

Abstract
We describe a microchip designed to quantify the levels of a dozen cytoplasmic and membrane proteins from single cells. We use the platform to assess protein–protein interactions associated with the EGF-receptor-mediated PI3K signaling pathway. Single-cell sensitivity is achieved by isolating a defined number of cells (n = 0–5) in 2 nL volume chambers, each of which is patterned with two copies of a miniature antibody array. The cells are lysed on-chip, and the levels of released proteins are assayed using the antibody arrays. We investigate three isogenic cell lines representing the cancer glioblastoma multiforme, at the basal level, under EGF stimulation, and under erlotinib inhibition plus EGF stimulation. The measured protein abundances are consistent with previous work, and single-cell analysis uniquely reveals single-cell heterogeneity, and different types and strengths of protein–protein interactions. This platform helps provide a comprehensive picture of altered signal transduction networks in tumor cells and provides insight into the effect of targeted therapies on protein signaling networks.
Although signal transduction inhibitors occasionally offer clinical benefit for cancer patients (1), signal flux emanating from oncogenes is often distributed through multiple pathways (2), potentially underlying the failure of most such inhibitors (3). Measuring signal flux through multiple pathways, in response to signal transduction inhibitors, may help uncover network interactions that contribute to therapeutic resistance and that are not predicted by analyzing pathways in isolation (4). The cellular and molecular complexity of a solid tumor microenvironment (5) suggests the need to study signaling in individual cancer cells.
Protein–protein interactions within signaling pathways are often elucidated by assessing the levels of relevant pathway proteins in model and tumor-derived cell lines and with various genetic and molecular perturbations. Such interactions, and the implied signaling networks, may also be elucidated via quantitative measurements of multiple pathway-related proteins within single cells (6). At the single-cell level, inhibitory and activating protein–protein relationships, as well as stochastic (single-cell) fluctuations, are revealed. However, most techniques for profiling signaling pathways (7, 8) require large numbers of cells. Single-cell immunostaining (9) is promising, and some flow cytometry (6) techniques are relevant, as discussed below.
We describe quantitative, multiplex assays of intracellular signaling proteins from single cancer cells using a platform called the single-cell barcode chip (SCBC). The SCBC is simple in concept: A single or defined number of cells is isolated within an approximately 2 nL volume microchamber that contains an antibody array (10) for the capture and detection of a panel of proteins. The SCBC design (11) permits lysis of each individual trapped cell.
Intracellular staining flow cytometry can assay up to 11 phosphoproteins from single cells (6). Our SCBC can profile a similar size panel, but only for approximately 100 single cells per chip. Each protein is assayed twice, yielding some statistical assessment for each experiment. The SCBC is a relatively simple platform and only requires a few hundred cells per assay.
We used the SCBC to study signal transduction in glioblastoma multiforme (GBM), a primary malignant brain tumor (12). GBM has been genetically characterized, yet the nature of signaling pathways downstream of key oncogenic mutations, such as epidermal growth factor receptor activating mutation (EGFRvIII) and phosphatase and tensin homolog (PTEN) tumor suppressor gene loss associated with receptor tyrosine kinase (RTK)/PI3K signaling, are incompletely understood (13–15). Single-cell experiments may also help resolve the characteristic heterogeneity of GBM.
We interrogated 11 proteins directly or potentially associated with PI3K signaling (see SI Appendix, Methods I) through three isogenic GBM cell lines: U87 (expressing wild-type p53, mutant PTEN, and low levels of wild-type EGFR, no EGFRvIII) (16, 17), U87 EGFRvIII (U87 cells stably expressing EGFRvIII deletion mutant), and U87 EGFRvIII PTEN (U87 cells coexpressing EGFRvIII and PTEN) (18). Fig. 1 diagrams this biology. Each cell line was investigated under conditions of standard cell culture, in response to EGF stimulation, and after erlotinib treatment followed by EGF stimulation. The proteins assayed represented RTKs and proteins signifying activation of PI3K and MAPK signaling. They were (p- denotes phosphorylation) p-Src, p-mammalian target of rapamycin (p-mTOR), p-p70 ribosomal protein S6 kinase (p-p70S6K), p-glycogen synthase kinase-3 (p-GSK-3α/β), p-p38 mitogen activated protein kinase (p-p38α), p-extracellular regulated kinase (p-ERK), p-c-Jun N-terminal kinase (p-JNK2), p-platelet derived growth factor receptor β (p-PDGFRβ), p-vascular endothelial growth factor receptor 2 (p-VEGFR2), tumor protein 53 (P53), and total EGFR.
