

Abstract
Aims
The first seconds of ventricular fibrillation (VF) are well organized and can consist of just one to two rotating waves (rotors). New rotors are spawned when local propagation block causes wave fragmentation. We hypothesized that this process, which leads to fully developed VF, begins at a consistent anatomic site.
Methods and results
We initiated VF with a stimulus timed to the local T-wave in 10 isolated pig hearts. Hearts were stained with a voltage-sensitive dye and four video cameras recorded electrical propagation panoramically across the epicardium. In each VF episode, we identified the position of the first wavebreak event that produced new rotor(s) that persisted for at least one cycle. The first such wavebreak occurred along the anterior right ventricular insertion (ARVI) in 26 of 32 VF episodes. In these episodes, wavebreak sites were 6 ± 4 mm from the midline of the ARVI. In the remaining 6 episodes, wavebreak sites were 24 ± 5 mm from the midline on either the LV or RV. During rapid pacing, conduction speed was locally depressed at the ARVI when waves crossed parallel to the midline. Action potential duration (APD) was slightly longer (2.2 ± 2.1 ms) at the ARVI compared with other sites (P< 0.01). Temporal APD alternans were small and not unique to the break site, suggesting that dynamic APD properties were not the cause of wavebreak.
Conclusion
The ARVI is the dominant site for wavebreak at the onset of VF in normal myocardium. This may be due to the anatomic complexity of the region.
Keywords: Arrhythmia, Re-entry, Rotor, Conduction block
1. Introduction
Sudden cardiac death is responsible for ∼12% of global mortality.1 Most of these deaths are caused by acute ventricular tachyarrhythmias, usually ventricular tachycardia, progressing to ventricular fibrillation (VF).2
VF starts as a relatively well-organized arrhythmia consisting of as few as one or two functionally re-entrant rotating waves (rotors). This stage is classically defined as Wiggers stage 1 VF. In a matter of seconds, it degenerates into a more complex, less coordinated activation pattern that is classically defined as Wiggers stage 2 VF.3 This process is thought to involve wavebreak, wherein wavefronts are split into two or more pieces by local propagation block. The dangling ends of the broken wavefronts may become the centres of new rotors.4 This progression towards more complex activation as VF develops over the first seconds has also been observed in more recent cardiac mapping studies.5,6
Moe et al.7 proposed that wavebreak was driven by patchy regions of refractoriness in the tissue. For example, a segment of a wavefront that passes through a region of prolonged refractoriness will be blocked. As the region recovers, the remaining wavefront segments can re-enter the tissue and create two counter-rotating rotors. These heterogeneous regions are static properties of the myocardium, i.e. they do not change beat-to-beat, and can result from local anatomical features8–10 or regional differences in cellular properties.11 More recently, computational models and experimental data have shown that dynamic changes in the cardiac action potential duration (APD) can produce and sustain VF, even in otherwise homogeneous tissue.4 During rapid pacing, beat-to-beat oscillations (alternans) of APD can produce spatially non-uniform refractoriness that leads to wavebreak.12
Anatomic structures have also been shown to affect wavefront dynamics, specifically, the papillary muscles,9 large coronary blood vessels,13 and a discontinuity of fibre orientation at the swine right ventricular epicardium.14 Differently oriented fibre fields converge in a space of several millimetres at the junction of the left and right ventricles,15 and wavebreaks frequently occur in this region during well developed VF in pig hearts in vivo.16 Computer modelling10 and recent experimental data17 have shown that regions of sharp fibre curvature can modulate wavefront conduction velocity.
In the present study, we initiated a simple functionally re-entrant arrhythmia and used panoramic optical mapping of nearly the entire ventricular epicardium to record the transition to developed VF. We hypothesized that the first wavebreak event that produced new rotors during this transition would occur at a consistent anatomical site. We found that this was the case with the event occurring at the anterior insertion of the right ventricle (RV) in 81% of episodes.
2. Methods
The investigation conforms with the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health). All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham (APN 060906873).
2.1. Isolated heart preparation
The isolated heart was prepared as described previously.18,19 Briefly, 10 mixed-breed pigs weighing 22.5 ± 2.1 kg were anaesthetized with intramuscular telazol (4.4 mg/kg), xylazine (2.2 mg/kg), and atropine (0.04 mg/kg) and maintained by isoflurane (1.5–2.5%) in 100% oxygen. The adequacy of anaesthesia was assured by the pedal withdrawal response. Euthanasia was by exsanguination accompanying heart excision. The hearts were cannulated through the aortic root, suspended, and perfused with warm (37°C) oxygenated modified Tyrode's solution recirculating at 200 mL/min. Hearts were stained with the fluorescent dye di-4-ANEPPS (Invitrogen) by injecting a 5–10 mL bolus of 15 μmol/L solution into the aortic cannula. Fluorescence produced by this indicator is proportional to membrane potential. Hearts were periodically re-stained throughout the study if fluorescence faded. Heart contraction was prevented with butanedione monoxime (20 mmol/L, Sigma).
