

Abstract
Rho kinase (ROCK), a downstream effector of Rho GTPase, is a serine/threonine protein kinase that regulates many crucial cellular processes via control of cytoskeletal structures. The C-terminal PH-C1 tandem of ROCKs has been implicated to play an autoinhibitory role by sequestering the N-terminal kinase domain and reducing its kinase activity. The binding of lipids to the pleckstrin homology (PH) domain not only regulates the localization of the protein but also releases the kinase domain from the close conformation and thereby activates its kinase activity. However, the molecular mechanism governing the ROCK PH-C1 tandem-mediated lipid membrane interaction is not known. In this study, we demonstrate that ROCK is a new member of the split PH domain family of proteins. The ROCK split PH domain folds into a canonical PH domain structure. The insertion of the atypical C1 domain in the middle does not alter the structure of the PH domain. We further show that the C1 domain of ROCK lacks the diacylglycerol/phorbol ester binding pocket seen in other canonical C1 domains. Instead, the inserted C1 domain and the PH domain function cooperatively in binding to membrane bilayers via the unconventional positively charged surfaces on each domain. Finally, the analysis of all split PH domains with known structures indicates that split PH domains represent a unique class of tandem protein modules, each possessing distinct structural and functional features.
Split pleckstrin homology (PH)3 domains are a unique subclass of PH domains in which the domain is split into two halves by the insertion of one or more autonomously folded protein modules. To date, only six proteins with diverse functions have been found to contain split PH domains in the eukaryotic genome as follows: the syntrophin family scaffold proteins; the second messenger metabolizing enzymes phospholipase Cγ (PLCγ); the Vps36 subunit of the yeast ESCRT-II sorting machinery; the neuronal GTPase PI 3-kinase enhancer (PIKE, also named AGAP and GGAP); the actin filament-based molecular motor myosin X; and the ROCK family serine/threonine protein kinases (supplemental Fig. 1A). Recent structural studies of α-syntrophin, PLCγ, Vps36, and PIKE showed that the split PH domains in these proteins all fold into a canonical PH domain conformation with or without the insertions of various protein domains (1-4). The functional significance of such domain organization has also been evaluated. It was demonstrated in α-syntrophin that the PDZ domain insertion functions synergistically with the split PH domain in binding to lipid membranes (1). Similarly, the nuclear localization sequence (NLS) insertion in the split PH domain of PIKE was found to coordinate with the split PH domain in the membrane attachment of the protein, and the PHN-NLS-PHC supramodule may function as a cytoplasmic/nuclear shuttling switch to regulate the subcellular localization of both PIKE-L and PIKE-A (4). Although the SH2-SH2-SH3 tandem insertion in the split PH domain of PLCγ was implicated in the regulation of the lipase activity of the enzyme, the mechanistic basis of this regulation is still unclear (2, 5, 6).
Rho kinases (ROCKs) are the major downstream effectors of Rho GTPase. They are involved in many aspects of cellular processes, such as stress fiber and focal adhesion formation, cell morphology, cadherin-mediated cell-cell adhesion, cytokinesis, membrane ruffling, smooth muscle contraction, cell migration, and neurite outgrowth (7, 8). Two ROCK isoforms, ROCK I (also known as ROKβ and p160 ROCK) and ROCK II (also known as ROKα), have been identified, and they share 65% amino acid sequence identity and 95% homology (9-11). Among protein kinase neighbors, ROCKs are most closely related to myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK) and citron Rho-interacting kinase (CRIK) (12, 13). All of these kinases share the same domain organization, which consists of an N-terminal kinase domain, a central coiled-coil region, and various functional motifs at their respective C termini. In ROCKs, these motifs include a Rho binding (RB) domain and a PH domain that is split into two halves by an internal cysteine-rich C1 domain (also referred to as the CRD, supplemental Fig. 1A). MRCK and CRIK also each contain a C-terminal PH and C1 domain, but the two domains are arranged next to each other instead of in the split PH domain arrangement (supplemental Fig. 1B). Another common feature of these kinases is that their catalytic activities are tightly regulated by autoinhibitory mechanisms. In MRCK, the kinase inhibitory motif acts as a negative regulator by directly binding to the kinase catalytic domain (14). Additionally, the C1 and PH domains have also been implied to inhibit the activity of MRCK (15). It was proposed that the binding of diacylglycerol (DAG)/phorbol ester to the C1 domain somehow releases the kinase inhibition by the kinase inhibitory motif (16, 17). CRIK has a similar C1 domain that more closely resembles the MRCK C1 domain than the ROCK C1 domain, and the CRIK C1 domain might play a regulatory role to the kinase activity of the enzyme (13, 18, 19). The regulatory mechanism of the kinase activity of ROCK is somewhat better studied. The C-terminal RB domain and the PH-C1 domain of ROCK sequester its N-terminal kinase domain and thus suppress its kinase activity (20). The deletion of the C-terminal RB and PH-C1 domains renders the mutant kinase constitutively active in vitro (9, 21). In vivo, ROCK I can be activated by the caspase-3-mediated cleavage of its C-terminal domains (22). The binding of GTP-bound active Rho to the RB domain also releases the autoinhibition of the kinase (11). Finally, lipids, such as arachidonic acid and PI(3,4,5)P3, have been reported to activate ROCK as well as to regulate its subcellular localization, presumably by binding to its PH domain (23, 24). However, it is not known how the ROCK PH domain interacts with the lipids. Amino acid sequence analysis indicated that the ROCK PH domain does not contain the signature phosphoinositide-binding motif found in typical lipid binding PH domains (25-28). Additionally, it is not known whether the C1 domain of ROCK can also bind to DAG/phorbol ester and thereby regulate its kinase activity, as the ROCK C1 domain is distinct from the C1 domains of MRCK and CRIK. Finally, the functional implications and their underlying molecular mechanisms of the splitting of the PH domain by the C1 insertion in ROCK are not known.