Fig. 1.

The PI3K pathway activated by EGF-stimulated EGFR or by the constitutively activated EGFRvIII. All proteins in light blue with central yellow background were assayed. Orange background proteins were expressed in the cell lines U87 EGFRvIII or U87 EGFRvIII PTEN. The oval, yellow background components are the investigated molecular perturbations.
Results
The SCBC for Quantitative, Multiplex Measurement of Intracellular Signaling Proteins.
The SCBC is comprised of a two-layer microfluidic network (11) (Fig. 2A and SI Appendix, Methods II). Valves isolate the chip into 120 microchambers for cell compartmentalization, cell lysis, and protein assays (Fig. 2 B and C). Upon cell loading, each microchamber contains zero to a few cells, which are counted through the transparent chip. Cells are lysed via diffusion of lysis buffer from the neighboring chamber (Fig. 2C and SI Appendix, Methods II). Capturing the (transient) levels of phosphorylated proteins is a key objective. After testing literature recipes (6–9) (see SI Appendix, Fig. S1), a protocol was developed. Following cell stimulation, the cells were trypsinized for 5 min and all subsequent steps were done at or near ice temperatures. The cells were centrifuged, resuspended in PBS, and introduced into a prechilled SCBC within 15 min of stimulation. Within 30 min of stimulation, the cells had been lysed. The lysis buffer contains phosphatase and protease inhibitors.
Fig. 2.

The single-cell barcode chip. (A) A photograph of an SCBC. The flow layer (red) and the control valve layer (blue) are delineated with food dyes. A photograph (B) and a drawing (C) of a single microchamber, with critical parts labeled. A cell is isolated in the cell chamber by the valves. The neighboring chamber contains cell lysis buffer. The duplicate DNA barcode copies are converted into an antibody array prior to cell loading, counting, and lysis. Also see SI Appendix, Fig. S1.
The SCBC antibody arrays begin as 20-μm-wide DNA barcodes (10). After SCBC assembly, they are converted into antibody barcodes using DNA-encoded antibody libraries (Fig. 2C and SI Appendix, Methods I). Captured proteins are developed by applying biotinylated detection antibodies and fluorophore-labeled streptavidin. A barcode contains 11 antibody stripes, an alignment stripe, and a control stripe. All ssDNA and antibody reagents are provided in SI Appendix, Table S1. The uniformity of the DNA barcodes was evaluated through the use of fluorophore-labeled complimentary DNAs. The barcodes exhibited uniform DNA loading, with coefficients of variation ≤ 11% (SI Appendix, Methods III).
The antibody pairs were selected to detect only phosphorylated proteins (excepting p53 and EGFR). The barcode protein assays exhibited sensitivities and dynamic ranges comparable to the commercial ELISAs. See SI Appendix, Methods III for calibration and cross-reactivity data, as well as coefficients of variation for all antibodies in the barcodes. A 2-h incubation used here reaches > 95% of maximal intensity for all assays (SI Appendix, Methods III).
For the single-cell assays, experimental variation can also arise from the location of cells in the microchambers prior to cell lysis, because of the competition between the antibody/antigen binding kinetics relative to protein diffusion times. The duplicate barcode assays in each chamber, coupled with a Monte Carlo simulation, allowed for estimation of this experimental variation to be < 15% (see SI Appendix, Methods III). The uncertainties are small compared to the measured variation in protein levels, as described below. The biologic variation can be extracted from  .
.