2.2. Panoramic optical mapping
The panoramic optical mapping system was configured as described previously.18,20 Briefly, excitation light was provided by 32 blue (470 nm) LEDs surrounding the heart. Fluorescence was recorded at 750 frames/s with four synchronized CCD video cameras equipped with 2.2 mm f/1.0 video lenses and 590 nm long-pass filters. The cameras were calibrated to provide a mapping between points in 3D space and each camera's image plane. The spatial resolution of each camera was set to 64 × 128 pixels. A fifth calibrated video camera (the geometry camera) orbited the heart and acquired images every 5°. The heart was backlit with electroluminescent film to form strong silhouettes, which were segmented and used to reconstruct the 3D epicardial geometry. The reconstructed 3D epicardial models were triangular meshes with ∼1.5 mm spacing, centroid-to-centroid (Figure 1A and B).
Figure 1.

Model of epicardial geometry. (A) Geometry camera image. (B) Corresponding view of the 3D epicardial mesh. The ARVI midline is white and test sites are black. (C) The same mesh flattened to 2D with a Hammer map projection.
The fluorescence signal assigned to each triangular face (‘mesh site’) in the model was a weighted average of the signals from the camera pixels that imaged that triangle.20 Signals assigned to mesh sites were median filtered (width = 40 ms) and smoothed with a 6.7 ms wide averaging filter. The result of this process was an optical mapping data set covering nearly the entire ventricular epicardium (data were missing from the apex, which was not well imaged; see, for example, Supplemental Video S1 in ref. 18).
2.3. VF initiation
VF episodes were induced by pacing the heart from one ventricular site (S1) and delivering stimuli timed to the local T-wave to a different ventricular site (S2). This procedure is well known to induce rotors.21,22 Details of the induction protocol are given in the ‘VF Initiation’ section of the Supplementary material online, Methods. Epicardial wavefronts were optically mapped for at least 300 ms prior to the S2 stimulus, and for at least 3 s after the S2. The epicardial geometry was scanned with the geometry camera within a few minutes of each VF recording. The heart was then defibrillated using paddle electrodes (LIFEPAK 12 defibrillator, Physio-Control).
2.4. Pacing protocol for APD analysis
After the last VF episode, the hearts were paced for at least 30 s and then mapped for 2.6 s at each of the following successive cycle lengths: 300, 250, 220, 200, 180, 170, 160, 150, and 140 ms. These recordings were used to find APD across the ventricular epicardium at the fastest pacing rate that produced 1:1 capture. We refer to runs at this rate (which differed from heart to heart) as rapid pacing. After these runs, the heart's geometry was scanned again.
2.5. Wavefront, waveback, and phase singularity tracking
To identify depolarization upstrokes and repolarization downstrokes, membrane potential signals were represented as a phase variable.23 Briefly, a phase plane was constructed with a detrended fluorescence signal on the abscissa and the temporal integral of this signal on the ordinate. During each action potential, the phase plane trajectory completes an orbit about the origin. The phase value at each time was computed by converting the phase plane to a polar coordinate system and taking the angular coordinate.
The phase value −π/2 corresponds to the upstroke of the action potential; at any time point, spatial isolines of this phase in the panoramic recordings identify contiguous wavefronts. Epicardial wavefronts were identified and tracked through space and time using our custom software.18,23 Following Banville et al.,24 wavebacks were similarly identified as isolines of a phase value that occurs during repolarization; we chose +π. The APD at a site for a particular beat was calculated as the difference in arrival times of a wavefront and the following waveback. Figure 2 shows examples of signals recorded during VF initiation and rapid pacing along with times of activation and repolarization identified by the phase method.
Figure 2.

Optical membrane potential before (grey) and after (black) filtering. Activation and repolarization times (dots and triangles, respectively) were identified by phase values. (A) VF initiation. (B) 180-ms pacing with ||APD alternans|| = 2.3 ms. (C) 180-ms pacing with ||APD alternans|| = 12.0 ms.
Phase singularities (PS) are points surrounded by all phases of activation. They occur at the intersection of wavefronts and wavebacks and are formed by wavebreak events. As the central pivot point of a rotor, they are the hallmark of functional re-entry.25,26 We identified PSs in each frame and tracked them through time as previously described.23 It is common for local propagation block to occur near the tip of a rotor in such a way that the original PS is annihilated, but the surviving part of the wavefront continues to rotate about a new PS in the same direction. In our previous publication, we termed such a succession of wavefronts rotating about a succession of PSs a compound rotor and described an algorithm for finding them.18 In the remainder of this article, rotors referred to in our data are compound rotors.
