8.21 (s, 2H), 8.14 (s, 1H), 7.70 (s, 1H), 4.09 (bs, 1H), 1.91-1.87 (m, 2H), 1.69-1.66 (m, 2H), 1.57-1.54 (m, 1H), 1.34-1.14 (m, 5H); MS+(APCI) 371.00 [M + H]+
1-(4-Methoxy-phenyl)-3-phenyl-thiourea (5d)26
65%, colorless solid: mp 159–161 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.61 (s, 1H), 9.59 (s, 1H), 7.46-7.43 (m, 2H), 7.32-7.27 (m, 4H), 7.10 (dt, J = 8, 1.2 Hz, 1H), 6.88 (dd, J = 8, 1.2 Hz, 2H), 3.76 (s, 3H); MS+(APCI) 258.97 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-phenyl-thiourea (5f)
86%, colorless solid: mp 141–143 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.29 (s, 1H), 10.20 (s, 1H), 8.23 (s, 2H), 7.79 (s, 1H), 7.43-7.34 (m, 4H), 7.17 (t, J = 7.2 Hz, 1H); MS+(APCI) 364.90 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-(4-methoxy-phenyl)-thiourea (5g)
91%, colorless solid: mp 148–150 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.12 (s, 1H), 10.01 (s, 1H), 8.23 (s, 2H), 7.76 (s, 1H), 7.11 (dd, J = 144.0, 8.8 Hz, 4H), 3.73 (s, 3H); MS+(APCI) 394.89 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-p-tolyl-thiourea (5h)
64%, colorless solid: mp 153–154 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.22 (s, 1H), 10.10 (s, 1H), 8.23 (s, 2H), 7.76 (s, 1H), 7.22 (dd, J = 48.8, 8.0 Hz, 4H), 3.30 (s, 3H); MS+(APCI) 378.89 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-o-tolyl-thiourea (5i)
83%, colorless solid: mp 153–154 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.05 (s, 1H), 9.88 (s, 1H), 8.25 (s, 2H), 7.76 (s, 1H), 7.28-7.17 (m, 4H), 3.33 (s, 3H); MS+(APCI) 378.92 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-(2-isopropyl-phenyl)-thiourea (5j)
82%, colorless solid: mp 148–150 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 10.29 (bs, 1H), 9.87 (s, 1H), 8.26 (s, 2H), 7.77 (s, 1H), 7.37-7.20 (m, 4H), 3.09 (septa, J = 7.2 Hz, 3H), 1.15 (d, J = 7.2 Hz, 6H); MS+(APCI) 406.93 [M + H]+
1-(3,5-Bis-trifluoromethyl-phenyl)-3-(2-tert-butyl-phenyl)-thiourea (5k)
65%, colorless solid: mp 137–138 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.89 (bs, 1H), 9.75 (bs, 1H), 8.30 (s, 2H), 7.77 (s, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.31-7.23 (m, 2H), 7.18 (d, J = 7.2 Hz, 1H), 1.26 (s, 9H); MS+(APCI) 420.94 [M + H]+
(4-Cyclohexyl-4H-[1,2,4]triazol-3-yl)-(4-methoxy-phenyl)-amine (6b)
75%, colorless solid; 1HNMR (DMSO-d6, 300 MHz) δ 8.08(s, 1H), 7.02 (dd, J = 138.3, 9.0 Hz, 4H), 5.94(d, J = 8.4), 3.44-3.33 (m, 1H), 1.82-1.76 (m, 2H), 1.66-1.63 (m, 2H), 1.54-1.51 (m, 1H), 1.33-1.25(m, 2H), 1.20-1.10 (m, 3H); MS+(APCI) 272.98 [M + H]+
(3,5-Bis-trifluoromethyl-phenyl)-(4-cyclohexyl-4H-[1,2,4]triazol-3-yl)-amine (6c)
44%, colorless solid; 1HNMR (DMSO-d6, 500 MHz) δ 9.35 (s, 1H), 8.48 (s, 1H), 8.35 (s, 2H), 7.54 (s, 1H), 4.12 (tt, J = 4.0, 11.5 Hz, 1H), 1.98-1.96 (m, 2H), 1.86-1.83 (m, 2H), 1.71-1.61 (m, 3H), 1.45-1.37 (m, 2H), 7.20 (tq, J = 4.0, 13.5 Hz, 1H); 13CNMR (DMSO-d6, 100 MHz) δ 148.7, 143.2, 138.8, 130.9, 130.8, 130.6, 130.5, 126.7, 124.5, 122.37, 120.1, 116.2, 112.5, 52.5, 32.8, 25.1, 24.5; MS+(APCI) 378.95 [M + H]+ Anal. Calcd for C16H16N4F6: C, 50.80; H, 4.26; N, 14.81. Found: C, 50.89; H, 4.43; N, 15.00.
