

Abstract
Aims
Fibrosis is known to promote cardiac arrhythmias by disrupting myocardial structure. Given recent evidence that myofibroblasts form gap junctions with myocytes at least in co-cultures, we investigated whether myofibroblast–myocyte coupling can promote arrhythmia triggers, such as early afterdepolarizations (EADs), by directly influencing myocyte electrophysiology.
Methods and results
Using the dynamic voltage clamp technique, patch-clamped adult rabbit ventricular myocytes were electrotonically coupled to one or multiple virtual fibroblasts or myofibroblasts programmed with eight combinations of capacitance, membrane resistance, resting membrane potential, and gap junction coupling resistance, spanning physiologically realistic ranges. Myocytes were exposed to oxidative (1 mmol/L H2O2) or ionic (2.7 mmol/L hypokalaemia) stress to induce bradycardia-dependent EADs. In the absence of myofibroblast–myocyte coupling, EADs developed during slow pacing (6 s), but were completely suppressed by faster pacing (1 s). However, in the presence of myofibroblast–myocyte coupling, EADs could no longer be suppressed by rapid pacing, especially when myofibroblast resting membrane potential was depolarized (−25 mV). Analysis of the myofibroblast–myocyte virtual gap junction currents revealed two components: an early transient-outward Ito-like current and a late sustained current. Selective elimination of the Ito-like component prevented EADs, whereas selective elimination of the late component did not.
Conclusion
Coupling of myocytes to myofibroblasts promotes EAD formation as a result of a mismatch in early vs. late repolarization reserve caused by the Ito-like component of the gap junction current. These cellular and ionic mechanisms may contribute to the pro-arrhythmic risk in fibrotic hearts.
Keywords: Arrhythmia, Early afterdepolarization, Myofibroblasts, Ventricular myocytes, Dynamic clamp
1. Introduction
The risk of ventricular tachycardia and fibrillation (VT/VF) is increased by tissue fibrosis. Traditionally, increased VT/VF risk in fibrotic hearts has been attributed to structural disruption by collagen bundles, resulting in reduced connectivity between myocytes. In normal adult hearts, quiescent fibroblasts substantially outnumber myocytes, and in response to haemodynamic stress or injury, differentiate into myofibroblasts that proliferate, secrete collagen, and synthesize new proteins such as smooth muscle α-actin, stretch-sensitive ion channels, and connexins.1 When co-cultured with cardiac myocytes, fibroblasts differentiate into myofibroblasts and form gap junctions between each other and with myocytes.2,3 Since myofibroblasts have a less negative resting membrane potential, they can depolarize myocytes sufficiently to induce spontaneous pacemaking4 when the myofibroblast population exceeds 15% in co-cultures.5 These studies raise the possibility that myofibroblast–myocyte coupling may alter myocyte electrophysiological properties directly6–8 and play an active role in arrhythmogenesis.
In intact native ventricular muscle, however, the evidence that fibroblasts or myofibroblasts form gap junctions with myocytes is equivocal, with some investigators believing it to be an artefact of cell culture. This controversy notwithstanding, the question of how myofibroblast–myocyte coupling may potentially directly alter myocyte electrophysiology to promote arrhythmias is interesting, and could be a potentially important issue for cell therapy, in which endogenous myofibroblasts in the fibrotic heart interact with exogenously delivered cultured cardiac cells.
For these reasons, we undertook this study to investigate the arrhythmogenic potential of myofibroblast–myocyte coupling, specifically focusing on early afterdepolarization (EAD), a common trigger of polymorphic ventricular tachycardia and Torsades de pointes which can lead to VF. Because it is difficult to regulate gap junction formation between myocytes and myofibroblasts in an experimentally controllable manner, we adopted a hybrid biological–computational approach using the dynamic clamp technique,9 in which a real ventricular myocyte was coupled to a virtual fibroblast or myofibroblast with programmable features. In the dynamic clamp technique, the voltage of the real myocyte interacts in real time and bidirectionally with the computed membrane voltage of the virtual myofibroblast, such that a gap junction current between the myocyte and virtual myofibroblast is continuously calculated in real time and injected into the myocyte. Using prior experimental data to estimate the physiologically relevant ranges of myocyte–myofibroblast gap junction coupling resistances and myofibroblast membrane properties, we found that when myocyte repolarization reserve was already reduced by oxidative or ionic stress, myofibroblast–myocyte coupling further enhanced EAD formation. The mechanism depended on a transient outward component of the gap junction current flowing out of the myocyte.
2. Methods
For full details of Methods, please refer to our Supplementary material online.