2.6. Identifying the first new wavebreak in the transition to fully developed VF
The one or two initial rotors produced by the tachyarrhythmia-initiating S2 stimulus were identified in each tachyarrhythmia episode. We refer to these rotors as initial VF (VF0) sources. Each mapping data set was then examined to identify the time and location of the first wavebreak event that produced at least one new rotor that persisted for at least one complete cycle of re-entry (or, in terms of our wavefront tracking model, long enough for the succession of wavefronts associated with the new compound rotor to activate an epicardial site more than once18,23). We required this persistence before registering the wavebreak event because (i) it indicates that the newly formed rotor(s) are wavefront sources and (ii), it prevents the appearance of transient PSs due to recording noise from being registered as the first new wavebreak. This was particularly important because the anterior right ventricular insertion (ARVI), is overlaid with epicardial fat, which can locally reduce signal quality. We refer to the PSs created during this wavebreak event as first transitional PSs (PST1).
PST1 sites were identified on the epicardial model that was specific to that VF recording. Because geometric models differed slightly from run to run, the PST1 sites from each VF episode were mapped onto the final recorded geometry of each heart (which we term the heart's common model). This was done by first scaling the models along the z-axis so that the apices were co-terminous and then projecting each PST1 site along its triangle's surface normal vector to the common model's surface.
We identified the region of the ARVI in each heart's common model by visually selecting the mesh points along the midline of the interventricular groove and drawing a curve through those points with cubic interpolation. The groove was readily identified by manipulating the model in 3D on a computer monitor and referring to the position of the left anterior descending coronary artery in images from the geometry camera. Mesh triangles within a left ventricular wall thickness (15 mm)27 of this line were considered to be in the ARVI region.
2.7. APD analysis
To compare APD and APD dynamics during rapid pacing at different sites across the epicardium, clusters of epicardial mesh sites were selected for analysis in each heart at each of the following four test sites: The ARVI, posterior RV insertion (PRVI), lateral LV, and lateral RV. Most or all PST1 sites within a heart were clustered in a compact region. One test site in each heart was centred on this region and the remaining three test sites were placed at the same distance from the base. In eight hearts, the test site centred on the PST1 sites was in the ARVI region; in the remaining two hearts, it was on the LV free wall. Test sites were expanded from their centre by successively adding neighbouring mesh triangles until 40 recording sites were included. Mesh triangles at which APD standard deviation (computed over all beats in each recording with 1:1 capture) exceeded 50 ms were deemed noisy and were not included in the test sites. The test sites on one epicardial model are shown in Figure 1B and C.
The magnitude of APD alternans (||APD alternans||) was estimated for all rapid pacing signals in each test site. ||APD alternans|| was defined as the absolute value of the difference between mean even-beat and mean odd-beat APDs during steady-state pacing.
2.8. Wavefront conduction speed measurement
Apparent epicardial conduction speed during pacing was computed over the epicardium using an extension of the method of Bayly et al.28 (see Supplementary material online, Methods for details). Briefly, quadratic functions were fit to activation times across overlapping regions ∼6 mm in diameter. Local speed across each region was then computed from the gradient of this function.
3. Results
Thirty-two tachyarrhythmia episodes were initiated and successfully mapped in 10 pig hearts (2–5 per heart). The VF0 sources resulting from S2 stimulation were either a single rotor, two counter-rotating rotors, or a fast breakthrough pattern consistent with an intramural rotor. We characterized global dynamics by computing the total number of compound rotors present on the epicardium after the S2 as a function of time. Figure 3 shows three examples, each with a different type of VF0 source. Only rotors with lifetimes >100 video frames were counted to prevent the signal from being obscured by transient PSs caused by recording noise.
Figure 3.

Global VF dynamics indicated by the number of compound rotors vs. time. (A) The VF0 source is a rotor pair on the LV (heart 4; same episode as Figure 4 and Supplementary material online, Video 1). (B) The VF0 source is a repetitive breakthrough on the LV (heart 6; same episode as Supplementary material online, Video 2). (C) The VF0 source is a rotor pair on the RV (heart 9; same episode as Supplementary material online, Video 3). Time 0 corresponds to the end of the S2. The times of the PST1-inducing wavebreaks are indicated by arrows.
3.1. VF0 sources
In the 24 episodes in which the S2 site was precisely known (those resulting from epicardial S2 stimulation), the VF0 source site (see Supplementary material online) was located 14 ± 8 mm from the S2 electrode. VF0 cycle length (see Supplementary material online) progressively decreased from 202 ± 55 ms immediately after induction to 142 ± 26 ms in the last cycle before PST1 formation (P< 0.01 by paired t-test). In addition, the epicardial conduction time for waves leaving the source (see Supplementary material online) progressively increased. The epicardial conduction time of the first VF0 wavefront was 78 ± 27 ms. By the last cycle before PST1 formation, this had significantly increased to 168 ± 32 ms (P< 0.01 by paired t-test). VF0 sources persisted for 6.6 ± 3.8 cycles before producing PST1s.