(4-Methoxy-phenyl)-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (6d)
63%, pale yellow oil; 1HNMR (DMSO-d6, 400 MHz) δ 8.77 (s, 1H), 7.65-7.60 (m, 6H), 7.15 (dd, J = 170, 8.8 Hz, 4H), 3.74 (s, 3H); MS+(APCI) 267.03 [M + H]+
[4-(4-Methoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-phenyl-amine (7d)
56%, colorless solid; mp 211–212 ºC; 1HNMR (DMSO-d6, 500 MHz) δ 8.37 (s, 1H), 8.36 (bs, 1H), 7.44-7.39 (m, 4H), 7.20 (t, J = 12 Hz, 2H), 7.11-7.07 (m, 2H), 6.83 (t, J = 12 Hz, 1H), 3.82 (s, 3H); 13CNMR (DMSO-d6, 125 MHz) δ 159.4, 150.3, 141.8, 141.5, 128.6, 127.1, 125.9, 120.2, 116.6, 114.9, 55.5; MS+(APCI) 267.03 [M + H]+
[4-(4-Chloro-phenyl)-4H-[1,2,4]triazol-3-yl]-phenyl-amine (7e)
20%, colorless oil; 1HNMR (DMSO-d6, 500 MHz) δ 8.68 (s, 1H), 8.49 (s, 1H), 7.59-7.56 (m, 2H), 7.52-7.49 (m, 5H), 7.26 (d, J = 8.5 Hz, 2H), 13CNMR (DMSO-d6, 125 MHz) δ 158.9, 149.7, 141.5, 140.7, 133.1, 129.9, 128.9, 128.4, 125.4, 123.8, 118.3; MS+(APCI) 271.4 [M + H]+; Anal. Calcd for C14H11ClN4: C, 62.11; H, 4.10; N, 20.70 found: C, 61.68; H, 3.86; N, 20.33.
(3,5-Bis-trifluoromethyl-phenyl)-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (6f)
84%, colorless solid; mp 191–192 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.40 (s, 1H), 8.54 (s, 1H), 8.31 (s, 2H), 7.62-7.53 (m, 5H), 7.50 (s, 1H); 13CNMR (DMSO-d6, 100 MHz) δ 149.2, 143.1, 141.7, 132.7, 131.2, 130.8, 130.5, 130.2, 129.9, 129.3, 127.5, 126.0, 124.8, 122.1, 119.4, 116.6, 112.7; MS+(APCI) 372.93 [M + H]+ Anal. Calcd for C16H10F6N4: C, 51.62; H, 2.71; N, 15.05 found: C, 51.57; H, 2.60; N, 14.93
(3,5-Bis-trifluoromethyl-phenyl)-[4-(4-methoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-amine (6g)
77%, colorless solid; mp 212–213 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.27 (s, 1H), 8.45 (s, 1H), 8.35 (s, 2H), 7.51 (s, 1H), 7.47 (dd, J = 9.2, 2.8 Hz, 2H), 7.13 (dd, J = 9.2, 2.8 Hz, 2H), 3.82 (s, 3H); MS+(APCI) 403.02 [M + H]+ Anal. Calcd for C14H12N4·0.5CH3OH: C, 50.25; H, 3.37; N, 13.39. Found: C, 50.49; H, 3.56; N, 13.57
(3,5-Bis-trifluoromethyl-phenyl)-(4-p-tolyl-4H-[1,2,4]triazol-3-yl)-amine (6h)
55%, colorless solid; mp 230–231 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.33 (s, 1H), 8.48 (s, 1H), 8.32 (s, 2H), 7.51 (s, 1H), 7.41 (dd, J = 13.6, 8 Hz, 4H), 2.39 (s, 3H); MS+(APCI) 386.85 [M + H]+ Anal. Calcd for C17H12F6N4: C, 52.86; H, 3.13; N, 14.50. Found: C, 52.62; H, 3.07; N, 14.32
(3,5-Bis-trifluoromethyl-phenyl)-(4-o-tolyl-4H-[1,2,4]triazol-3-yl)-amine (6i)