In this study, we solved the three-dimensional structures of the split PH domain and the C1 domain of ROCK II. Structural analysis revealed that the C1 domain of ROCK adopts an atypical structure and that the domain cannot bind to DAG/phorbol ester. Instead, the C1 domain functions synergistically with the PH domain in binding to membrane bilayers with a mechanism distinct from all other known lipid binding PH domains.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The ROCK I PHN-C1-PHC tandem (residues 1119-1319), the ROCK II PHN-C1-PHC tandem (residues 1142-1342), the joined PHN-PHC (residues 1142-1227, 1312-1342), the C1 domain (residues 1228-1311), and various mutants of the PHN-C1-PHC tandem were PCR-amplified from the rat hippocampal cDNA library with specific primers. The amplified DNA fragments were inserted into a modified version of the pET32a vector (Novagen) in which the DNA sequences encoding the S tag and thioredoxin were removed. The resulting proteins each contained a His6 tag in their N termini. The C1-PHN-PHC and PHN-PHC-C1 mutants were constructed, respectively, by connecting the C1 domain and the joint PHN-PHC domain with a 10-residue “Gly-Ser-Gly-Gly-Ser-Gly-Gly-Ser-Gly-Ser” linker. The recombinant plasmids harboring the respective target genes were individually transformed into Escherichia coli BL21 (DE3) host cells for large scale protein production. Uniformly 15N- and 15N/13C-labeled proteins were prepared by growing bacteria in M9 minimal medium containing 15NH4Cl with or without [13C6]glucose as stable isotope sources and 20 μm ZnCl2. The His-tagged fusion proteins were purified under native conditions using nickel-nitrilotriacetic acid-agarose (Qiagen) affinity chromatography. After dialysis in 30 mm MES buffer (pH 5.5, with 75 mm Na2SO4, 20 μm ZnCl2, and 10 μm β-mecaptoethanol) overnight, the proteins were purified using size-exclusion chromatography (Hiload 26/60 Superdex 200, preparation grade). The N-terminal His-tagged peptide fragment was then cleaved by digesting the fusion protein with 3C protease, and the proteins were purified by another step of size-exclusion chromatography.
NMR Structure Determination—The NMR samples used contained ∼1.0 mm of the joint PHN-PHC or C1 domain in 30 mm MES (pH 5.5, with 1 mm dithiothreitol, and 75 mm Na2SO4). All NMR spectra were acquired at 25 °C on Varian Inova 500- and 750-MHz spectrometers. The backbone and side-chain resonance assignments of the protein were obtained by standard heteronuclear correlation experiments (29). The side chains of aromatics were assigned using 1H two-dimensional total correlation spectroscopy/NOESY experiments (30).
Interproton distance restraints were derived from the NOESY spectra (two-dimensional 1H NOESY, three-dimensional 15N-NOESY, and three-dimensional 13C-NOESY). Hydrogen bonding restraints were generated from the standard secondary structures of the protein based on the NOE patterns and backbone secondary chemical shifts. The backbone dihedral angle restraints (ϕ and ψ angles) were derived from the chemical shift analysis program TALOS (31). CYANA was used to calculate 20 structures from random starting conformers. The calculations of the C1 domain structures were first performed in the absence of zinc ligation restraints. The conformer with the lowest penalty function value was then used as input for refinement using torsion angle-simulated annealing calculations in CNS (32). Zincs were incorporated into the calculations by the introduction of the covalent restraints to maintain their tetrahedral geometry. Sγ-Zn and Nε2-Zn bond lengths were constrained to 2.3 and 2.0 Å, respectively, with force constants of 250 kcal mol-1Å-2. Bond angles defining the zinc coordination sites were constrained to the following values: 112° for Sγ-Zn-Sγ bond angles, 108° for Cβ-Sγ-Zn angles, 111° for Sγ-Zn-Nε2 angles, and 102° for Nε2-Zn-Nε2 angles. A total of 200 structures were calculated with the final set of restraints, and the 20 structures with the lowest NOE energies were selected.
Lipid Binding Assay—A liposome stock consisting of total bovine brain lipids was prepared by resuspending bovine brain lipid extracts (Folch fraction I, Sigma B1502) at 10 mg/ml in a buffer containing 50 mm HEPES, pH 7.5, 75 mm Na2SO4. The sedimentation-based assay followed the method described earlier (1). Defined liposomes were reconstituted from synthetic l-α-phosphatidylcholine (PC) and l-α-phosphatidylserine (PS) (Avanti Polar Lipids) with or without various PIPs (Echelon Biosciences; see Ref. 1 for details).
RESULTS AND DISCUSSION
Split PH Domain of the PHN-C1-PHC Tandem from ROCK II Adopts the Same Fold with or without the C1 Domain Insertion—To evaluate the effect of the cysteine-rich C1 domain insertion on the conformation of the PH domain in ROCK II, we compared the structures of the PH domains with and without the insertion of the C1 domain using NMR spectroscopy. To achieve this, we produced and purified the PHN-C1-PHC tandem (residues 1142-1342, referred to hereafter as PHN-C1-PHC), the isolated PH domain (PHN-PHC, residue 1142-1227 and 1312-1342), and the C1 domain (residue 1227-1311) of ROCK II. Each of the three recombinant proteins was eluted at their molecular mass indicative of a stable monomer when analyzed by analytical gel filtration chromatography (data not shown). Furthermore, each of the three proteins had a well dispersed 1H and 15N HSQC spectrum, indicating that they are well folded (Fig. 1A). The HSQC spectra of the joined PHN-PHC domain (Fig. 1A, red peaks) and C1 domain (Fig. 1A, green peaks) overlap very well with the vast majority of the peaks from the HSQC spectrum of the PHN-C1-PHC tandem (Fig. 1A, black peaks). Only a small set of peaks did not overlap in this analysis. After assigning the chemical shifts of the PHN-C1-PHC tandem as well as the isolated PH and C1 domains, we plotted the chemical shift differences of backbone amides as a function of residue number between the PHN-C1-PHC tandem and the isolated PH and C1 domains (Fig. 1B). The largest chemical shift differences were located in the linker regions that connect the two halves of the PH domain with the C1 domain, an observation that is reasonable considering that the C1 domain was removed from the PHN-C1-PHC tandem. Minimal chemical shift changes were observed in the C1 domain, indicating that insertion of the C1 domain in the middle of PH domain does not cause appreciable conformational changes to the C1 domain. The shift changes in the β4/β5-loop of PH domain might result from the intrinsic rigidity of this loop and its close proximity to the split site in the β6/β7-loop (Fig. 1C). To further confirm that the folded C1 domain has no influence on the structure of the PH domain, we treated the PHN-C1-PHC tandem with EDTA (Fig. 1D). In the presence of EDTA, the removal of Zn2+ ions led to the complete unfolding of the C1 domain. Correspondingly, the peaks representing the folded C1 domain (Fig. 1A, green peaks) completely disappeared. Concomitantly, a new set of peaks (Fig. 1D, blue peaks) appeared at the random coil region of the spectrum. In contrast, the peaks corresponding to the split PH domain showed no significant changes upon EDTA treatment, indicating that the inserted C1 domain, folded or unfolded, does not affect the structure of the PH domain. Taken together, analogous to what have been observed in the split PH domains of α-syntrophin, PLCγ, and PIKE (1, 2, 4), our biochemical and NMR spectroscopic data indicate that the two complementary parts of the split PH domain in ROCK II interact intramolecularly to fold into a stable structure and that the inserted C1 domain does not alter the structure of the split PH domain.
FIGURE 1.