3.2. First wavebreak in the transition to fully developed VF
The predominant pattern we observed during the first wavebreak event was a wavefront propagating away from the VF0 source catching up to the waveback from the previous cycle as it crossed the ARVI. This resulted in wavebreak and the formation of PST1s. An example is shown in Figure 4. This is the same episode shown in Figure 3A and is also animated in the Supplementary material online, Video 1. Two additional examples that correspond to the episodes shown in Figure 3B and C are animated in the Supplementary material online, Videos 2 and 3, respectively.
Figure 4.

The first wavebreak event in the transition to developed VF. (A) Each subpanel displays a snapshot of wavefronts (black) and wavebacks (grey) on the ventricular epicardium using a Hammer map projection. The ARVI midline is white; the RV is to the left of the ARVI and the LV to the right. Panels are not equally spaced in time. Time 0 is the end of the S2 stimulus. At t = 132, the LV contains two counter-rotating rotors; arrows point to the two VF0 phase singularities. The left-most phase singularity is rotating counter-clockwise and the right-most is clockwise. At t = 260, the rotors have completed a full rotation. At t = 323, the wavefront approaching the ARVI impinges upon the waveback of the previous cycle and the wavefront breaks (arrows). This is preceded by the appearance of a short wavefront segment in the block region at t = 311. This segment arises from small fluorescence deflections (D) and is likely an artefact of the phase algorithm, which does not reject all low-amplitude deflections when computing the phase.23 The broken wavefront ends soon rejoin (t = 369), but small fluorescence deflections in the block region (D) result in a small, slow re-entrant wavefront that, like the previous wavefront at this site, may be an artefact (t = 384–409). On the following cycle, the wavebreak becomes more pronounced (t = 429; arrows) resulting in two new well-defined rotors (t = 437). This episode is from animal 4 and is also shown in Figure 3A and Supplementary material online, Video 1. (B–E) De-trended fluorescence (black) and corresponding phase (grey) at sites indicated in the first subpanel of (A). Black circles show phase-indicated depolarization times. Scale bars are 10% of the amplitude of the last S1-induced action potential at the respective site.
PST1 formation at the ARVI was predominant, but not universal (Table 1). In 5 of the 10 hearts, in all episodes, PST1s formed in the ARVI region in a manner similar to Figure 4. In three additional hearts, this pattern was observed in most episodes (2/3 or 3/4). In the remaining two hearts, it occurred in only one episode.
Table 1.
VF initiation sites (S2), and number of episodes with PST1s in the ARVI region
| Heart number | S2 site | Episodes with PST1s in ARVI region |
|---|---|---|
| 1 | LV (posterior endocardium) | 1 of 3 |
| 2 | LV (lateral endocardium) | 5 of 5 |
| 3 | LV (anterior epicardium) | 3 of 4 |
| 4 | LV (posterior epicardium) | 4 of 4 |
| 5 | LV (lateral epicardium) | 3 of 3 |
| 6 | LV (lateral epicardium) | 2 of 2 |
| 7 | RV (lateral epicardium) | 2 of 3 |
| 8 | RV (anterior epicardium near base) | 2 of 3 |
| 9 | RV (anterior epicardium) | 3 of 3 |
| 10 | ARVI (epicardium) | 1 of 2 |
Figure 5 shows the sites of all PST1s relative to the ARVI region and S2 site in each heart. In three episodes, the wavefront broke in two places simultaneously. In these cases, all PST1s were registered. The 54 PST1s inside the ARVI region were 6 ± 4 mm from the ARVI midline. The 15 PST1s outside were 24 ± 5 mm from the midline. The ARVI region occupied 26 ± 2% of the area of the epicardial model. If PST1 formation was independent of location, the probability of 54/69 PST1s occurring in an area of this size by chance is <10−8 by χ2 analysis.
Figure 5.

PST1 sites in all 10 hearts. Sites from all episodes in a heart are displayed on the common epicardial model for that heart (with a Hammer projection). In each model, the ARVI region is outlined. The RV is in the centre of each model, to the left, and above the ARVI, and the LV is on either edge. S1 sites are labelled ‘S1’ and S2 sites are indicated by stimulus symbols. PST1 sites in the ARVI region are black and all others are white. Heart numbers correspond to heart numbers in Table 1. The six hearts with S2 delivery at an LV site are in the upper left panel. The three hearts with RV S2 delivery are in the upper right panel. The one heart with S2 delivery on the ARVI is in the lower panel.