50%, colorless solid; mp 239–240 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.26 (s, 1H), 8.43 (s, 1H), 8.40 (s, 2H) 7.51 (s, 1H), 7.49 - 7.41 (m, 4H), 2.05 (s, 3H); MS+(APCI) 386.97 [M + H]+ Anal. Calcd for C17H12F6N4: C, 52.86; H, 3.13; N, 14.50. Found: C, 52.71; H, 2.92; N, 14.60
(3,5-Bis-trifluoromethyl-phenyl)-[4-(2-isopropyl-phenyl)-4H-[1,2,4]triazol-3-yl]-amine (6j)
45%, colorless solid; mp 216 – 217 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.21 (s, 1H), 8.46 (s, 1H), 8.43 (s, 2H), 7.60 (m, 2H), 7.53 (s, 1H), 7.42 - 7.40 (m, 2H), 2.45 (septa, J = 6.8 Hz, 1H), 1.08 (dd, J = 6.8, 1.8 Hz, 6H); MS+(APCI) 414.90 [M + H]+ Anal. Calcd for C19H16F6N4: C, 55.08; H, 3.89; N, 13.52. Found: C, 54.82; H, 3.76; N, 13.66
(3,5-Bis-trifluoromethyl-phenyl)-[4-(2-tert-butyl-phenyl)-4H-[1,2,4]triazol-3-yl]-amine (6k)
(following general procedure III) 49%, colorless solid; mp 249–250 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 9.11 (s, 1H), 8.49 (s, 2H), 8.48 (s, 1H), 7.73 (dd, J = 7.6, 1.2 Hz, 1H), 7.58 (dt, J = 6.8, 1.6 Hz, 1H), 7.52 (s, 1H), 7.40 (dt, J = 7.6, 1.2 Hz, 1H), 7.23 (dd, J = 7.6, 1.2 Hz, 1H), 2.45 (septa, J = 6.8 Hz, 1H), 1.15 (s, 9H); MS+(APCI) 428.98 [M + H]+ Anal. Calcd for C20H18F6N4: C, 56.08; H, 4.24; N, 13.08. Found: C, 55.94; H, 4.09; N, 13.00
1-Benzyl-1-(4-methoxy-phenyl)-3-phenyl-thiourea (8a)
93%, off-white solid: mp 87–88 ºC; 1HNMR (DMSO-d6, 500 MHz) δ 8.69 (s, 1H), 7.34-7.26 (m, 8H), 7.23 (t, J = 6.5 Hz, 1H), 7.15-7.10 (m, 1H), 7.03 (dd, J = 110, 9.0 Hz, 4H), 5.50 (s, 2H), 3.72 (s, 3H); 13CNMR (DMSO-d6, 125 MHz) δ 182.5, 158.3, 140.7, 137.5, 134.4, 128.7, 128.2, 127.8, 127.0, 126.3, 124.9, 114.9, 57.6, 55.2; MS+(APCI) 348.96 [M + H]+; RT = 10.84
1-Benzyl-1-(4-chloro-phenyl)-3-phenyl-thiourea (8b)
85%, 1HNMR (CDCl3, 500 MHz) δ 7.38-7.27 (m, 10H), 7.22-7.19 (m, 1H), 7.06 (dd, J = 8.5, 2.0 Hz, 1H), 6.95 (bs, 1H), 5.55 (s, 2H); 13CNMR (DMSO-d6, 125 MHz) δ 181.9, 139.2, 138.9, 136.7, 134.9, 130.7, 129.4, 128.6, 128.4, 127.6, 126.2, 125.8, 58.1; MS+(APCI) 352.98 [M + H]+
1-Benzyl-3-(4-methoxy-phenyl)-1-phenyl-thiourea (9)
77%, beige solid: mp 141–142 ºC; 1HNMR (DMSO-d6, 400 MHz) δ 8.70 (s, 1H), 7.36-7.13 (m, 12H), 6.82 (d, J = 9 Hz, 2H), 5.49 (s, 2H), 3.69 (s, 3H); MS+(APCI) 348.86 [M + H]+; RT = 10.60
Benzyl-(4-methoxy-phenyl)-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (10a)
20%, colorless solid; 1HNMR (DMSO-d6, 400 MHz) δ 8.57 (s, 1H), 7.38 (d, J = 7.2 Hz, 2H), 7.32-7.18 (m, 9H), 6.60 (dd, J = 27.0, 9.0 Hz, 4H), 4.84 (s, 2H), 3.55 (s, 3H); MS+(APCI) 357.03 [M + H]+; RT = 8.86
Benzyl-(4-chloro-phenyl)-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (10b)