Quantitative protein abundances (copy numbers for a given protein) are utilized herein. Some standard proteins (used for calibrations) were not commercially available (e.g., p-VEGFR2 and EGFRvIII), and so relative fluorescence intensities are used. Furthermore, standard proteins may differ from the cellular proteins (e.g., glycosylation levels may vary). Calibrations utilized standard protein spiked into buffer, whereas SCBC protein levels are measured in a more complex environment. Background levels from microchambers containing zero cells, correlations of protein signal strengths with numbers of cells (SI Appendix, Methods III), and comparison of SCBC assays with traditional (Western blotting) assays, as well as with flow cytometry (19) have all been measured to further validate the SCBC assays.
PI3K Pathway-Associated Protein Assays from Single GBM Cells and Bulk Cell Lysates.
Here we give protocols for stating whether a protein was detected, and we provide comparisons of the SCBC protein assays to assays from bulk cell lysate, including literature results.
Fig. 3 shows heat map data from SCBC experiments on U87 EGFRvIII PTEN cells and from measurements on bulk populations of those cells. Individual microchamber data are shown in SI Appendix, Fig. S2. The detection threshold for a given protein and set of conditions (numbers of cells, cell line, stimulation conditions) was defined by a signal/noise (S/N)≥2. The signal was the average reading from the duplicate barcode assays from each microchamber, averaged over all experiments for a given set of conditions. The noise was estimated from the negative control DNA stripe within each barcode. See SI Appendix, Table S2 for the calculated average S/N. For example, for single U87 EGFRvIII PTEN cells stimulated with EGF, we observed nine proteins (S/N levels are included after each protein name): EGFR (130), p53 (13), p-VEGFR2 (14), p-ERK (12), p-p38α (11), p-GSK3α/β (12), p-p70S6K (10), p-mTOR (8), and p-Src (11), thus indicating that we detect both membrane and cytoplasmic proteins. All 11 assayed proteins were detected in the single-cell experiments, except that p-PDGFRβ was detected only at low levels (S/N = 2) or not at all; p-JNK2 was only detected consistently in U87 EGFRvIII cells (S/N of 2–6 for single-cell assays); p-mTOR and p-70S6K were not detected for U87 EGFRvIII PTEN cells with erlotinib + EGF.
Fig. 3.

SCBC and bulk cell measurements of U87 EGFRvIII PTEN cells. (A) Heat maps of SCBC protein level assays. Each column represents one microchamber assay; each row represents a protein. (B) Protein assays from a population of U87 EGFRvIII PTEN cells under EGF stimulation. The contrast of these images has been equally adjusted, and the intensity of EGFR is divided by five in the heat maps.
Fig. 3B shows protein assays measured from a population of EGF-stimulated U87 EGFRvIII PTEN cells. These assays used similar cell lysis and assay protocols as the SCBC assays. Comparison across Fig. 3 A and B reveals that the bulk assays and SCBC measurements are self-consistent. Comparisons between bulk cell assays (SI Appendix, Fig. S3), SCBC single-cell measurements, and literature results (18, 20–23) were also done to detect distinct phosphorylation states of EGFR under the influence of EGF and erlotinib stimulation. Those results again formed a self-consistent dataset.
Data, such as shown in Fig. 3, were first averaged to recapitulate measurements of proteins from cell populations for comparison with known biology. It was then more fully analyzed to yield a statistical representation of fluctuations at the single-cell level.
Bulk-Like Protein Profiles Collected from SCBC Data.
Fig. 4A presents the protein abundances (averaged over all three-cell experiments), measured for each cell line and for all conditions (mean intensities and standard deviations are presented in SI Appendix, Table S3). We compared these SCBC results with literature findings that used conventional bulk cell assays, as well as with our own Western blot assays (Fig. 4). In the following discussion, literature citations following the protein names provide validation of our SCBC results.
Fig. 4.