3.3. APD analysis
The areas of the epicardial test sites for the ARVI, PRVI, LV, and RV were 67.4 ± 14.0, 67.3 ± 13.3, 67.0 ± 12.2, and 65.6 ± 9.9 mm2, respectively. The fastest pacing rate with 1:1 capture (rapid pacing) was 177.0 ± 33.3 ms. Rapid pacing APDs were 132.1 ± 25.3, 130.8 ± 24.5, 129.4 ± 23.0, and 129.7 ± 23.0 ms for the ARVI, PRVI, LV, and RV test sites, respectively. There was a significant difference among APDs at these sites by repeated measures ANOVA (P< 0.01). Post hoc analysis indicated that the APD was 2.2 ± 2.1 ms longer at the ARVI than the mean of the other sites.
||APD alternans|| during rapid pacing were 4.1 ± 2.1, 2.6 ± 1.4, 3.2 ± 1.7, and 3.0 ± 3.1 ms for the ARVI, PRVI, LV, and RV test sites, respectively. These values were not significantly different (P= 0.49). ||APD alternans|| for the signals in Figure 2B and C are 2.3 and 12.0 ms, respectively. To place these values in context, we randomly shuffled the APD values from each signal and recomputed ||APD alternans||. This should destroy any alternating structure present in the signals and yield values close to 0. ||APD alternans|| values after shuffling were 2.7 ± 1.1, 2.0 ± 0.7, 2.8 ± 1.6, and 2.1 ± 0.9 ms at the same sites, respectively. These values are significantly different from ||APD alternans|| in ordered beats (P< 0.01), but the size of the difference is small.
3.4. Wavefront conduction velocity during rapid pacing
Mean epicardial conduction speed during rapid pacing was highly variable across the epicardium. In addition, patterns of fast and slow regions were not consistent from heart to heart, probably partly due to the variation in pacing site among hearts. In four of the hearts, the paced wavefronts crossing the ARVI in the vicinity of the PST1 sites were relatively straight and parallel to the ARVI. In these hearts (hearts 3–6), all 24 PST1s that were located in the ARVI region were co-localized with regions with locally slowed conduction, although the regions were not necessarily the slowest on the epicardium. Figure 6A shows an example of one of these hearts (heart 6). In the remaining hearts, paced propagation across the ARVI near PST1 sites was more complex, typically involving collisions between the wavefront propagating from the pacing site and wavefronts breaking through to the epicardium from below. In these hearts, the activation sequence was likely a more important determinant of apparent epicardial conduction speed than tissue properties. Accordingly, the relationship between PST1 sites and conduction speed patterns was variable: PST1s in the ARVI region were co-localized with either locally slowed conduction (n= 10), locally elevated conduction (n= 11), or missing speed data (n= 9). An example of a heart with complex conduction at the ARVI is shown in Figure 6B (heart 8). Very similar results were obtained during 300 ms pacing (data not shown).
Figure 6.
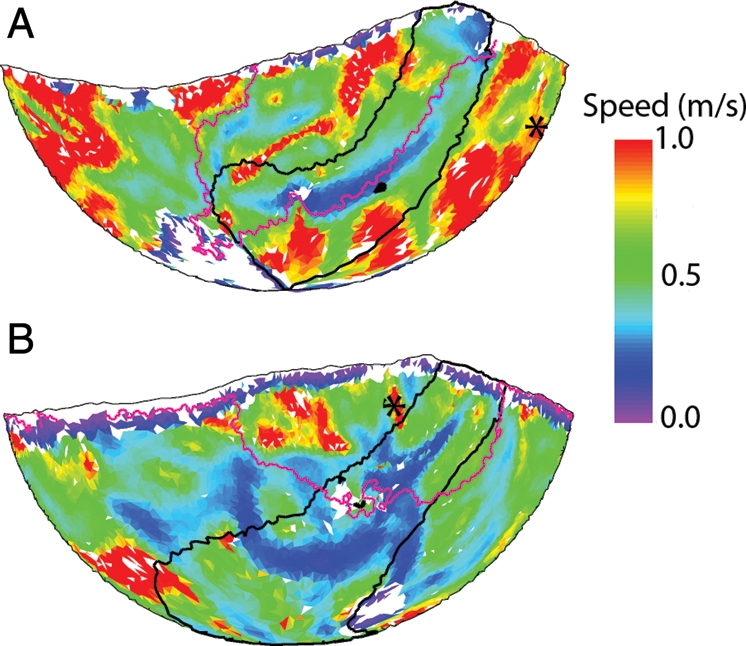
Relation between PST1 sites and conduction speed during the fastest pacing rate with 1:1 capture. A black outline identifies the ARVI region. Black asterisks indicate the pacing site (S1). The magenta line indicates the position of the paced wavefront as it traverses the ARVI near PST1 sites. White triangles indicate missing speed data; black triangles are PST1 sites. (A) Propagation is smooth and parallel to the ARVI. There is a speed trough co-localized with the PST1 sites. (B) Propagation across the ARVI is complex involving the collision between an epicardial wave and a wave breaking through to the epicardium from the septum. PST1s at the ARVI do not correspond to local speed depression in this case.