Comparison of the structures of the split PH and C1 domains in the ROCK II PHN-C1-PHC tandem and in their respective isolated states. A, superposition plots of 1H, 15N HSQC spectra of the PHN-C1-PHC tandem (black), the isolated C1 domain (green), and the joint PHN-PHC domain (red). B, plot of chemical shift differences as a function of the residue number of the split PH and C1 domains in the tandem and in their respective isolated forms. The combined 1H and 15N chemical shift changes are defined as follows: Δppm = ((ΔδHN)2 + (ΔδN × αN)2)½, where ΔδHN and ΔδN represent chemical shift differences of amide proton and nitrogen chemical shifts between the PHN-C1-PHC tandem and the isolated PH and C1 domains, respectively. The scaling factor αN used to normalize the 1H and 15N chemical shifts is 0.17. The domain organization of the PHN-C1-PHC tandem is indicated at the top of the plot. C, mapping of the chemical shift changes of the PH and C1 domains onto the three-dimensional structure of each domain as a result of separating the two domains from the tandem. The coloring scheme is represented using a horizontal bar at the top. D, superposition plots of the 1H, 15N HSQC spectra of the PHN-C1-PHC tandem in the presence of 4 molar ratios of EDTA (blue) and the joint PHN-PHC domain (magenta). Figures were generated using PYMOL, MOLMOL (43), and GRASP (44).
Structure of the Isolated PHN-PHC Domain of ROCK II—The PHN-C1-PHC tandem aggregates heavily and is prone to precipitation at high concentrations (>0.5 mm; data not shown), and thus the protein is not friendly for NMR-based structural determination. Because both PH and C1 domains fold into the same structures in tandem and in their isolated states (Fig. 1), we decided to solve the three-dimensional structures of the joined PHN-PHC and the isolated C1 domains using NMR spectroscopy (Fig. 2A and Fig. 3A and Table 1).
FIGURE 2.
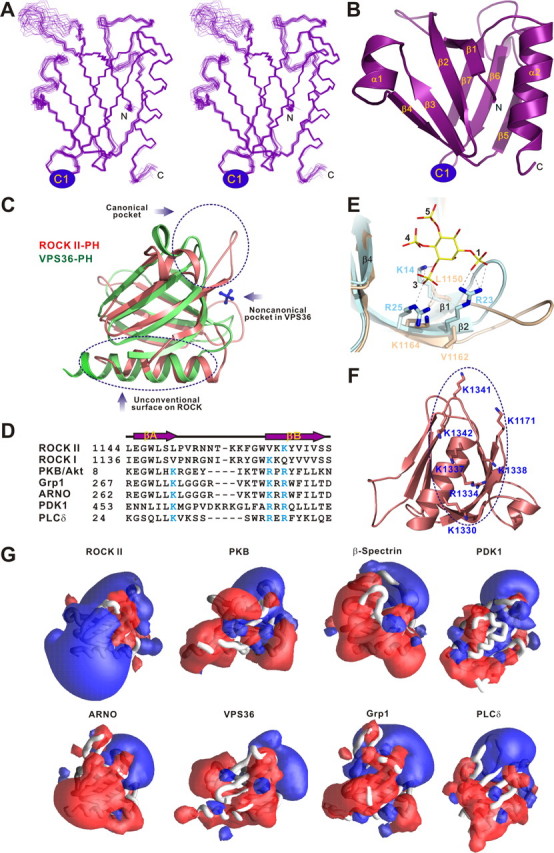
Structure of the ROCK II joint PHN-PHC domain. A, stereo view showing the backbones of 20 superimposed NMR-derived structures of the joint PHN-PHC. B, ribbon diagram of a representative NMR structure of the joint PH domain. The insertion of the C1 domain in the β6/β7-loop of the split PH domain is indicated. C, structural comparison of the split PH domains from ROCK II (pink) and VPS36 (green, PDB code 2CAY). The sulfate anion (blue sticks) bound to the noncanonical pocket formed by residues from the β5/β6- and β7/α2-loops of VPS36 split PH domain is indicated. D, structure-based sequence alignment of the β1/β2-loop of the ROCK split PH domains and the β1/β2-loops from some specific lipid-binding PH domains. The basic residues from the signature phosphoinositide-binding motifs are highlighted in cyan. E, comparison of the PIP lipid head binding pocket of the PKB/Akt PH domain (cyan, PDB code 1H10) with the same region of the ROCK II PHN-PHC domain (light yellow). The critical residues of the lipid head-binding pocket are drawn using the explicit atom representation. F, residues forming the flat, positively charged surface of the ROCK II split PH domain. G, comparison of the surface electrostatic properties of the ROCK II split PH domain with those of representative phosphoinositide-binding PH domains (Btk, PDB code, 1B55; DAPP1, PDB code 1FAO; β-spectrin, PDB code 1BTN; Grp1, PDB code 1FGY; PKB/Akt, PDB code 1H10; PLCδ1, PDB code 1MAI; ARNO, PDB code 1U27; PDK1, PDB code 1W1D; VPS36, PDB code 2CAY). The PH domains are shown in worm models. Positive (blue) and negative (red) electrostatic potentials are contoured at +3 and -3 kT, respectively. The orientations of the domains are similar to that in Fig. 2C. Electrostatic potentials were calculated with GRASP (44).
FIGURE 3.
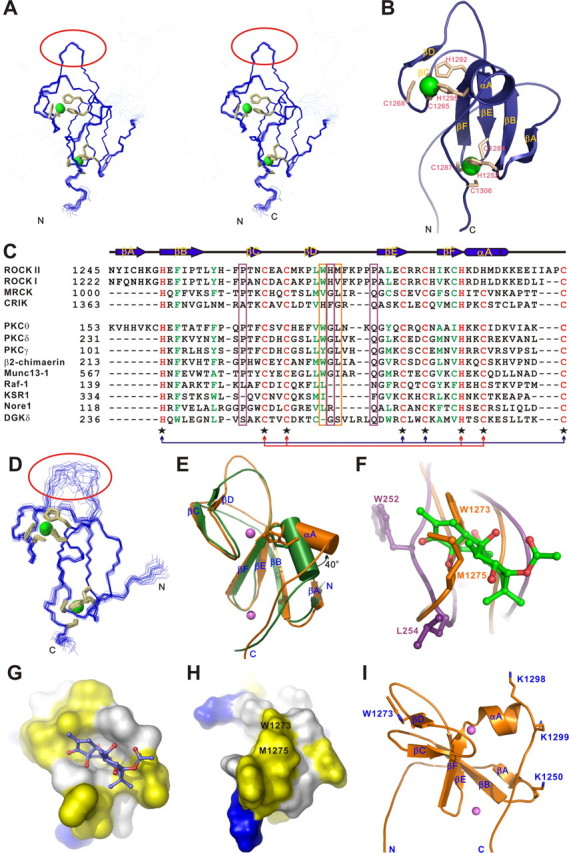
Structure of the ROCK II C1 domain. A, stereo view plot of 20 superimposed NMR structures of the isolated C1 domain. The cysteine and histidine residues involved in the Zn2+ coordination are shown as orange sticks, and the two Zn2+ ions are depicted as green spheres. The rigid βD/βE-loop is highlighted with a red circle. B, ribbon diagram drawing of the C1 domain structure. C, amino acid sequence alignment of the C1 domains of the ROCK family kinases (upper panel) and the structurally based sequence alignment of all C1 domains with known structures (lower panel). The absolutely conserved amino acids are shown in red, the highly conserved residues in green, and the variable residues in black. The residues involved in Zn2+ binding are indicated with black star below the sequences. The two sets of Zn2+-binding motifs are highlighted with arrows colored red and blue, respectively. The residues in the position homologous to Pro-241, Gly-253, and Gln-257 of PKCδ are highlighted with a purple box, and the residues in the positions homologous to Trp-1273 and Met-1275 of ROCK II are highlighted with an orange box. D, superimposed NMR structures of the C1A domain of PKCθ (PDB code 2ENN). The flexible βD/βE-loop is highlighted with a red circle. E, structure comparison of the C1 domains from ROCK II (orange) and PKCθ (green). The α-helix is shown as cylinder. The side chains of the two discriminating residues in the CCHC- and CCHH-type zinc fingers are also shown. F, close-up view of the potential DAG/phorbol ester-binding sites in ROCK II C1 domain (orange) and the interaction of PKCδ C1B domain with phorbol ester (purple, PDB code 1PTR). The phorbol ester is shown as green sticks. G, surface diagram of the PKCθ C1B domain. The orientation of the C1B domain is similar to that in Fig. 3F. The positively charged amino acids are highlighted in blue, the negatively charged residues in red, the hydrophobic residues in yellow, and the others in white. The phorbol ester is shown in sticks. H, surface diagram of the ROCK II C1 domain. The orientation of the C1 domain is similar to that in Fig. 3F. The hydrophobic Trp-1273 and Met-1275 that occlude phorbol ester from binding to the domain are labeled. I, several basic residues are clustered at one side of the ROCK II C1 domain.