24%, colorless solid; 1HNMR (CDCl3, 500 MHz) δ 7.69 (d, J = 8.0 Hz, 2H), 7.22-7.19 (m, 3H), 7.13-7.10 (m, 2H), 7.10-6.95 (m, 5H), 6.91 (m, 3H) 5.35 (s, 2H); 13CNMR (DMSO-d6, 125 MHz) δ141.9, 140.1, 135.7, 134.4, 132.3, 129.5, 128.64, 128.60, 128.53, 128.52, 126.8, 126.6, 126.1, 120.5 ; MS+(APCI) 361.02 [M + H]+
Benzyl-(4-methoxy-phenyl)-(4-phenyl-4H-[1,2,4]triazol-3-yl)-amine (11)
18%, colorless solid; 1HNMR (DMSO-d6, 400 MHz) δ 8.67 (s, 1H), 7.40 (d, J = 8 Hz, 2H), 7.28-7.19 (m, 5H), 7.04 (t, J = 7.2 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.71 (t, J = 6.8 Hz, 1H), 6.61 (d, J = 8.4 Hz, 2H), 4.82 (s, 2H), 3.71 (s, 3H); MS+(APCI) 356.89 [M + H]+; RT = 9.07
Supplementary Material
Acknowledgments
We thank Drs. Daniel C. Tosteson and Magdalena Tosteson for their continuous encouragement and support. This work was supported in part by NIH grant NCDDG 5 U19 CA87427.
Footnotes
SUPPORTING INFORMATION: General procedures for synthesis, 1HNMR of 3a, 3c, 4a, 4b, 4c, 4k, 6c, 6d, 7d and 6f; 13CNMR of 3a, 4a, 4b, 4k, 6c, 7d and 6f; LCMS of 3c, 4c, 6d–7d regioisomeric mixture and 6e–7e regioisomeric mixture and NOESY and TOCSY of 6c, 6f and 6g. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Dumas J. Curr Opin Drug Discov Devel. 2002;5:718–27. [PubMed] [Google Scholar]
- 2.De Clercq E. Med Res Rev. 2002;22:531–65. doi: 10.1002/med.10021. [DOI] [PubMed] [Google Scholar]
- 3.DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Comb Chem High Throughput Screen. 2004;7:473–94. doi: 10.2174/1386207043328544. [DOI] [PubMed] [Google Scholar]
- 4.Smith RA, Barbosa J, Blum CL, Bobko MA, Caringal YV, Dally R, Johnson JS, Katz ME, Kennure N, Kingery-Wood J, Lee W, Lowinger TB, Lyons J, Marsh V, Rogers DH, Swartz S, Walling T, Wild H. Bioorg Med Chem Lett. 2001;11:2775–8. doi: 10.1016/s0960-894x(01)00571-6. [DOI] [PubMed] [Google Scholar]
- 5.Cheung M, Harris PA, Hasegawa M, Ida S, Kano K, Nishigaki N, Sato H, Veal JM, Washio Y, West RI. Glaxo Group Ltd. 2002. [Google Scholar]
- 6.Jain J, Almquist SJ, Shlyakhter D, Harding MW. J Pharm Sci. 2001;90:625–37. doi: 10.1002/1520-6017(200105)90:5<625::aid-jps1019>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Baraldi PG, Tabrizi MA, Preti D, Bovero A, Romagnoli R, Fruttarolo F, Zaid NA, Moorman AR, Varani K, Gessi S, Merighi S, Borea PA. J Med Chem. 2004;47:1434–47. doi: 10.1021/jm0309654. [DOI] [PubMed] [Google Scholar]
- 8.Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S, Morishima H. J Med Chem. 2001;44:4615–27. doi: 10.1021/jm0103256. [DOI] [PubMed] [Google Scholar]