4. Discussion
4.1. Major findings
In this study, we optically mapped cardiac wave propagation during early VF over nearly the entire ventricular epicardium. Our major finding is that in normal isolated swine hearts, the ARVI was the favoured site for the first wavebreak event in the transition from the organized tachyarrhythmia we induced to more complex, fully developed VF (81% of episodes). This occurred both when wavefronts crossed from the left to the right ventricle and vice versa. This finding is consistent with Qin et al.,16 who recorded frequent wavebreaks along the ARVI during fully developed VF. The evident importance of this region raises the possibility that it could be specifically targeted in anti-VF therapy.
4.2. Limitations
This study is subject to a number of limitations. Although we were able to image nearly the entire epicardium simultaneously using recently developed optical mapping technology, we could not map events within the ventricular walls. Focused 3D mapping of the ARVI using plunge needle electrodes or optrodes29 might help explain the arrhythmogenicity of this region.
To characterize spatial and temporal APD patterns, we used rapid pacing as a surrogate for VF0 activation. This is because VF0 activation progressively accelerated, masking APD alternans.
To enable electrical activation and recovery patterns to be mapped with high spatial and temporal resolution over the entire epicardium, we used isolated swine hearts that were treated with BDM to suppress contractions. VF activation patterns in this preparation differ somewhat from in situ patterns;19 however, we believe it is unlikely that the preparation would affect the heart heterogeneously such that wavebreaks were more likely to occur at the ARVI in the isolated heart than in vivo.
4.3. Potential wavebreak mechanisms
Classically, wavebreak occurs when a wave encounters a region with locally delayed repolarization.7 More recently, it has been proposed that variations in APD can form dynamically even in tissue that is spatially uniform. The hallmark of such variability is the presence of beat-to-beat APD alternans.30 The evidence that spatial or temporal APD variability was responsible for wavebreak events at the ARVI in our study is weak. First, the APD at the ARVI test sites was significantly longer than at the other test sites during rapid pacing, but only by ∼2–3 ms. Second, although temporal APD alternans could be detected during rapid pacing, they were very small: Their magnitudes were in the low single digits and only ∼1 ms longer than the magnitudes of alternans obtained when the APDs associated with each signal were randomly shuffled. In addition, the magnitude of alternans did not differ among the test sites. We therefore believe it is unlikely that spatial or temporal APD dynamics contributed to PST1 formation at the ARVI.
An alternative possibility is that differences in micro- or macroscopic anatomy at the ARVI relative to other regions affects coupling and promotes propagation failure. Our propagation speed data indicate that this might be the case. In four of our hearts, paced wavefronts crossing the ARVI were relatively straight, smooth, and approximately parallel to the insertion. Under these conditions, epicardial conduction speed as waves traverse the insertion is largely a function of tissue properties (as opposed to wavefront shape). In these hearts (hearts 3–6), paced propagation was markedly slowed at the ARVI, and when PST1s formed at the ARVI (12 of the 13 episodes observed in these hearts) they were co-local with the velocity trough (Figure 6A). This suggests that conduction in this region is depressed relative to surrounding regions. As the VF0 rotors accelerated, the excitable gap between wavefronts became progressively narrower everywhere, but was first squeezed out completely at the ARVI, causing a wavebreak. In the remaining hearts, complex activation patterns at the ARVI during pacing (e.g. collision and epicardial breakthrough; Figure 6B) made the apparent epicardial conduction speed more a function of wave shape and orientation relative to the epicardium than of electrical coupling in the tissue. We believe that this is why PST1s at the ARVI did not consistently co-localize with local speed minima in these hearts.
Conduction slowing and wavebreak at the ARVI might be explained by macroscopic electrical loading effects. The junction of the ventricles can be thought of as a tissue expansion, which is well known to inhibit propagation and promote wavebreak.8 However, this explanation would predict that propagation would be more likely to fail when waves cross from the thin-walled RV to the thick-walled LV than the other way around, which was not the case in our data. It has also been suggested that blood vessels can act as current sinks and inhibit conduction.13,16 Thus, it is possible that the left anterior descending coronary artery, which overlies the ARVI, could have played a role in our results.
We believe that the most likely cause for our results is the complex muscle fibre architecture where the RV free wall merges with the LV. Even in normal hearts, the RV insertions are characterized by sharp changes in fibre orientation including cells abutting at angles near 90°.31 In hypertrophic cardiomyopathy, such structure, known as myocardial disarray, is more extensive and is associated with arrhythmia and sudden death.32 Previous studies have shown that the insertions of trabeculae and papillary muscles, which also feature abrupt changes in fibre orientation, can promote wavebreak and re-entry during VF in swine hearts.9,33 We have shown both experimentally17 and computationally10 that steep gradients of fibre orientation—even in the absence of abrupt discontinuities—can depress conduction, potentially causing wavebreak. In 2D cell cultures that mimicked the cross-sectional geometry of mouse hearts, including realistic fibre orientation, wavebreaks were produced at the junctions of the LV and RV regions along the steepest local gradient in the fibre angle.34 Although these findings suggest that fibre anatomy plays an important role in promoting wavebreak at the ARVI, further research is necessary to confirm this hypothesis and to clarify why the anterior side is favoured over the posterior side.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (HL64184); and the National Science Foundation (CBET 0756117).