2.1. Ventricular myocyte isolation
This study was approved by the UCLA Chancellor's Animal Research Committee (ARC 2003-063-23C) and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication No. 85-23, revised 1996) and with UCLA Policy 990 on the Use of Laboratory Animal Subjects in Research (revised 2010). Young adult (3–4-month-old) New Zealand white male rabbits (1.7–2.0 kg) were euthanized by an intravenous injection of heparin sulfate (1000 U) and sodium pentobarbital (100 mg/kg); adequacy of anaesthesia was confirmed by the lack of pedal withdrawal reflex, corneal reflex, and motor response to pain stimuli by scalpel tip. Using the standard Langendorff retrograde perfusion method, hearts were perfused at 22 mL/min, 37°C, with Ca2+-free Tyrode's solution for 4 min followed by enzyme solution for 28 min, and then with 0.2 mmol/L Ca2+ Tyrode's solution. Hearts were subsequently removed from the perfusion apparatus and gently agitated to dissociate the myocytes. The Ca2+ concentration was gradually increased to 1.8 mmol/L over 30 min. Freshly isolated myocytes were used within 8 h.
2.2. Solutions
Typical standard Tyrode's solution was used for cell isolation and extracellular perfusion in patch clamp studies unless otherwise indicated (see Supplementary material online). EADs were induced by adding 0.1 or 1 mmol/L hydrogen peroxide (H2O2) or reducing K+ to 2.7 mmol/L (HypoK) in the bath solution. The pipette solution for whole cell recordings contained 0.0 or 0.1 mmol/L EGTA.
2.3. Patch clamp
Action potentials (APs) were elicited by 2-ms current pulses at twice threshold using borosilicate glass electrodes (tip resistance 2–3 MΩ) and standard whole-cell patch clamp methods in the current clamp mode. Corrections were made for liquid junction potentials. Data were acquired (Axopatch 200B patch-clamp amplifier; Digidata 1200 acquisition board; and Clampex 8.0, Axon Instruments, Inc.) and filtered at 2 kHz.
2.4. Dynamic clamp
A patch-clamped rabbit ventricular myocyte was bi-directionally coupled in real-time to a virtual myofibroblast using the dynamic clamp technique.9 The dynamic clamp processed the myocyte membrane voltage signal Vm from the patch clamp amplifier and injected back into the myocyte a predicted virtual gap junction current (Ij) proportional to the voltage difference and gap junction coupling conductance (Gj) between the real myocyte (Vm) and the virtual myofibroblast (Vf) (Supplementary material online, Figure S1), using a real-time Linux-based software (Real-Time eXperiment Interface, www.rtxi.org).
2.5. Mathematical models
Virtual fibroblast and myofibroblast models in the dynamic clamp experiments were modified from MacCannell et al.'s10 ‘active’ fibroblast model, adding a non-selective cation current Insc to adjust Ef to between −50 and −25 mV. Virtual fibroblasts and myofibroblasts had identical ionic currents and were distinguished solely by cell capacitance (6.3 pF for fibroblasts; 50.0 pF for myofibroblasts). We generically use the term ‘myofibroblast’ to refer to both fibroblasts and myofibroblasts.
We performed simulations using the Luo-Rudy 1 ventricular myocyte AP model11 coupled to a ‘passive’ myofibroblast model devoid of ionic currents.12 EADs in the AP model were generated as described previously (see Supplementary material online); under these conditions, EAD threshold was defined as the largest value of 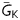 (maximal K conductance) at which EADs still occurred.
(maximal K conductance) at which EADs still occurred.
2.7. Data analysis and statistical methods
Electrophysiological data were analysed using Clampfit 9.2 (Axon instruments, Inc) and Origin 7.5 (Microcal Software, Inc). The statistical significance was assessed by Fisher's exact test. To measure the effect size of Ef, Gj, and Cf on EAD incidence, we used the hazard ratio (HR), i.e. the fraction of cells with EADs in the ‘high’ condition for that factor (Ef −25 mV; Gj 3.0 nS; Cf 50.0 pF), divided by the same fraction in the ‘low’ condition (Ef −50 mV; Gj 0.3 nS; Cf 6.3 pF). Confidence intervals (CI) were determined by Monte Carlo methods.13,14 Briefly, for each study, a sample with the same group sizes as those in the original study was drawn with replacement from the original sample, and its HR was calculated. This process was repeated 1000 times, generating a set of 1000 estimates of the variability of that HR due to sampling variations. A 95% CI around the observed HR was then calculated from these 1000 estimates by a bias-corrected accelerated CI method14 (MATLAB, Natick, MA, USA).
3. Results
3.1. Induction of bradycardia-dependent EADs
Under control conditions, isolated young adult rabbit ventricular myocytes, patch-clamped in the whole cell mode and superfused with normal Tyrode's solution (34–36°C), exhibited normal APs at all pacing cycle lengths (PCL) tested from 1 to 6 s (Figure 1A). Upon addition of H2O2 (0.1–1.0 mmol/L) to generate oxidative stress15 or exposure to hypokalaemia (HypoK, 2.7 mmol/L) to generate ionic stress,16 EADs developed consistently within 10 min during slow pacing at PCL 6 s, occurred irregularly at PCL 4 or 3 s, and were always suppressed completely at PCL ≤ 2 s (Figure 1B and C).