TABLE 1.
Structural statistics for the family of 20 structures of the joined PHN-PHC domain and the isolated C1 domain
None of the structures exhibits distance violations greater than 0.3 Å or dihedral angle violations greater than 4°. r.m.s. indicates root mean square.
| PHN-PHC | C1 | |
|---|---|---|
| Restraint statistics | ||
| Distance restraints | ||
| Intraresidue (i − j = 0) | 725 | 412 |
| Sequential (|i − j| = 1) | 664 | 520 |
| Medium range (2 ≤ |i − j| ≤ 4) | 458 | 183 |
| Long range (|i − j| > 5) | 1016 | 704 |
| Hydrogen bonds | 56 | 32 |
| Total | 2919 | 1851 |
| Dihedral angle restraints | ||
| ϕ | 70 | 33 |
| ψ | 69 | 32 |
|
Total
|
139
|
65
|
| Structure statistics | ||
| Mean r.m.s. deviations from the experimental restraints | ||
| Distance (Å) | 0.005 ± 0.000 | 0.006 ± 0.000 |
| Dihedral angle (°) | 0.139 ± 0.017 | 0.231 ± 0.014 |
| Mean r.m.s. deviations from idealized covalent geometry | ||
| Bond (Å) | 0.001 ± 0.000 | 0.002 ± 0.000 |
| Angle (°) | 0.312 ± 0.004 | 0.827 ± 0.016 |
| Improper (°) | 0.172 ± 0.010 | 0.207 ± 0.005 |
| Mean energies (kcal mol−1) | ||
| ENOEa | 6.37 ± 0.16 | 4.99 ± 0.29 |
| Ecdiha | 0.17 ± 0.04 | 0.21 ± 0.03 |
| EL-J | −465.93 ± 12.51 | −234.81 ± 11.53 |
| Ramachandran plotb (%) | ||
| Most favorable regions | 79.7 | 71.4 |
| Additional allowed regions | 19.5 | 27.2 |
| Generously allowed regions | 0.7 | 1.3 |
|
Disallowed regions
|
0.1
|
0.2
|
| Coordinate precision | ||
| Atomic r.m.s. difference (Å)c | ||
| Backbone heavy atoms (N, Cα, and C′) | 0.29 | 0.19 |
| Heavy atoms | 0.72 | 0.64 |
The final values of the square-well NOE and dihedral angle potentials were calculated with force constants of 50 kcal mol−1 Å−2 and 200 kcal mol−1 rad−2, respectively.
The program Procheck (45) was used to assess the overall quality of the structures. For C1, the unstructured N-terminal (residues 1228-1242) is excluded.
The precision of the atomic coordinates is defined as the average r.m.s. difference between 20 final structures and the mean coordinates of the protein. Residues 1143-1152, 1160-1227, and 1312-1342 are for PHN-PHC. Residues 1245-1308 are for C1.
Except for an additional short α-helix in the β3/β4-loop, the PHN and PHC fragments interact with each other to form a canonical PH domain fold containing seven β-strands and a characteristic C-terminal α-helix (Fig. 2B). The PHN half is composed of six β-strands (β1-β6) and the short α-helix (α1), and the PHC half contains the remaining β-strand (β7) and the C-terminal α-helix (α2). Because of the short linking sequence (7 residues to be precise) connecting the β6- and β7-strands, the β6/β7-loop of the joint PH domain is relatively rigid and well defined. Among all known split PH domains, this is only the second example of a PH domain that is split into two halves by the insertion of a protein module between the β6- and β7-strands of the domain (the other is the split PH domain of VPS36) (1-4, 33). The structural comparison of the split PH domains from ROCK II and VPS36 reveals that except for the flexible loop regions and the length of their C-terminal α-helices, these two split PH domains have very similar overall conformations (Fig. 2C).
The binding of lipids to the PH domain of ROCK has been implicated to activate ROCK and regulate its subcellular localization (23, 24). To identify the potential phospholipid-binding sites, we first analyzed the residues located in the β1/β2-loop of the split PH domain, which are known to form a positively charged pocket and to play a critical role in binding to phosphoinositides in known lipid binding PH domains (25-28). Although it contains several positively charged residues, the β1/β2-loop of the of ROCK II (as well as ROCK I) PH domain does not contain the signature “KXn(K/R)XR” phosphoinositide-binding motif, where the first Lys is located at the penultimate position of the β1-strand, and the “(K/R)XR” sequence corresponds to residues 2-4 of the β2-strand (Fig. 2, D and E). In the ROCK II PH domain, the penultimate residue in the β1-strand is a Leu instead of a Lys, and the second and the fourth residues in the β2-strand are Val and Lys, respectively. Because two out of the required positively charged residues in the canonical phosphoinositol lipid-binding motif are absent in the split PH domain of ROCK II, we predicted that it would not be able to bind to lipids using its canonical binding pocket.