Averaged responses for all three cell lines to EGF and erlotinib (eb) + EGF exposures. (A, Left) Measured protein expression profiles, in fluorescent intensity units, averaged over the three-cell measurements (n of approximately 20). The signal of EGFR/EGFRvIII is divided by 10. Results are shown as mean ± SD. SD represents the combined experimental error and intrinsic biological variation, but is dominated by the biological variation. (Right) Western blot analysis of p-EGFR, p-ERK, p-mTOR, p-Src, p-p70S6K, p-GSK3β, and p-Akt expression in U87 EGFRvIII and U87 EGFRvIII PTEN cell lines at the basal, EGF stimulation, and erlotinib + EGF treatment states. (B) Heat map of relative expression fold changes of proteins, normalized by unperturbed U87 cells. (C, Left) Mean fold change of phosphorylation levels of p-ERK and p-mTOR in different cell lines and conditions, relative to unperturbed U87 cells. (E, EGF; e + E, erlotinib + EGF). (Right) Western blot analysis of p-ERK and p-mTOR expression in response to EGF stimulation in U87 EGFRvIII and U87 EGFRvIII PTEN cell lines. These cells were cultured in DMEM medium containing 10% FBS for 24 h, then in serum-free medium for 24 h or (+) erlotinib (10 μM) treatment in serum-free medium, followed by stimulation (+) with EGF (20 ng/mL) for 15 min. Cells were lysed and the listed proteins were detected by Western blotting.
At basal level, U87 cells (Fig. 4A, Top) showed low EGFR phosphorylation (23) (SI Appendix, Fig. S3) and modest activation of signaling proteins, including p-Src (22), p-mTOR (24), p-p70S6K (23, 25), p-GSK3α/β (23, 26, 27), p-p38α (24), and p-ERK (18, 22, 26), whereas p-JNK2 was not detected (23). U87 EGFRvIII cells (Fig. 4A, Middle) exhibited increased baseline levels of phosphorylation compared with cells expressing wild-type EGFR, including p-Src (22), p-mTOR, p-p70S6K (25), p-ERK (18), and p-JNK2 (28). In U87, EGFRvIII PTEN cells (Fig. 4A, Bottom), PTEN coexpression diminished baseline phosphorylation of p-Src, p-mTOR, p-p70S6K (25), p-ERK (18), and p-JNK2 compared with U87 EGFRvIII.
EGF stimulation induced EGFR phosphorylation (18, 27, 28) (SI Appendix, Fig. S3) and promoted downstream pathway activation in all three cell lines, irrespective of PTEN status, including activation of p-p70S6K (25) and p-ERK (18). The increase of levels of p-ERK in response to EGF stimulation in U87 EGFRvIII and U87 EGFRvIII PTEN cells is demonstrated by the Western blots shown in Fig. 4A. The level of p-GSK3α/β in response to EGF stimulation was increased in U87 (27) and U87 EGFRvIII cells, but remained relatively unchanged in U87 EGFRvIII PTEN cells (consistent with Western blots in Fig. 4A).
Erlotinib inhibition + EGF stimulation diminished phosphorylation of both EGFR and EGFRvIII (18) (SI Appendix, Fig. S3) relative to EGF stimulation. It led to decreased phosphorylation levels in U87, although those levels are higher than in the unstimulated cells. One previously identified example of this effect is p-p70S6K (14, 18). Erlotinib + EGF showed little impact on U87 EGFRvIII cells, indicating that PTEN loss confers resistance to EGFR tyrosine kinase inhibitors (14, 21). The phosphoprotein expression levels decrease, but are above the unstimulated levels. Representative proteins include p-Src (22) and p-p70S6K (14, 18, 25). Erlotinib significantly diminished phosphorylation levels of p-ERK, p-p70S6K (14, 18), p-mTOR, and p-Src only for the U87 EGFRvIII PTEN cells. Those phosphorylation levels are below those observed for unperturbed cells; p-p70S6k and p-mTOR drop to below the detection limit. These results are consistent with previous findings that coexpression of EGFRvIII and PTEN protein by GBM cells is associated with clinical response to EGFR kinase inhibitor therapy (14).