Figure 1.

Induction of bradycardia-dependence of EAD by hypokalaemia or H2O2. A patch-clamped rabbit myocyte was superfused with Tyrode's solution containing normal 5.4 mmol/L K+ (A), reduced 2.7 mmol/L K+ (B), or 1.0 mmol/L H2O2 (in normal 5.4 mmol/L K+) (C). Both hypokalaemia and oxidative stress with H2O2 caused EADs (*) at long pacing cycle lengths (PCL) but not at PCL 1 s.
3.2. Effects of myofibroblast–myocyte coupling on EADs: loss of bradycardia dependence
We next investigated the effects of coupling a virtual myofibroblast to an isolated patch-clamped ventricular myocyte by activating the dynamic clamp. We simulated a range of conditions using the MacCannell ‘active’ fibroblast model, corresponding to typical experimentally measured fibroblast and myofibroblast properties by varying capacitance Cf (6.3 pF for fibroblasts; 50 pF for myofibroblasts),17 uncoupled resting membrane potential Ef (−25 or −50 mV, based on a reported physiologic range of −60 to −5 mV18,19), gap junction coupling conductance Gj (0.3 or 3.0 nS, based on a reported physiologic range of 0.3–8 nS),3 and myocyte:myofibroblast coupling ratio (1:1; 1:2; 1:3; 1:4). In this active myofibroblast model, with Ef at −50 mV, myofibroblast membrane conductance Gf was 26 pS/pF, whereas with Ef at −25 mV, Gf was 123 pS/pF. In all, eight combinations of myofibroblast–myocyte coupling parameters were studied (Table 1). Uncoupled myocytes typically had a capacitance of 100–130 pF and resting membrane potential of −80 mV (control or H2O2) or −95 mV (HypoK).
Table 1.
Electrophysiological effects of myofibroblast coupling on myocyte resting membrane potential (RMP) and action potential duration (APD90).
| Combination | Cf (pF) | Ef (mV) | Gj (nS) | ΔRMP (mV, n) | ΔAPD90 (ms, n) |
|---|---|---|---|---|---|
| 1 (fibroblast) | 6.3 | −50 | 0.3 | +0.2 ± 0.1 (40) | −5 ± 5 (25) |
| 2 (fibroblast) | 6.3 | −50 | 3.0 | +0.3 ± 0.2 (35) | −21 ± 15 (29) |
| 3 (fibroblast) | 6.3 | −25 | 0.3 | +0.9 ± 0.4 (25) | −10 ± 9 (20) |
| 4 (fibroblast) | 6.3 | −25 | 3.0 | +2.8 ± 1.6 (50) | −6 ± 6 (24) |
| 5 (myofibroblast) | 50.0 | −50 | 0.3 | +0.5 ± 0.3 (33) | −7 ± 6 (27) |
| 6 (myofibroblast) | 50.0 | −50 | 3.0 | +1.3 ± 0.7 (23) | −45 ± 15 (31) |
| 7 (myofibroblast) | 50.0 | −25 | 0.3 | +1.0 ± 0.2 (23) | −2 ± 2 (13) |
| 8 (myofibroblast) | 50.0 | −25 | 3.0 | +2.4 ± 1.6 (41) | −12 ± 13 (35) |
Coupling of rabbit ventricular myocytes to a virtual fibroblast or myofibroblast by dynamic clamp under control conditions using eight different coupling combinations slightly depolarized the myocyte resting membrane (less negative RMP) and shortened APD90 during PCL 1 s. Results are reported as mean ± SD.
During superfusion with normal Tyrode's solution, none of the eight combinations of myocyte–myofibroblast coupling parameters induced EADs at any PCL studied. Depending on the specific combination, however, coupling resulted in a mild depolarization of myocyte resting membrane ranging from 0.2 to 3 mV, and shortened APD from 2 to 45 ms (Table 1), in general agreement with previous simulation results by MacCannell et al.10 After exposure to oxidative stress or hypokalaemia, however, myofibroblast–myocyte coupling exacerbated EAD formation at all PCL testing, even at PCL 1 s, which was fast enough to suppress EADs completely in the absence of coupling in all myocytes tested (Figure 2B and C). The incidence of EAD reappearance at PCL 1 s was greater when Ef was −25 mV, but also occasionally occurred when Ef was −50 mV (Figure 2B and C). Superimposition of AP traces in Figure 2 shows that myofibroblast coupling detectably lowered the voltage level of the early AP plateau. In addition to EADs, delayed afterdepolarizations (DADs) and triggered activities were also observed (arrows in Figure 2B).
Figure 2.