A characteristic feature of phosphoinositide binding PH domains is their strong surface electrostatic polarity; one end of the domain, including the C-terminal α-helix, is rich in acidic residues, whereas the opposite end, including the β1/β2-, β3/β4-, and β6/β7-loops, is clustered with an array of positively charged residues and is responsible for lipid binding (Fig. 2G) (34). In sharp contrast, surface charge potential analysis of the ROCK II PH domain shows that the domain possesses a prominent positive lobe at the end containing the C-terminal α-helix, and this relatively flat, positively charged surface is formed by a total of seven basic amino acids (Fig. 2, C, F, and G). It is tempting to hypothesize that this flat, positive surface of ROCK II PH domain might function as a distinct lipid membrane binding site (see below for more details).
Structure of the Isolated C1 Domain of ROCK II—The cysteine-rich domain of ROCK II folds into a C1 domain structure, composed of two anti-parallel β-sheets and a C-terminal helix (αA) (Fig. 3B). The first β-sheet forms the core of the C1 structure and consists of four β-strands (βA, βB, βE, and βF), and the second contains the two remaining β-strands (βC and βD). Each C1 domain coordinates two Zn2+ ions. The N terminus of the C1 domain is not well defined, as the residues in this region lack any detectable medium and long range NOEs.
There are two distinct structural properties of the C1 domain in ROCK II. First, its βΑ-strand is absent in all other C1 domains with known structures except for that of protein kinase Cθ (PKCθ) (Fig. 3, C-E). The residues from the βA-strand extensively contact those from βB- and αA-strand and presumably enhance the packing of the core structure and stabilize the folding of the C1 domain. A structural comparison of the C1 domains from ROCK II and PKCθ shows that the two domains are very similar. The most significant differences are the flexibilities of their βD/βE-loops and the orientations of their αA-helices. The conformation of the βD/βE-loop in the ROCK II C1 domain is highly rigid, whereas the same loop in the PKCθ C1 domain is much more flexible (Fig. 3, A and C). The αA-helix of the ROCK II C1 domain is rotated ∼40°anti-clockwise with respect to the corresponding α-helix in the PKCθ C1 domain (Fig. 3E). The other unique feature of ROCK C1 is that, unlike in other C1 domains, of which both of the Zn2+-binding motifs are of the CCHC-type (i.e. each composed of three cysteines and one histidine), one of its Zn2+-binding sites is composed of an atypical CCHH-type Zn2+-binding motif (i.e. formed by two cysteines and two histidines) (Fig. 3, B and C). The substitution of a small cysteine with a bulky histidine pushes the αA-helix away and leads the helix to rotate outward by ∼40° (Fig. 3E). The combination of the CCHC- and CCHH-type zinc fingers is conserved in the C1 domain of ROCK I and II, but not in the homologues kinases MRCK or CRIK and not in PKCθ (Fig. 3C). Further structural and biochemical investigations are required to uncover whether this kind of peculiar organization has a unique function (such as regulation of C1 domain folding and subsequent function).
A well known function of C1 domains is their capacity in binding to phorbol esters or DAG. The crystal structure of the PKCδ C1B in complex with phorbol ester reveals that phorbol ester fits snugly into a cavity formed by the residues from the βB/βC- and βD/βE-loops (Fig. 3, F and G). This phorbol ester binding presents a continuous hydrophobic cap that allows the region to be buried into the lipid bilayer, thus stabilizing its membrane insertion. Because the C1 domain of MRCK was found to bind to DAG/phorbol ester, and DAG/phorbol ester binding was shown to activate the kinase (16, 17), by homology ROCKs were hypothesized to employ the same DAG/phorbol ester binding-induced kinase activation mechanism, although this had not been directly tested prior to this study. To test this possibility, we first analyzed the DAG/phorbol ester binding properties of the ROCK II C1 domain. Sequence alignment reveals that the ROCK II C1 domain does not contain the consensus residues (Pro-241, Gly-253, and Gln-257 in the PKCδ C1B domain) required to form the DAG-binding site (35) (Fig. 3C). The replacement of Gly with His in this motif likely decreases the flexibility of the βD/βE-loop in the ROCK II C1 domain (Fig. 3, A and D). The rigidity of the βD/βE-loop is also conferred by the hydrophobic interaction between the side chains of Trp-1273 and Met-1275 (Fig. 3F). The rigid side chains of Trp-1273 and Met-1275 in ROCK II C1 occlude DAG/phorbol ester from binding to the ROCK II C1 domain (Fig. 3, G and H). Therefore, we believe that the ROCK II C1 domain is not likely to be able to bind to DAG/phorbol ester. Our biochemically based binding assay using phorbol ester confirmed this prediction (data not shown). Interestingly, further structural analysis revealed that several basic residues are clustered at one side of the ROCK II C1 domain, leading to a polarized surface charge distribution on the domain (Fig. 3I). It is possible that the positively charged surface of the ROCK II C1 domain might interact with negatively charged membranes (see below for more details).
ROCK II PHN-C1-PHC Tandem Functions as a Supramodule with Distinct Lipid Membrane Binding Properties—Next, we directly compared the lipid membrane binding properties of the PHN-C1-PHC tandem with those of the two isolated domains by assaying their binding to liposomes prepared from total bovine brain lipid extracts. Consistent with our prediction above, the PHN-C1-PHC tandem was found to bind efficiently to these liposomes in a dose-dependent manner (Fig. 4A). To our surprise, the joined PHN-PHC domain and the C1 domain both showed much weaker binding to liposomes than the PHN-C1-PHC tandem, even though they each fold into the same structures alone and in the PHN-C1-PHC tandem (Fig. 4B, 2nd and 3rd panels). This phenomenon is very similar to what was observed in the split PH domains of α-syntrophin and PIKE, where the split PH domain and inserted sequence function synergistically in binding to lipids (1, 4). Thus, the PHN-C1-PHC tandem of ROCK II represents another example of which the insertion of a protein module (C1 domain in this case) in the middle of a PH domain produces a supramodule with distinct biological functions. To rule out the possibility that the enhanced lipid binding property of the PHN-C1-PHC tandem is just a simple additive effect of two weak lipid binders, we created two mutants of the PHN-C1-PHC tandem by placing the C1 domain either in front of or after the joined PHN-PHC domain. A 10-residue flexible linker (“Gly-Ser-Gly-Gly-Ser-Gly-Gly-Ser-Gly-Ser”) was inserted between the C1 domain and the joined PHN-PHC domain in both mutants to avoid artificial conformational restraints. Both mutants displayed weaker lipid binding than the PHN-C1-PHC tandem (Fig. 4C), further supporting our notion that the PHN-C1-PHC tandem functions as a supramodule with distinct lipid binding properties.
FIGURE 4.