Fig. 4B shows the heat map of relative mean fold changes in the expression levels of proteins and phosphoproteins for the different cell lines and conditions, normalized by the protein levels measured from unperturbed U87 cells. This plot was calculated as follows. For a microchamber i containing n cells, the fluorescence levels recorded from the two barcode assays for a given protein ρ were averaged to yield ρi,n. The fluorescence intensity for ρ, averaged over all zero-cell measurements, was subtracted as background: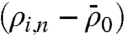 . This value was then normalized against the background-subtracted, fluorescence levels of ρ averaged over all n cell measurements for unperturbed U87:
. This value was then normalized against the background-subtracted, fluorescence levels of ρ averaged over all n cell measurements for unperturbed U87: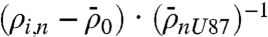 . These fold changes were then averaged over all microchambers containing two to five cells, and were combined to produce the heat map of Fig. 4B. This map provides a relative comparison of the pathway activation states in different cell lines and conditions, but it also emphasizes that the phosphorylation of p-ERK (representative of MAPK signaling) exhibits correlation with the phosphorylation of mTOR (PI3K signaling).
. These fold changes were then averaged over all microchambers containing two to five cells, and were combined to produce the heat map of Fig. 4B. This map provides a relative comparison of the pathway activation states in different cell lines and conditions, but it also emphasizes that the phosphorylation of p-ERK (representative of MAPK signaling) exhibits correlation with the phosphorylation of mTOR (PI3K signaling).
Recent work suggests cross-talk between the Rat scarcoma (Ras)/MAPK and PI3K signaling pathways (3, 29). Recent work has also uncovered a negative regulatory feedback loop by which mTOR complex 1 signaling through S6K1 suppresses PI3K-mediated activation of MAPK activity, so that inhibition of mTOR signaling through S6K1 can activate MAPK (30, 31). This implied correlation between PI3K and MAPK signaling can be estimated by comparing the phosphorylation levels of ERK and mTOR in varying genetic contexts that regulate PI3K signaling and in response to ligand stimulation and/or inhibition. The mean fold changes of p-ERK and p-mTOR in U87 EGFRvIII and U87 EGFRvIII PTEN cell lines are shown in Fig. 4C, Left. In U87 EGFRvIII cells, the fold change of p-ERK under basal level, EGF stimulation, and erlotinib + EGF treatment are statistically lower than that of p-mTOR (SI Appendix, Table S3). However, in U87 EGFRvIII PTEN cells, the situation is reversed. Obviously, PTEN expression sensitizes GBM cells to MAPK signaling stimulated by EGF. This preferential activation of MAPK signaling pathways in response to EGF activation in GBM cells containing PTEN was validated by immunoblot analysis (Fig. 4C, Right) and is consistent with recent findings that minimal levels of ERK signaling are required for optimal EGFRvIII-mediated tumor cell growth in PTEN null glioblastomas (15). These data demonstrate that SCBC measurements can uncover feedback loops and pathway cross-talk in situations where the connectivity is less well defined.
Single-Cell Protein Profiles, Protein–Protein Correlations, and Correlation Networks.
Profiles that reveal the relative importance of the measured biological fluctuations versus the experimental errors are shown in SI Appendix, Fig. S4 A and B. Two points are relevant for comparing bulk cell assays and single-cell measurements. SI Appendix, Fig. S4A, which plots P53 intensity versus experiment number, for the sets of one, two, and three cell experiments, illustrates how a small fraction of cells can dominate an assay. SI Appendix, Fig. S4B provides histograms of the number of p-ERK molecules detected, versus frequency of detection, for single U87 EGFRvIII PTEN cells under all three conditions. Those histograms may be compared against the averaged p-ERK intensities presented at the bottom of Fig. 4A. According to Fig. 4A, the p-ERK level for the unperturbed cells is only slightly higher than for the EGF + erlotinib exposed cells. However, the coefficient of variation of p-ERK levels is much larger (57%) than in the EGF + erlotinib perturbed cells (28%). This effect, which is not captured in bulk assays, may represent an increased amount of regulation for p-ERK in the EGF + erlotinib perturbed cells (32).
The levels of several proteins associated with PI3K signaling should exhibit coordinated behaviors (6). A typical protein–protein positive correlation (p-mTOR vs. p-p70S6K for unstimulated U87 EGFRvIII cells) and an anticorrelation (p-GSK3α/β vs. p-ERK for unstimulated U87 EGFRvIII PTEN) are shown in SI Appendix, Fig. S4 C and D. The positive correlation is independent of the numbers of cells per microchamber assay, whereas the negative correlation begins to be masked for populations as low as three cells.