Promotion of EADs by myofibroblast–myocyte coupling. (A) Coupling a patch-clamped myocyte superfused with normal Tyrode's solution to a virtual fibroblast did not induce EADs at PCL 1 s. Superimposed traces at a faster time scale to the right illustrate that coupling lowers the AP plateau and shortens ADP. (B and C) In myocytes exposed to hypokalaemia or H2O2 to induce bradycardia-dependent EADs at PCL 6 s (not shown), pacing at PCL 1 s suppressed EADs completely (top traces). However, coupling the myocyte to a virtual fibroblast (Cf 6.3 pF, Gj 3.0 nS) caused EADs to reappear, which were more prominent when Ef was −25 mV (bottom traces) than −50 mV (middle traces). In addition to EADs, DADs (some triggering APs) were also frequently observed (arrows). Lower panels show superimposed traces of control vs. coupled for the Ef −50 and −25 mV cases, respectively, at a faster time scale. Note that coupling lowers the early AP plateau voltage before EAD onset.
Among the Ef, Cf, and Gj combinations studied (Table 1), Ef had the largest effect at promoting EADs [effect size based on HR 3.3; 95% CI (2.2, 5.54)], when compared with Gj [HR 1.6; 95% CI (1.0, 2.0)] or Cf [HR 1.4; 95% CI (1.0, 2.1)]. The EAD-promoting effects of Ef and Gj were both significant (P< 0.0001 and P= 0.01, respectively, one-tailed Fisher's exact test), while the Cf effect was not (P= 0.05, one-tailed Fisher's exact test). At Ef −50 mV, all combinations of Cf and Gj produced EADs, varying in effect from 10 to 27% of H2O2-treated myocytes paced at PCL 1 s (Figure 3); this was overall statistically significant (P< 0.01) compared with control uncoupled conditions, in which no EADs occurred. At Ef −25 mV, both fibroblasts and myofibroblasts were even more effective, causing EADs to reappear in 42–80% of H2O2-treated myocytes paced at PCL 1 s (P< 0.0001 compared with Ef −50 mV, one-tailed Fisher's exact test). At Ef −25 mV, higher Gj and higher Cf induced more EADs (P= 0.005 and 0.05, respectively, one-tailed Fisher's exact test). If a myocyte was coupled to more than one myofibroblast (coupling ratio 1:2, 1:3, or 1:4), the incidence of EAD induction increased, particularly when Ef was −25 mV (Figure 4).
Figure 3.

Data summary of EAD reappearance at PCL 1 s induced by myocyte–myofibroblast 1:1 coupling. Patch-clamped myocytes were exposed to H2O2 to induce bradycardia-dependent EADs at PCL 6 s, which were then suppressed at PCL 1 s. Bars indicate the fraction of myocytes in which EADs reappeared when the myocyte was coupled to a virtual myofibroblast, for each of the eight different coupling parameter sets listed in Table 1. The total number of ventricular myocytes tested for each coupling parameter set is indicated inside each bar, with the corresponding probability of EAD reappearance shown above.
Figure 4.

Higher probability of EAD induction by coupling multiple myofibroblasts to a single myocyte. A patch-clamped myocyte was exposed to H2O2 to induce bradycardia-dependent EADs at PCL 6 s, which were then suppressed at PCL 1 s. Coupling the myocyte (MC) to one myofibroblast (MF, with Cf 50.0 pF, Gj 3.0 nS, Ef −25 mV) depolarized the myocyte and shortened APD (middle trace), but did not cause EADs to reappear. However, when the myocyte was coupled to two myofibroblasts in parallel (effectively increasing Cf to 100 pF and Gj to 6 nS), EADs and repolarization failure ensued (lower trace).
In the MacCannell ‘active’ fibroblast model,10 the parameter adjustments made to increase Ef from −50 mV (‘low’) to −25 mV (‘high’) also increased Gf from 26 (‘low’) to 123 pS/pF (‘high’). To determine whether the increased EAD occurrence at Ef −25 mV was due to the change in Ef or Gf, we simulated myocyte–myofibroblast coupling12 using the Luo-Rudy 1 ventricular AP model11 coupled to a simplified ‘passive circuit’ myofibroblast model in which Ef and Gf could be varied independently instead of being restricted to the low Ef–low Gf and high Ef–high Gf combinations (black/grey bars in Figure 5). We then characterized how various combinations, including low Ef–high Gf and high Ef–low Gf (blue bars in Figure 5) as well as low Ef–low Gf and high Ef–high Gf, affected the EAD threshold, defined as the largest value of 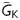 in the AP model at which EADs still occurred (see Supplementary material online). With the AP model uncoupled from the myofibroblast, the threshold value of
in the AP model at which EADs still occurred (see Supplementary material online). With the AP model uncoupled from the myofibroblast, the threshold value of 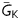 was 0.323 mS/μF. Upon coupling, a shift to a higher
was 0.323 mS/μF. Upon coupling, a shift to a higher 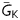 indicated increased EAD sensitivity (i.e. the myocyte AP required more K conductance to prevent EADs), and vice versa. This analysis showed that high Ef (−25 mV), rather than high Gf (123 pF), was the major factor promoting EAD reappearance during pacing at 1 s.
indicated increased EAD sensitivity (i.e. the myocyte AP required more K conductance to prevent EADs), and vice versa. This analysis showed that high Ef (−25 mV), rather than high Gf (123 pF), was the major factor promoting EAD reappearance during pacing at 1 s.