The PHN-C1-PHC supramodule binds to lipid with enhanced avidity. A, dose-dependent binding between the ROCK II PHN-C1-PHC tandem and liposomes prepared from bovine brain lipid extracts. In this assay, the amount of the PHN-C1-PHC tandem is fixed at 12.5 μm, and the concentration of liposome varies. S and P denote proteins recovered in the supernatants and pellets, respectively, in the centrifugation-based liposome binding assays. B, comparison of the brain liposome bindings of the ROCK II PHN-C1-PHC tandem and its isolated domains. The concentration of liposome was fixed at 0.75 mg/ml in the assay. The right panel shows the quantitation of the binding assays. C, binding of the two mutants of the ROCK II PHN-C1-PHC tandem to the brain liposomes. In these two mutants, the C1 domain was placed either at the front (C1-PHN-PHC) or after (PHN-PHC-C1) the split PH domain. D, interactions of ROCK II PHN-C1-PHC tandem with various PIPs (5%) reconstituted into the defined PC/PS (75/20%) liposomes assayed by the sedimentation method. The ratio of proteins recovered in the pellet and supernatant in each assay is also plotted. E, interaction of ROCK I PHN-C1-PHC tandem with various PIPs (5%) reconstituted into the defined PC/PS (75/20%) liposomes assayed by the same sedimentation method. In the graphed plots, all measured bindings are means ± S.D. of at least three different experiments.
To test where the PHN-C1-PHC supramodule might recognize specific phosphoinositides embedded in membrane bilayers, we assayed its lipid binding using reconstituted liposomes with various PIPs. The results showed that the PHN-C1-PHC supramodules of both ROCK I and II robustly bind to reconstituted PC/PS liposomes containing PI(3,4,5)P3, PI(3,4)P2, and PI(3,5)P2, and weakly interact with PC/PS liposomes containing other phosphoinositides (Fig. 3, D and E). The binding of the PHN-C1-PHC tandem to PC/PS only liposomes is at the assay background level. The high binding avidity of the ROCK PHN-C1-PHC supramodule toward 3′-phosphate-phosphoinositides indicates that it might act as a PI(3,4,5)P3 sensor, as cellular concentrations of PI(3,4)P2 and PI(3,5)P2 are generally very low. Consistent with this notion, ROCK II activity was recently shown to be down-regulated by PI 3-kinase inhibition (24). It is possible that binding of lipids (such as PI(3,4,5)P3 and arachidonic acid) to the PHN-C1-PHC tandem of ROCK releases the auto-inhibited conformation of the enzyme (20, 22-24).
As mentioned in previous sections, the PHN-C1-PHC tandem contains a flat, positively charged surface on each of its split PH and C1 domains (Fig. 2F and Fig. 3I). All the residues forming these positively charged surfaces are evolutionarily conserved in both ROCK I and ROCK II (Fig. 5A), pointing to their potential functional significance. To test this hypothesis, we mutated several residues from the positively charged surfaces of the PHN-C1-PHC tandem and tested the lipid membrane binding avidities of the mutants. As expected, the mutations of residues in the positively charged surfaces of either the C1 domain (Lys-1250, Lys-1298, and Lys-1299 to Ala) or the PH domain (Lys-1171, and/or Arg-1334, Lys-1337, and Lys-1338 to Ala) significantly compromised the lipid membrane binding capacity of the protein. Mutations of the positively charged residues from both the PH and C1 domains further decreased the lipid membrane binding of the protein (Fig. 5B). As a control, we also mutated two positively charged residues (Arg-1153 and Lys-1164), either individually or combined, in the β1/β2-loop of the split PH domain (Fig. 2D), which we predicted were not involved in the lipid binding. Consistent with our prediction, the substitution of Arg-1153 and/or Lys-1164 with Ala did not reduce the lipid binding capacity of the PHN-C1-PHC tandem. The slightly increased lipid binding avidity of K1164A is probably because of the mutation-induced instability (prone to precipitation) of the protein (Fig. 5B). Taken together, we conclude that both the split PH domain and the C1 domain contribute to the binding of ROCK to lipid membranes with two unconventional positively charged surfaces.
FIGURE 5.
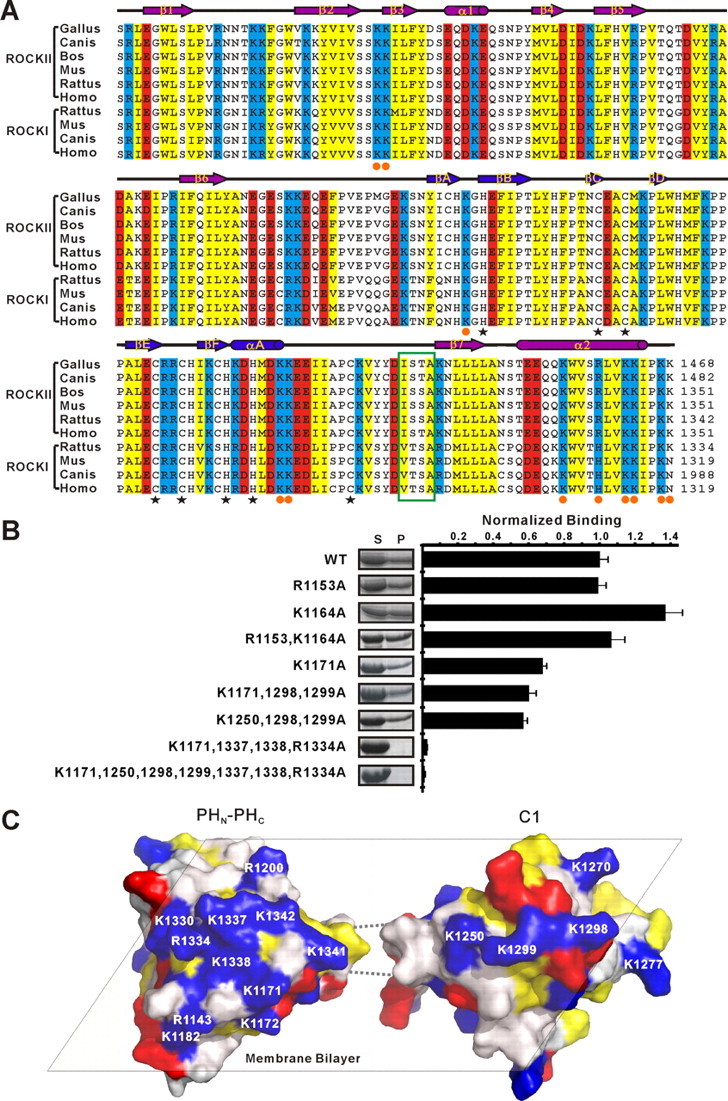
The ROCK II split PH and C1 domains function cooperatively in binding to membrane bilayers. A, amino acid sequence alignment of the PHN-C1-PHC tandem of the mammalian ROCK family proteins. In this alignment, the conserved positively charged amino acids are highlighted in blue, the negatively charged residues in red, and the hydrophobic residues in yellow. The cysteine and histidine residues involved in Zn2+ binding are indicated with black star below the sequences. The basic residues forming the positively charged surfaces shown in C are indicated with orange circles. The 4 residues linking the rigid C-terminal end of the C1 domain and theβ7-strand of the PH domain are highlighted with a green box. B, sedimentation-based liposome binding assay investigating the roles of the basic residues from the potential lipid binding surfaces of the PH and C1 domains in lipid membrane binding. The concentration of liposome was fixed at 0.75 mg/ml in the assay. The right panel shows the quantitation of the assay. The measured bindings are mean ± S.D. of at least three different experiments. WT, wild type. C, model showing the potential synergetic actions of the split PH and the C1 domains in the PHN-C1-PHC supramodule in binding to membranes.