Fig. 5 provides nine SCBC-derived protein correlation networks. The line weight defines the strength of the correlation (see key). We used the Bonferroni method (33), which limits correlations to those that exhibit a p value ≤ 0.05; correlation coefficients above 0.4 or below -0.4 are significant. Perturbation by ligand stimulation and/or receptor inhibition reveal new relationships and the genetic context of those relationships. EGF stimulation of EGFRvIII-expressing GBM cells greatly enhances network connectivity in a way that is very different from what would be expected from simply summing the effects of EGF treatment (U87 + EGF, Fig. 5, Top Center) and EGFRvIII expression (U87 EGFRvIII, Middle Left). This represents a clinically and biologically relevant result because wild-type EGFR is always present in EGFRvIII-expressing cells (14). The greatly enhanced network interconnectivity for the EGF-stimulated U87 EGFRvIII cells may suggest a mechanism underlying the difficulty of inhibiting downstream signaling in EGFRvIII-expressing, PTEN null tumor cells, potentially providing one mechanism for their striking tumorigenicity and their established role in promoting therapeutic resistance. This observation is consistent with the clinical failure and the lack of p70S6K inhibition observed in EGFRvIII-expressing, PTEN-deficient GBM patients treated with erlotinib (14), and suggests that clinically relevant insights may potentially be derived from these types of single-cell experiments.
Fig. 5.

Protein correlation maps under different genetic and environmental perturbations. All indicated correlations pass a Bonferroni corrected p-value test (p = 0.05). Underlined proteins are below the detection limit.
Classical genetics is also often used to combine perturbations and phenotypic responses to infer functional relationships between genes (34), but specific interactions are difficult to extract because intermediate interacting partners may contribute combinations of positive and/or negative interactions.
Discussion
The SCBC provides certain advantages for assaying cytoplasmic proteins. The ability to normalize protein levels to numbers of cells permits for the SCBC data to recapitulate qualitative protein measurements from bulk cell populations, but in a quantitative fashion. One example relates toward interrogating cross-talk between the Ras/MAPK and RTK/PI3K signaling in GBM (3, 29, 30). Using the SCBC, we found that, for U87 EGFRvIII PTEN cells, stimulation with EGF (associated with RTK/PI3K signaling) led to a sharp increase in levels or p-ERK (associated with the Ras/MAPK pathway), a result that was confirmed using Western blot analysis of the bulk cell lines. Exposure of those same cells to erlotinib + EGF kept the p-ERK levels near the level of unstimulated U87 EGFR vIII PTEN cells.
A second advantage relates to the assessment of the single-cell fluctuations, defined by the distribution of the levels of a given protein, measured across many SCBC assays. The measured biological variation that arises from the functional heterogeneity of a genetically identical cell population is significantly higher than the experimental error and varies across proteins. These fluctuations provide a gauge of the heterogeneity of the cell population and can be used to predict the thermodynamic stability of specific proteins toward perturbations (32).
The SCBC barcodes could potentially be expanded to 35–40 proteins, depending upon the availability of antibody pairs, but even for just 11 intracellular proteins, the correlation networks extracted from SCBC data already provide interesting parallels with the tumorigenecity and therapeutic resistance of EGFRvIII positive, PTEN null tumors. Expanding the protein panel will permit a more complete mapping of the connectivity between known GBM signaling pathways and how that connectivity may be influenced by molecular (i.e., therapeutic) or physical (i.e., hypoxia) perturbations. A further significant challenge will be to extend this platform toward the analysis of clinical specimens.
Materials and Methods
Cell Lines, Antibodies and Regents.
The human GBM cell line U87 was purchased from American Tissue Culture Collection. U87 EGFRvIII and U87 EGFRvIII PTEN cells were constructed as previously described (14, 18). Cell lines were routinely maintained in DMEM (American Type Culture Collection) containing 10% fetal bovine serum in a humidified atmosphere of 5% CO2, 95% air at 37 °C. See SI Appendix, Table S1 for DNA and antibody reagents. Other reagents were obtained as follows: phosphatase inhibitor cocktail, bovine serum albumin, and n-dodecyl-β-d-maltoside, Sigma-Aldrich; Cy5-conjugated straptavidin, eBioscience; human EGF, Prospec; cell lysis buffer, Cell Signaling; complete protease inhibitor cocktail, Roche.