Figure 5.

Effects of Ef vs. Gf on EAD threshold. Threshold for EAD formation (see text) of the myofibroblast-coupled state was compared with that of the control myofibroblast-uncoupled state. (A) Coupling a passive myofibroblast model to a Luo-Rudy 1 myocyte AP model to simulate the same eight coupling combinations tested experimentally in Table 1 lowered EAD threshold much more prominently for high Ef–high Gf (Ef −25 mV/Gf 123 pS) cases than for low Ef–low Gf (Ef −50 mV/Gf 26 pS) cases. (B) When Ef was kept low at −50 mV for all eight parameter combinations, Gf had little effect or increased EAD threshold. (C) When Ef was held high at −25 mV for all eight parameter combinations, Gf decreased EAD threshold in all cases, indicating that high Ef is more important than high Gf at lowering EAD threshold. Numbers indicate the per cent change in EAD threshold.
3.3. Ionic mechanism of EAD potentiation by myocyte–myofibroblast coupling
When an H2O2-treated myocyte paced at PCL 1 s was coupled to a virtual myofibroblast (as in Figure 2), a gap junction current (Ij) was generated by the dynamic clamp and injected into the myocyte (Figure 6A). Ij had two components: (i) a rapid early transient outward Ito-like component beginning with the AP upstroke, related to charging the capacitance of the myofibroblast; (ii) a smaller sustained component that was initially outward and became inward at a later point during the AP plateau, related to the leak current through the myofibroblast membrane. To assess which component was more critical for EAD induction, we selectively uncoupled the virtual myofibroblast from the myocyte during the first 100 ms of the AP to eliminate the transient outward component of Ij, while leaving coupling intact during the remaining 900 ms before the next stimulus (for PCL 1 s): EADs disappeared. Conversely, if coupling to the virtual myofibroblast was left intact during the initial 100 ms to preserve the transient outward component of Ij and uncoupled during the remaining 900 ms to eliminate the late sustained component of Ij, EADs persisted. This finding indicates that EAD formation depends principally on the early Ito-like component of Ij. Similar results were obtained when the Luo-Rudy 1 ventricular AP model was coupled to a myofibroblast model (Figure 6B and C). The finding that an early outward current promotes EAD formation may seem paradoxical, but is consistent with theoretical analyses of EAD dynamics20 (see Discussion).
Figure 6.

The early Ito-like component of Ij promotes EADs. (A) A patch-clamped myocyte was exposed to 1.0 mmol/L H2O2 to induce bradycardia-dependent EADs at PCL 6 s, which were then suppressed at PCL 1 s (not shown). Coupling the myocyte to a virtual fibroblast (Cf6.3pF, Ef −25 mV, Gj 0.3 nS) (left upper trace) caused EADs to reappear. The virtual gap junction current Ij (left lower trace) consisted of an early transient outward Ito-like component followed by a sustained component. When coupling was allowed only during the first 100 ms of the AP, the EAD persisted (middle upper trace). However, when coupling was allowed only during the last 900 ms, the EAD disappeared (right upper trace). (B) Corresponding simulation: same protocol as in (A), but with the real myocyte replaced by the Luo-Rudy 1 ventricular AP model modified to produce bradycardia-induced EADs. EADs reappear at PCL 1 s when coupling to the fibroblast is present throughout the cardiac cycle (left) or only the first 100 ms of the AP (middle), but not when uncoupled during the first 100 ms of the AP (right). Bottom row shows expanded traces of the corresponding gap junction currents Ij.
4. Discussion
Using a hybrid biological–computational approach, we made the following observations: (i) coupling a real rabbit ventricular myocyte by dynamic clamp to a virtual myofibroblast with physiologically realistic membrane properties and gap junction coupling conductance had relatively minor electrophysiological effects under control conditions (Table 1). (ii) However, when the same myocyte was exposed to oxidative stress (H2O2) or hypokalaemia to induce bradycardia-dependent EADs, coupling the myocyte to a myofibroblast potentiated EAD occurrence, such that EADs reappeared at short PCLs which completely suppressed EADs in the absence of coupling. (iii) The resting membrane potential Ef was more important than myofibroblast membrane capacitance Cf and conductance Gf or gap junction coupling conductance Gj in potentiating EAD formation, with more depolarized Ef having stronger effects. (iv) Among the two components of the gap junction current Ij flowing between the myocyte and the myofibroblast, the early Ito-like component played the most critical role in EAD formation, with the late sustained component having minor effects.