Supplementary Material
Acknowledgements
We thank Frank Vance and Reuben Collins for their help with animal preparation.
Conflict of interest: none declared.
References
- 1.Mehra R. Global public health problem of sudden cardiac death. J Electrocardiol. 2007;40:S118–S122. doi: 10.1016/j.jelectrocard.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 2.Huikuri H, Castellanos A, Myerburg R. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–1482. doi: 10.1056/NEJMra000650. [DOI] [PubMed] [Google Scholar]
- 3.Wiggers CJ. Studies of ventricular fibrillation caused by electric shock: cinematographic and electrocardiographic observations of the natural process in the dog's heart: its inhibition by potassium and the revival of coordinated beats by calcium. Am Heart J. 1930;5:351–365. doi: 10.1046/j.1542-474X.2003.08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss JN, Chen PS, Qu Z, Karagueuzian HS, Garfinkel A. Ventricular fibrillation: how do we stop the waves from breaking? Circ Res. 2000;87:1103–1107. doi: 10.1161/01.res.87.12.1103. [DOI] [PubMed] [Google Scholar]
- 5.Huang J, Rogers JM, Killingsworth CR, Singh KP, Smith WM, Ideker RE. Evolution of activation patterns during long-duration ventricular fibrillation in dogs. Am J Physiol Heart Circ Physiol. 2004;286:H1193–H1200. doi: 10.1152/ajpheart.00773.2003. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Rogers JM, KenKnight BH, Rollins DL, Smith WM, Ideker RE. Evolution of the organization of epicardial activation patterns during ventricular fibrillation. J Cardiovasc Electrophysiol. 1998;9:1291–1304. doi: 10.1111/j.1540-8167.1998.tb00105.x. [DOI] [PubMed] [Google Scholar]
- 7.Moe GK, Reinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J. 1964;67:200–220. doi: 10.1016/0002-8703(64)90371-0. [DOI] [PubMed] [Google Scholar]
- 8.Fast VG, Kleber AG. Role of wavefront curvature in propagation of cardiac impulse. Cardiovasc Res. 1997;33:258–271. doi: 10.1016/s0008-6363(96)00216-7. [DOI] [PubMed] [Google Scholar]
- 9.Kim YH, Xie F, Yashima M, Wu TJ, Valderrabano M, Lee MH, et al. Role of papillary muscle in the generation and maintenance of reentry during ventricular tachycardia and fibrillation in isolated swine right ventricle. Circulation. 1999;100:1450–1459. doi: 10.1161/01.cir.100.13.1450. [DOI] [PubMed] [Google Scholar]
- 10.Rogers JM, McCulloch AD. Nonuniform muscle fiber orientation causes spiral wave drift in a finite element model of cardiac action potential propagation. J Cardiovasc Electrophysiol. 1994;5:496–509. doi: 10.1111/j.1540-8167.1994.tb01290.x. [DOI] [PubMed] [Google Scholar]
- 11.Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293:H2024–H2038. doi: 10.1152/ajpheart.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- 13.Gibb M, Bishop M, Burton R, Kohl P, Grau V, Plank G., et al. The role of blood vessels in rabbit propagation dynamics and cardiac arrhythmias. In: Ayache N, Delingette H, Sermesant M, editors. Functional Imaging and Modeling of the Heart. Heidelberg: Springer Berlin; 2009. pp. 268–276. [Google Scholar]
- 14.Vetter FJ, Simons SB, Mironov S, Hyatt CJ, Pertsov AM. Epicardial fiber organization in swine right ventricle and its impact on propagation. Circ Res. 2005;96:244–251. doi: 10.1161/01.RES.0000153979.71859.e7. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen PM, Le Grice IJ, Smaill BH, Hunter PJ. Mathematical model of geometry and fibrous structure of the heart. Am J Physiol. 1991;260:H1365–H1375. doi: 10.1152/ajpheart.1991.260.4.H1365. [DOI] [PubMed] [Google Scholar]
- 16.Qin H, Huang J, Rogers JM, Walcott GP, Rollins DL, Smith WM, et al. Mechanisms for the maintenance of ventricular fibrillation: the nonuniform dispersion of refractoriness, restitution properties, or anatomic heterogeneities? J Cardiovasc Electrophysiol. 2005;16:888–897. doi: 10.1111/j.1540-8167.2005.40650.x. [DOI] [PubMed] [Google Scholar]