Microchip Fabrication.
The SCBCs were assembled from a DNA barcode microarray glass slide and a polydimethylsiloxane (PDMS) slab containing the microfluidic circuit, as fully described in SI Appendix, Methods II. The PDMS SCBC chip was fabricated using a two-layer soft lithography, with a control layer and a flow layer (11). The control layer PDMS chip was aligned onto the flow layer and bonded for 60 min at 80 °C. The two-layer PDMS chip was then cut off, access holes were drilled, and then it was thermally bonded onto the barcoded glass slide to yield an SCBC.
Cell Stimulation and Erlotinib Treatment.
For EGF stimulation, cells were serum starved for 24 h and then stimulated by EGF at 50 ng/mL for 10 min before harvest. For erlotinib treatment, serum-starved cells were treated with 10 μM erlotinib for 24 h, followed by EGF stimulation (50 ng/mL) for 10 min before harvest. The treated cells were dissociated with trypsin and EDTA and suspended in cold PBS with a concentration of 1,000 cells per microliter prior to loading to the device.
Cytoplasmic Protein Measurement Using SCBCs.
All SCBC microchannels were blocked with blocking buffer for 60 min. A cocktail of all DNA-antibody conjugates was flowed through the channels for 60 min, transforming the DNA barcode microarrays into antibody microarrays. Unbound conjugates were removed with washing buffer. Then 3× lysis buffer was loaded into the lysis buffer chambers, and cells were loaded in the cell chamber while keeping the valves between these chambers closed. The valves were opened to allow on-chip diffusion of lysis buffer to the neighboring cell chambers for 30 min on ice. The SCBC was then incubated 30 min on ice and 1 h at room temperature with gentle shaking. Cell lysate was quickly removed by washing buffer, followed by flowing biotin-labeled detection antibodies and fluorescent dye-labeled streptavidin for visualization. The barcoded glass slide was then detached for scanning.
Data Analysis and Statistics.
Axon GenePix 4400A (Molecular Devices) was used to obtain the fluorescence images at laser power 80% (635 nm) and 10% (532 nm), optical gains 600 (635 nm) and 400 (532 nm), brightness 80, and contrast 83. Fluorescence line profiles were generated by ImageJ (National Institutes of Health). A custom Excel macroextracted average fluorescence signal for all bars within a given barcode and the barcode profiles were compared to the number of cells using the same program. Heatmap profiles were generated using Treeview (Stanford), and R software was used for computing Pearson correlation coefficients. Comparisons in phosphorylation levels of mTOR and ERK were performed with a one-tailed t test. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments.
We thank Bruz Marzolf and Pamela Troisch for printing DNA microarrays, and the University of California, Los Angeles nanolab for photomask fabrication. This work was funded by the National Cancer Institute Grant 5U54 CA119347 (to J.R.H., principal investigator), The Ben and Catherine Ivy Foundation, the Goldhirsch Foundation, and the Grand Duchy of Luxembourg.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110865109/-/DCSupplemental.