It is noteworthy that the EAD-promoting effects of coupling a single myofibroblast to a myocyte had only small effects on myocyte resting membrane potential or APD (Table 1). In contrast, a much greater degree of resting membrane depolarization (averaging +5 to +23 mV) induced by myofibroblast coupling was required to induce automaticity in heterocellular cultures.5 However, coupling of multiple myofibroblasts to a single myocyte increased the incidence of EAD induction (Figure 4), suggesting that the latter situation is even more arrhythmogenic.
Our findings were similar for EADs induced by either oxidative stress or hypokalaemia, implying that the specific ionic mechanism underlying EAD formation is not critical. H2O2 promotes EAD formation primarily via oxidative activation of Ca–calmodulin kinase II (CaMKII), which potentiates the L-type Ca current (ICa,L)15 and late Na current,21 whereas hypokalaemia primarily reduces K conductance to decrease repolarization reserve.22
4.1. Mechanism of EAD promotion by myocyte–myofibroblast coupling
When a myofibroblast is coupled to a myocyte, the myofibroblast acts like a leaky capacitor. Charging of the myofibroblast capacitance during the AP upstroke causes a transient outward Ito-like component of the gap junction current Ij, followed by a sustained current proportional to the myofibroblast membrane conductance Gf and driving force (Em–Ef). This predominance of outward current until late in the AP when (Em–Ef) reverses tends to lower the AP plateau and generally shorten APD, as found previously in simulations by MacCannell et al.10 We confirmed this purely theoretical prediction when a real rabbit ventricular myocyte was coupled to a virtual myofibroblast (Figure 2). In a study simulating a one-dimensional cable of mouse ventricular AP model cells coupled with fibroblasts, Jacquemet and Henriquez23 also reported a decrease in AP amplitude, although APD was prolonged rather than shortened in the triangular mouse AP. Conduction velocity was not significantly slowed until 10 or more fibroblasts with Cf of 4.5 pF (a rough equivalent of one myofibroblast of 50 pF in our study) were coupled to each myocyte (153.4 pF). Extrapolating from these results, we do not expect myofibroblast coupling to have much effect on conduction velocity per se, and attribute conduction slowing in fibrotic hearts mainly to disruption of myocyte–myocyte coupling by collagen deposition.
It may seem counterintuitive that outward current flow from the myocyte is the major factor causing EADs to emerge during coupling to a myofibroblast (Figure 6). However, our results are consistent with our previous report that the native Ito in rabbit myocytes promoted EAD formation during exposure to H2O2 by lowering the AP plateau voltage into the range which allowed reactivation of ICa,L (<0 mV).24 The present findings also agree with previous reports that AP triangulation was more potent at inducing EADs than AP prolongation per se,25 since AP triangulation lowered the voltage into the range allowing ICa,L reactivation. Theoretical simulations predict that strong early repolarization reserve (i.e. an Ito-like current) coupled with weak late repolarization reserve is more important for EAD induction than an overall reduction in repolarization reserve.26,27
Despite the importance of the early Ito-like current component of Ij in EAD formation, its amplitude and kinetics were not significantly affected by Ef, the most potent myofibroblast parameter influencing EAD formation. This is because the Ito-like component reflects the charge movement required to depolarize the fibroblast membrane capacitance Cf from the coupled resting potential to the AP plateau. Since coupling to a single myofibroblast, with Ef at −25 or −50 mV, displaces the resting potential of the myocyte by only a few mV (Table 1), the voltage jump to the peak of the AP upstroke is similar in both cases, consequently Ito is similar. During the later AP plateau, however, the sustained component of Ij remains outward until the coupled voltage becomes more negative than Ef. It is presumably the earlier reversal of this outward current component when Ef is −25 mV that enhances EAD formation, indicating that the late component of Ij strongly modulates the effectiveness of the early component at inducing an EAD.
4.2. Limitations
The heart rate at which myofibroblast coupling caused EADs to reappear was unphysiologically slow (60bpm) for rabbit, but is typical of the normal to bradycardic range at which EADs typically occur in humans. The most compelling evidence for myocyte–myofibroblast gap junction coupling is based on data from heterocellular co-cultures.2,7 Whether myocyte–myofibroblast coupling in native normal or diseased cardiac muscle occurs and is significant enough to exert the pro-arrhythmogenic effects described in this study remains to be demonstrated.28 It is beyond the scope of this study to resolve this controversy, but hopefully our findings will stimulate further interest in this issue, given the potential importance to arrhythmogenesis in fibrotic hearts.