- 9.Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, Klaus B, Madwed J, Moriak M, Moss N, Pargellis C, Pav S, Proto A, Swinamer A, Tong L, Torcellini C. J Med Chem. 2002;45:2994–3008. doi: 10.1021/jm020057r. [DOI] [PubMed] [Google Scholar]
- 10.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Nat Struct Biol. 2002;9:268–72. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 11.Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S, Morishima H. J Med Chem. 2001;44:4628–40. doi: 10.1021/jm010326y. [DOI] [PubMed] [Google Scholar]
- 12.Patani GA, LaVoie EJ. Chem Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 13.Kucukguzel I, Kucukguzel SG, Rollas S, Kiraz M. Bioorg Med Chem Lett. 2001;11:1703–7. doi: 10.1016/s0960-894x(01)00283-9. [DOI] [PubMed] [Google Scholar]
- 14.Street LJ, Baker R, Davey WB, Guiblin AR, Jelley RA, Reeve AJ, Routledge H, Sternfeld F, Watt AP, Beer MS, et al. J Med Chem. 1995;38:1799–810. doi: 10.1021/jm00010a025. [DOI] [PubMed] [Google Scholar]
- 15.Hitotsuyanagi Y, Motegi S, Fukaya H, Takeya K. J Org Chem. 2002;67:3266–71. doi: 10.1021/jo010904i. [DOI] [PubMed] [Google Scholar]
- 16.Hitotsuyanagi Y, Motegi S, Hasuda T, Takeya K. Org Lett. 2004;6:1111–4. doi: 10.1021/ol040005r. [DOI] [PubMed] [Google Scholar]
- 17.Brockunier LL, Parmee ER, Ok HO, Candelore MR, Cascieri MA, Colwell LF, Jr, Deng L, Feeney WP, Forrest MJ, Hom GJ, MacIntyre DE, Tota L, Wyvratt MJ, Fisher MH, Weber AE. Bioorg Med Chem Lett. 2000;10:2111–4. doi: 10.1016/s0960-894x(00)00422-4. [DOI] [PubMed] [Google Scholar]
- 18.Borg S, Vollinga RC, Labarre M, Payza K, Terenius L, Luthman K. J Med Chem. 1999;42:4331–42. doi: 10.1021/jm990197+. [DOI] [PubMed] [Google Scholar]
- 19.Jesberger M, Davis TP, Barner L. Synthesis. 2003:1929–1958. [Google Scholar]
- 20.Katritzky AR, Qi M, Feng D, Zhang G, Griffith MC, Watson K. Org Lett. 1999;1:1189–1191. [Google Scholar]
- 21.Larsen SD, DiPaolo BA. Org Lett. 2001;3:3341–3344. doi: 10.1021/ol016578a. [DOI] [PubMed] [Google Scholar]
- 22.Stocks MJ, Cheshire DR, Reynolds R. Org Lett. 2004;6:2969–71. doi: 10.1021/ol048863a. [DOI] [PubMed] [Google Scholar]
- 23.Levallet C, Lerpiniere J, Ko SY. Tetrahedron. 1997;53:5291–5304. [Google Scholar]
- 24.Yong YF, Kowalski JA, Lipton MA. Journal of Organic Chemistry. 1997;62:1540–1542. [Google Scholar]
- 25.Sohtome Y, Tanatani A, Hashimoto Y, Nagasawa K. Tetrahedron Letters. 2004;45:5589–5592. [Google Scholar]
- 26.Sahu M, Garnaik BK, Behera R. Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry. 1987;26B:779–81. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.