Because the rigid C-terminal end of the C1 domain is connected to the β7-strand of the PH domain by a highly conserved short stretch of linker sequence (four residues to be precise, see Fig. 5A), the orientations of the two domains in the PHN-C1-PHC tandem are likely to be restricted to a certain extent. We envision a model in which the split PH domain and C1 domain are preferentially orientated such that the two flat, positively charged surfaces face to the same side and collaborate with each other in binding to negatively charged membrane bilayers (Fig. 5C). It is possible that the 3′-phosphate-phosphoinositides generated by activated PI 3-kinases are recognized by the PHN-C1-PHC tandem of ROCK II, which could not only recruit the kinase to the specific membrane domain but may also release the kinase from its auto-inhibited conformation. Further investigations are required to determine whether such cross-talk between ROCK and PI 3-kinase signaling pathways is indeed mediated by the PHN-C1-PHC supramodule of ROCK.
Overall Features of the Split PH Domains with Known Structures—To date, the structures of five out a total of six known split PH domains (those from α-syntrophin, PLCγ1, VPS36, PIKE, and ROCK II) have been elucidated, and their functions have been partially characterized. The only remaining uncharacterized split PH domain is that of myosin X, which contains a PH domain split by an insertion of another intact PH domain (supplemental Fig. 1A). The structural and functional features of the five split PH domains with known structures show several salient features. First, PH domains can be split in various loop regions (e.g. β3/β4-, β5/β6-, or β6/β7-loop) by insertion sequences of various lengths (unstructured nuclear localization sequences, single folded domain, or multiple protein domains) with diverse functions (Fig. 6) (1-4, 33). Second, split PH domains together with their inserted domain(s) often form supramodules with distinct biological functions. For example, the split PH domains of α-syntrophin, PIKE, and ROCK II function cooperatively with their respective inserted domains in binding to lipid membranes. Preliminary studies of the myosin X split PH domain also show that the insertion of the second PH domain in the β3/β4-loop (predicted from sequence analysis) of the first PH domain creates a functional module with enhanced lipid binding avidity.4 In PLCγ1, the split PH domain together with its inserted SH2-SH2-SH3 domains collectively play an autoinhibitory role in the regulation of the lipase activity of the enzyme (5).4 Third, it remains as an open question whether partial PH domains indeed exist in proteins, and whether such hypothetical partial PH domains may complement the other half of PH domain either in the known split PH domains or somehow “hidden” in yet to be identified proteins (36, 37).
FIGURE 6.

Structural comparison of the known split PH domains. All split PH domains with known structures adopt canonical PH domain folds. For syntrophin and PLCγ, the domain splitting insertions are located in the β3/β4-loop; for VPS36 and ROCK, the domain insertions fall in the β6/β7-loop; and the domain insertion of PIKE is located in the β5/β6-loop.
Finally, the split PH domain is not the only case of an intact domain being split by the insertion of other domains/sequences. A statistical analysis has shown that ∼9% of nonredundant protein domains deposited in the Protein Data Bank contain domain insertions (38, 39). A good example related to this study is the catalytic X/Y box of PLCγ, which is split by insertion of a large PHN-SH2-SH2-SH3-PHC tandem (supplemental Fig. 1A). It has been well recognized that protein-protein interaction modules arranged in tandem are not just simple attachments of “beads on a string,” but often represent functional supramodules with distinct structural features and biological functions (40-42). This type of domain-splitting arrangements in proteins is not likely to be the result of accidental gene rearrangements, as such organizational patterns are often conserved throughout the evolution, and rearrangements of the relative positions of the inserted domains often alter the functions of the proteins (1, 2, 4). Rather, we hypothesize that such domain insertions have been selected to cater to the functional requirements of those proteins. The identification of such unique domain insertion organizations and the elucidation of their structural and functional significance represent important areas of future research.
Supplementary Material
Acknowledgments
We thank Anthony Zhang for careful reading of the manuscript. The NMR spectrometers used in this study were purchased with funds donated to the Biotechnology Research Institute by the Hong Kong Jockey Club.
The atomic coordinates and structure factors (codes 2ROV and 2ROW) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by the Research Grants Council of Hong Kong Grants HKUST6419/05M, 6442/06M, 663407, CA07/08.SC01, and AoE/B-15/01-II (to M. Z.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are: PH, pleckstrin homology domain; ROCK, Rho kinase; C1, protein kinase C homology 1 domain; MRCK, myotonic dystrophy kinase-related Cdc42-binding kinase; CRIK, Citron Rho-interacting kinase; PLC, phospholipase C; PIKE, PI3-kinase enhancer; NLS, nuclear localization sequence; RB, Rho-binding; DAG, diacylglycerol; PKC, protein kinase C; NOE, Nuclear Overhauser effect; HSQC, heteronuclear single quantum coherence; NOESY, nuclear Overhauser effect spectroscopy; TOCSY, total correlation spectroscopy; SH, Src homology; PDB, Protein Data Bank; PC, l-α-phosphatidylcholine; PS, l-α-phosphatidylserine; PIP, phosphatidylinositol phosphate; PI(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate; PI(3,4)P2, phosphatidylinositol 3,4-bisphosphate; PI(3,5)P2, phosphatidylinositol 3,5-bisphosphate; MES, 4-morpholineethanesulfonic acid.