We assumed that myofibroblast–myocyte coupling is mediated by heterocellular gap junction formation, but recently, coupling via large conductance membrane nanotubes has also been proposed.29 The parameter choices for our virtual myofibroblast and fibroblast models were based on a mixture of available data obtained from adult and neonatal3,6 ventricular fibroblasts, both in vivo and in situ,19 from freshly isolated17 or cultured conditions,17,18 and from a variety of species, and may not accurately reflect the properties of native human ventricular myofibroblasts. Additionally, the MacCannell model simulated basal conditions, whereas the interventions used here to induce EADs in the myocytes (H2O2 or hypokalaemia) may affect myofibroblast electrical properties in real cardiac tissue. In the beating heart, biomechanical inputs may also alter both myocyte and myofibroblast electrophysiology/coupling, but were not considered here. Partly mitigating these concerns, however, we deliberately examined a wide range of myofibroblast–fibroblast properties, varying Cf and Gj over an order of magnitude, and Ef over a 25 mV range. Finally, we focused our study mainly on the effects of coupling a single fibroblast/myofibroblast to a single myocyte, whereas the stoichiometry in intact heart muscle is uncertain.
4.3. Clinical implications
We emphasize that myocyte–myofibroblast coupling facilitated EADs only in myocytes that were already prone to EADs, but did not induce EADs de novo in myocytes with normal repolarization reserve. Thus, our findings are mainly relevant to conditions in which repolarization reserve is already compromised, such as by drugs, genetic channelopathies, or heart failure. In this context, it is intriguing to speculate that fibrosis may be a second factor that allows oxidative stress or hypokalaemia to cause EAD-mediated VT/VF in diseased hearts, both by promoting EADs at relatively fast heart rates as a result of myocyte–myofibroblast coupling as well as by disrupting myocardial architecture and creating favourable tissue source-sink relationships allowing EADs to emerge.24,30
Our observation that Ef strongly influences EAD formation could also be relevant to the episodic nature of ventricular arrhythmias, since myofibroblasts respond to both mechanical and humoural factors which alter their resting membrane potential.8,31 For example, in response to acute hypoxia, Ef of in situ adult rat atrial fibroblasts depolarized from −23 ± 5 to −5 ± 2 mV, and then transiently hyperpolarized to −60 ± 8 mV during reoxygenation.32 It is intriguing to speculate that ischaemia, stretch, autonomic tone, and neurohumoral factors may trigger arrhythmias in intact heart not only through direct electrophysiological effects on cardiac myocytes, but also by dynamically modulating myofibroblast resting membrane potential and membrane properties.
Finally, therapeutic interventions to suppress fibrosis have been proposed as a promising strategy to reduce arrhythmia risk and improve contractile function in diseased hearts. If myofibroblast–myocyte coupling is significant in diseased cardiac tissue, then our findings suggest that strategies to reduce fibrosis may have additional antiarrhythmic benefits, by suppressing EAD formation and EAD-mediated arrhythmias. In addition to promoting EADs, depolarization of myocyte resting membrane by myofibroblast coupling can also induce automaticity,28 and will likewise bring DADs closer to the threshold for inducing triggered activity. Thus, selective therapy directed at inhibiting myofibroblast–myocyte gap junction coupling may have general antiarrhythmic efficacy by reducing ectopic triggers via multiple mechanisms. Finally, if atrial myocytes behave similarly, the same considerations may apply to atrial fibrillation.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Heart Rhythm Society (to T.P.N.); the American Heart Association (to T.P.N.); the National Institutes of Health (HL078931 and HL103662 to J.N.W.); and the Laubisch and Kawata endowments (to J.N.W.).
Supplementary Material
Acknowledgements
We thank Dr David J. Christini and Jonathan Bettencourt for their invaluable technical assistance with the dynamic clamp.
Conflict of interest: none declared.