References
- 1.Reynoso D, Trent JC. Neoadjuvant and adjuvant imatinib treatment in gastrointestinal stromal tumor: Current status and recent developments. Curr Opin Oncol. 2010;22:330–335. doi: 10.1097/CCO.0b013e32833aaaad. [DOI] [PubMed] [Google Scholar]
- 2.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.She Q-B, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janes KA, Reinhardt HC, Yaffe MB. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell. 2008;135:343–354. doi: 10.1016/j.cell.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marusyk A, Polyak K. Tumor heterogeneity: Causes and consequences. Biochim Biophys Acta Rev Cancer. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308:523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 7.Ciaccio MF, Wagner JP, Chuu CP, Lauffenburger DA, Jones RB. Systems analysis of EGF receptor signaling dynamics with microwestern arrays. Nat Methods. 2010;7:148–155. doi: 10.1038/nmeth.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du JY, et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol. 2009;27:77–83. doi: 10.1038/nbt.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheong R, Wang CJ, Levchenko A. High content cell screening in a microfluidic device. Mol Cell Proteomics. 2009;8:433–442. doi: 10.1074/mcp.M800291-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin YS, et al. Chemistriesfor patterning robust DNA microbarcodes enable multiplex assays of cytoplasm proteins from single cancer cells. Chemphyschem. 2010;11:3063–3069. doi: 10.1002/cphc.201000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorsen T, Maerkl SJ, Quake SR. Microfluidic large-scale integration. Science. 2002;298:580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- 12.Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: Lessons learned and the road ahead. Neurotherapeutics. 2009;6:500–512. doi: 10.1016/j.nurt.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin L, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellinghoff IK, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 15.Huang PH, et al. Phosphotyrosine signaling analysis of site-specific mutations on EGFRvIII identifies determinants governing glioblastoma cell growth. Mol Biosyst. 2010;6:1227–1237. doi: 10.1039/c001196g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furnari FB, Lin H, Huang HJS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bostrom J, et al. Mutation of the PTEN (MMAC1) tumor suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q. Cancer Res. 1998;58:29–33. [PubMed] [Google Scholar]
- 18.Wang MY, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- 19.Ma C, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–744. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amos S, Martin PM, Polar GA, Parsons SJ, Hussaini IM. Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase C delta/c-Src pathways in glioblastoma cells. J Biol Chem. 2005;280:7729–7738. doi: 10.1074/jbc.M409056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan QW, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007;67:7960–7965. doi: 10.1158/0008-5472.CAN-07-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu KV, et al. Fyn and Src are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009;69:6889–6898. doi: 10.1158/0008-5472.CAN-09-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Premkumar DR, Arnold B, Jane EP, POllack IF. Synergistic interaction between 17-AAG and phosphatidylinositol 3-kinase inhibition in human malignant glioma cells. Mol Carcinog. 2006;45:47–59. doi: 10.1002/mc.20152. [DOI] [PubMed] [Google Scholar]
- 24.Aoki H, et al. Telomere 3′ overhang-specific DNA oligonucleotides induce autophagy in malignant glioma cells. FASEB J. 2007;21:2918–2930. doi: 10.1096/fj.06-6941com. [DOI] [PubMed] [Google Scholar]
- 25.Guo DL, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci USA. 2009;106:12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cournoyer P, Desrosiers RR. Valproic acid enhances protein L-isoaspartyl methyltransferase expression by stimulating extracellular signal-regulated kinase signaling pathway. Neuropharmacology. 2009;56:839–848. doi: 10.1016/j.neuropharm.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Servidei T, Riccardi A, Mozzetti S, Ferlini C, Riccardi R. Chemoresistant tumor cell lines display altered epidermal growth factor receptor and HER3 signaling and enhanced sensitivity to gefitinib. Int J Cancer. 2008;123:2939–2949. doi: 10.1002/ijc.23902. [DOI] [PubMed] [Google Scholar]
- 28.Fromm JA, Johnson SAS, Johnson DL. Epidermal growth factor receptor 1 (EGFR1) and its variant EGFRvIII regulate TATA-binding protein expression through distinct pathways. Mol Cell Biol. 2008;28:6483–6495. doi: 10.1128/MCB.00288-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Downward J. Targeting RAS and PI3K in lung cancer. Nat Med. 2008;14:1315–1316. doi: 10.1038/nm1208-1315. [DOI] [PubMed] [Google Scholar]
- 30.Carracedo A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinkade CW, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin YS, et al. Protein signaling networks from single cell fluctuations and information theory profiling. Biophys J. 2011;100:2378–2386. doi: 10.1016/j.bpj.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtin F, Schulz P. Multiple correlations and Bonferroni’s correction. Biol Psychiatry. 1998;44:775–777. doi: 10.1016/s0006-3223(98)00043-2. [DOI] [PubMed] [Google Scholar]
- 34.Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.