W. Wen, J. Yan, and M. Zhang, unpublished data.
References
- 1.Yan, J., Wen, W., Xu, W., Long, J. F., Adams, M. E., Froehner, S. C., and Zhang, M. (2005) EMBO J. 24 3985-3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen, W., Yan, J., and Zhang, M. (2006) J. Biol. Chem. 281 12060-12068 [DOI] [PubMed] [Google Scholar]
- 3.Teo, H., Gill, D. J., Sun, J., Perisic, O., Veprintsev, D. B., Vallis, Y., Emr, S. D., and Williams, R. L. (2006) Cell 125 99-111 [DOI] [PubMed] [Google Scholar]
- 4.Yan, J., Wen, W., Chan, L. N., and Zhang, M. (2008) J. Mol. Biol. 378 425-435 [DOI] [PubMed] [Google Scholar]
- 5.DeBell, K., Graham, L., Reischl, I., Serrano, C., Bonvini, E., and Rellahan, B. (2007) Mol. Cell. Biol. 27 854-863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poulin, B., Sekiya, F., and Rhee, S. G. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 4276-4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riento, K., and Ridley, A. J. (2003) Nat. Rev. Mol. Cell Biol. 4 446-456 [DOI] [PubMed] [Google Scholar]
- 8.Van Aelst, L., and D'Souza-Schorey, C. (1997) Genes Dev. 11 2295-2322 [DOI] [PubMed] [Google Scholar]
- 9.Leung, T., Chen, X. Q., Manser, E., and Lim, L. (1996) Mol. Cell. Biol. 16 5313-5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa, O., Fujisawa, K., Ishizaki, T., Saito, Y., Nakao, K., and Narumiya, S. (1996) FEBS Lett. 392 189-193 [DOI] [PubMed] [Google Scholar]
- 11.Ishizaki, T., Maekawa, M., Fujisawa, K., Okawa, K., Iwamatsu, A., Fujita, A., Watanabe, N., Saito, Y., Kakizuka, A., Morii, N., and Narumiya, S. (1996) EMBO J. 15 1885-1893 [PMC free article] [PubMed] [Google Scholar]
- 12.Leung, T., Chen, X. Q., Tan, I., Manser, E., and Lim, L. (1998) Mol. Cell. Biol. 18 130-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Cunto, F., Calautti, E., Hsiao, J., Ong, L., Topley, G., Turco, E., and Dotto, G. P. (1998) J. Biol. Chem. 273 29706-29711 [DOI] [PubMed] [Google Scholar]
- 14.Ng, Y., Tan, I., Lim, L., and Leung, T. (2004) J. Biol. Chem. 279 34156-34164 [DOI] [PubMed] [Google Scholar]
- 15.Chen, X. Q., Tan, I., Leung, T., and Lim, L. (1999) J. Biol. Chem. 274 19901-19905 [DOI] [PubMed] [Google Scholar]
- 16.Tan, I., Seow, K. T., Lim, L., and Leung, T. (2001) Mol. Cell. Biol. 21 2767-2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi, S. H., Czifra, G., Kedei, N., Lewin, N. E., Lazar, J., Pu, Y., Marquez, V. E., and Blumberg, P. M. (2008) J. Biol. Chem. 283 10543-10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madaule, P., Furuyashiki, T., Reid, T., Ishizaki, T., Watanabe, G., Morii, N., and Narumiya, S. (1995) FEBS Lett. 377 243-248 [DOI] [PubMed] [Google Scholar]
- 19.Yamashiro, S., Totsukawa, G., Yamakita, Y., Sasaki, Y., Madaule, P., Ishizaki, T., Narumiya, S., and Matsumura, F. (2003) Mol. Biol. Cell 14 1745-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amano, M., Chihara, K., Nakamura, N., Kaneko, T., Matsuura, Y., and Kaibuchi, K. (1999) J. Biol. Chem. 274 32418-32424 [DOI] [PubMed] [Google Scholar]
- 21.Ishizaki, T., Naito, M., Fujisawa, K., Maekawa, M., Watanabe, N., Saito, Y., and Narumiya, S. (1997) FEBS Lett. 404 118-124 [DOI] [PubMed] [Google Scholar]
- 22.Sebbagh, M., Renvoize, C., Hamelin, J., Riche, N., Bertoglio, J., and Breard, J. (2001) Nat. Cell Biol. 3 346-352 [DOI] [PubMed] [Google Scholar]
- 23.Feng, J., Ito, M., Kureishi, Y., Ichikawa, K., Amano, M., Isaka, N., Okawa, K., Iwamatsu, A., Kaibuchi, K., Hartshorne, D. J., and Nakano, T. (1999) J. Biol. Chem. 274 3744-3752 [DOI] [PubMed] [Google Scholar]
- 24.Yoneda, A., Multhaupt, H. A., and Couchman, J. R. (2005) J. Cell Biol. 170 443-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas, C. C., Deak, M., Alessi, D. R., and van Aalten, D. M. (2002) Curr. Biol. 12 1256-1262 [DOI] [PubMed] [Google Scholar]
- 26.Ferguson, K. M., Kavran, J. M., Sankaran, V. G., Fournier, E., Isakoff, S. J., Skolnik, E. Y., and Lemmon, M. A. (2000) Mol. Cell 6 373-384 [DOI] [PubMed] [Google Scholar]
- 27.Cronin, T. C., DiNitto, J. P., Czech, M. P., and Lambright, D. G. (2004) EMBO J. 23 3711-3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson, K. M., Lemmon, M. A., Schlessinger, J., and Sigler, P. B. (1995) Cell 83 1037-1046 [DOI] [PubMed] [Google Scholar]
- 29.Cavanagh, J., Fairbrother, W. J., Palmer, A. G., III, and Skelton, N. J. (1996) Protein NMR Spectroscopy: Principles and Practice, Academic Press, San Diego
- 30.Wüthrich, K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, Inc., New York
- 31.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR 13 289-302 [DOI] [PubMed] [Google Scholar]
- 32.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 33.Alam, S. L., Langelier, C., Whitby, F. G., Koirala, S., Robinson, H., Hill, C. P., and Sundquist, W. I. (2006) Nat. Struct. Mol. Biol. 13 1029-1030 [DOI] [PubMed] [Google Scholar]
- 34.Lemmon, M. A., and Ferguson, K. M. (2000) Biochem. J. 350 1-18 [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, G., Kazanietz, M. G., Blumberg, P. M., and Hurley, J. H. (1995) Cell 81 917-924 [DOI] [PubMed] [Google Scholar]
- 36.van Rossum, D. B., Patterson, R. L., Sharma, S., Barrow, R. K., Kornberg, M., Gill, D. L., and Snyder, S. H. (2005) Nature 434 99-104 [DOI] [PubMed] [Google Scholar]
- 37.Lemmon, M. A. (2005) Cell 120 574-576 [DOI] [PubMed] [Google Scholar]
- 38.Aroul-Selvam, R., Hubbard, T., and Sasidharan, R. (2004) J. Mol. Biol. 338 633-641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selvam, R. A., and Sasidharan, R. (2004) Nucleic Acids Res. 32 D193-D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng, W., Shi, Y., Li, M., and Zhang, M. (2003) Nat. Struct. Biol. 10 972-978 [DOI] [PubMed] [Google Scholar]
- 41.Long, J. F., Tochio, H., Wang, P., Fan, J. S., Sala, C., Niethammer, M., Sheng, M., and Zhang, M. (2003) J. Mol. Biol. 327 203-214 [DOI] [PubMed] [Google Scholar]
- 42.Long, J., Wei, Z., Feng, W., Yu, C., Zhao, Y. X., and Zhang, M. (2008) J. Mol. Biol. 375 1457-1468 [DOI] [PubMed] [Google Scholar]
- 43.Koradi, R., Billeter, M., and Wuthrich, K. (1996) J. Mol. Graphics 14 51-55 [DOI] [PubMed] [Google Scholar]
- 44.Nicholls, A., Sharp, K. A., and Honig, B. (1991) Proteins 11 281-296 [DOI] [PubMed] [Google Scholar]
- 45.Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R., and Thornton, J. M. (1996) J. Biomol. NMR 8 477-486 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.