References
- 1.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc Res. 2004;62:415–425. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 2.Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007;583:225–236. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rook MB, van Ginneken AC, de Jonge B, el Aoumari A, Gros D, Jongsma HJ. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am J Physiol. 1992;263:C959–C977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 4.Jacquemet V. Pacemaker activity resulting from the coupling with nonexcitable cells. Phys Rev E Stat Nonlin Soft Matter Phys. 2006;74:011908. doi: 10.1103/PhysRevE.74.011908. [DOI] [PubMed] [Google Scholar]
- 5.Miragoli M, Salvarani N, Rohr S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ Res. 2007;101:755–758. doi: 10.1161/CIRCRESAHA.107.160549. [DOI] [PubMed] [Google Scholar]
- 6.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 7.Zlochiver S, Munoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorval AD, Christini DJ, White JA. Real-Time linux dynamic clamp: a fast and flexible way to construct virtual ion channels in living cells. Ann Biomed Eng. 2001;29:897–907. doi: 10.1114/1.1408929. [DOI] [PubMed] [Google Scholar]
- 10.MacCannell KA, Bazzazi H, Chilton L, Shibukawa Y, Clark RB, Giles WR. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys J. 2007;92:4121–4132. doi: 10.1529/biophysj.106.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo CH, Rudy Y. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ Res. 1991;68:1501–1526. doi: 10.1161/01.res.68.6.1501. [DOI] [PubMed] [Google Scholar]
- 12.Xie Y, Garfinkel A, Weiss JN, Qu Z. Cardiac alternans induced by fibroblast-myocyte coupling: mechanistic insights from computational models. Am J Physiol Heart Circ Physiol. 2009;297:H775–H784. doi: 10.1152/ajpheart.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Efron B, Tibshirani R. Statistical data analysis in the computer age. Science. 1991;253:390–395. doi: 10.1126/science.253.5018.390. [DOI] [PubMed] [Google Scholar]
- 14.Manly BFJ. Randomization, Bootstrap, and Monte Carlo Methods in Biology. Boca Raton/FL: Chapman & Hall/CRC; 2007. p. 455. [Google Scholar]
- 15.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato D, Xie LH, Nguyen TP, Weiss JN, Qu Z. Irregularly appearing early afterdepolarizations in cardiac myocytes: random fluctuations or dynamical chaos? Biophys J. 2010;99:765–773. doi: 10.1016/j.bpj.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chilton L, Ohya S, Freed D, George E, Drobic V, Shibukawa Y, et al. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am J Physiol Heart Circ Physiol. 2005;288:H2931–H2939. doi: 10.1152/ajpheart.01220.2004. [DOI] [PubMed] [Google Scholar]
- 18.Kamkin A, Kiseleva I, Lozinsky I, Scholz H. Electrical interaction of mechanosensitive fibroblasts and myocytes in the heart. Basic Res Cardiol. 2005;100:337–345. doi: 10.1007/s00395-005-0529-4. [DOI] [PubMed] [Google Scholar]
- 19.Kiseleva I, Kamkin A, Pylaev A, Kondratjev D, Leiterer KP, Theres H, et al. Electrophysiological properties of mechanosensitive atrial fibroblasts from chronic infarcted rat heart. J Mol Cell Cardiol. 1998;30:1083–1093. doi: 10.1006/jmcc.1998.0673. [DOI] [PubMed] [Google Scholar]
- 20.Tran DX, Sato D, Yochelis A, Weiss JN, Garfinkel A, Qu Z. Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys Rev Lett. 2009;102:258103. doi: 10.1103/PhysRevLett.102.258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated Ca/Calmodulin Kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osadchii OE. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol. 2010;24:547–559. doi: 10.1111/j.1472-8206.2010.00835.x. [DOI] [PubMed] [Google Scholar]
- 23.Jacquemet V, Henriquez CS. Modelling cardiac fibroblasts: interactions with myocytes and their impact on impulse propagation. Europace. 2007;9(Suppl. 6)):vi29–vi37. doi: 10.1093/europace/eum207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie Y, Zhao Z, Weiss JN, Qu Z, Xie LH. The transient outward current Ito promotes early afterdepolarizations. Biophysical Society Meeting Abstracts. Biophys J. 2010;98(Suppl. 1):531a. [Google Scholar]
- 25.Hondeghem LM, Hoffmann P. Blinded test in isolated female rabbit heart reliably identifies action potential duration prolongation and proarrhythmic drugs: importance of triangulation, reverse use dependence, and instability. J Cardiovasc Pharmacol. 2003;41:14–24. doi: 10.1097/00005344-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 26.January CT, Chau V, Makielski JC. Triggered activity in the heart: cellular mechanisms of early after-depolarizations. Eur Heart J. 1991;12(Suppl. F):4–9. doi: 10.1093/eurheartj/12.suppl_f.4. [DOI] [PubMed] [Google Scholar]
- 27.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–1899. doi: 10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6:848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 29.He K, Shi X, Zhang X, Dang S, Ma X, Liu F, et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res. 2011;92:39–47. doi: 10.1093/cvr/cvr189. [DOI] [PubMed] [Google Scholar]
- 30.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamkin A, Kiseleva I, Isenberg G, Wagner KD, Gunther J, Theres H, et al. Cardiac fibroblasts and the mechano-electric feedback mechanism in healthy and diseased hearts. Prog Biophys Mol Biol. 2003;82:111–120. doi: 10.1016/s0079-6107(03)00009-9. [DOI] [PubMed] [Google Scholar]
- 32.Kamkin A, Kiseleva I, Wagner KD, Lozinsky I, Gunther J, Scholz H. Mechanically induced potentials in atrial fibroblasts from rat hearts are sensitive to hypoxia/reoxygenation. Pflugers Arch. 2003;446:169–174. doi: 10.1007/s00424-003-1032-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.