- 17.Bourgeois EB, Fast VG, Collins RL, Gladden JD, Rogers JM. Change in conduction velocity due to fiber curvature in cultured neonatal rat ventricular myocytes. IEEE Trans Biomed Eng. 2009;56:855–861. doi: 10.1109/TBME.2008.2007501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kay MW, Walcott GP, Gladden JD, Melnick SB, Rogers JM. Lifetimes of epicardial rotors in panoramic optical maps of fibrillating swine ventricles. Am J Physiol Heart Circ Physiol. 2006;291:H1935–H1941. doi: 10.1152/ajpheart.00276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin H, Kay MW, Chattipakorn N, Redden DT, Ideker RE, Rogers JM. Effects of heart isolation, voltage-sensitive dye, and electromechanical uncoupling agents on ventricular fibrillation. Am J Physiol, Heart Circ Physiol. 2003;284:H1818–H1826. doi: 10.1152/ajpheart.00923.2002. [DOI] [PubMed] [Google Scholar]
- 20.Kay MW, Amison PM, Rogers JM. Three-dimensional surface reconstruction and panoramic optical mapping of large hearts. IEEE Trans Biomed Eng. 2004;51:1219–1229. doi: 10.1109/TBME.2004.827261. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Y, Mowrey KA, Van Wagoner DR, Tchou PJ, Efimov IR. Virtual electrode-induced reexcitation: a mechanism of defibrillation. Circ Res. 1999;85:1056–1066. doi: 10.1161/01.res.85.11.1056. [DOI] [PubMed] [Google Scholar]
- 22.Frazier D, Wolf P, Wharton J, Tang A, Smith W, Ideker R. Stimulus-induced critical point. Mechanism for electrical initiation of reentry in normal canine myocardium. J Clin Invest. 1989;83:1039–1052. doi: 10.1172/JCI113945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers JM. Combined phase singularity and wavefront analysis for optical maps of ventricular fibrillation. IEEE Trans Biomed Eng. 2004;51:56–65. doi: 10.1109/TBME.2003.820341. [DOI] [PubMed] [Google Scholar]
- 24.Banville I, Gray RA. Effect of action potential duration and conduction velocity restitution and their spatial dispersion on alternans and the stability of arrhythmias. J Cardiovasc Electrophysiol. 2002;13:1141–1149. doi: 10.1046/j.1540-8167.2002.01141.x. [DOI] [PubMed] [Google Scholar]
- 25.Gray RA, Pertsov AM, Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature. 1998;392:75–78. doi: 10.1038/32164. [DOI] [PubMed] [Google Scholar]
- 26.Winfree AT. When Time Breaks Down. Princeton: Princeton University Press; 1987. [Google Scholar]
- 27.Rogers JM, Huang J, Pedoto RW, Walker RG, Ideker RE. Fibrillation is more complex in the left ventricle than the right. PACE. 2000;23:684. doi: 10.1046/j.1540-8167.2000.01364.x. [DOI] [PubMed] [Google Scholar]
- 28.Bayly PV, KenKnight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng. 1998;45:563–571. doi: 10.1109/10.668746. [DOI] [PubMed] [Google Scholar]
- 29.Byars JL, Smith WM, Ideker RE, Fast VG. Development of an optrode for intramural multisite optical recordings of Vm in the heart. J Cardiovasc Electrophysiol. 2003;14:1196–1202. doi: 10.1046/j.1540-8167.2003.03203.x. [DOI] [PubMed] [Google Scholar]
- 30.Weiss JN, Qu Z, Chen PS, Lin SF, Karagueuzian HS, Hayashi H, et al. The dynamics of cardiac fibrillation. Circulation. 2005;112:1232–1240. doi: 10.1161/CIRCULATIONAHA.104.529545. [DOI] [PubMed] [Google Scholar]
- 31.Kuribayashi T, Roberts WC. Myocardial disarray at junction of ventricular septum and left and right ventricular free walls in hypertrophic cardiomyopathy. Am J Cardiol. 1992;70:1333–1340. doi: 10.1016/0002-9149(92)90771-p. [DOI] [PubMed] [Google Scholar]
- 32.de Bakker JM, van Rijen HM. Continuous and discontinuous propagation in heart muscle. J Cardiovasc Electrophysiol. 2006;17:567–573. doi: 10.1111/j.1540-8167.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- 33.Valderrabano M, Lee MH, Ohara T, Lai AC, Fishbein MC, Lin SF, et al. Dynamics of intramural and transmural reentry during ventricular fibrillation in isolated swine ventricles. Circ Res. 2001;88:839–848. doi: 10.1161/hh0801.089259. [DOI] [PubMed] [Google Scholar]
- 34.Badie N, Scull JA, Klinger RY, Krol A, Bursac N. Conduction block in micropatterned cardiomyocyte cultures replicating the structure of ventricular cross-sections. Cardiovasc Res. 2012;93:263–271. doi: 10.1093/cvr/cvr304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.