

Abstract
Hsp70 escort proteins (Hep) have been implicated as essential for maintaining the function of yeast mitochondrial hsp70 molecular chaperones (mtHsp70), but the role that escort proteins play in regulating mammalian chaperone folding and function has not been established. We present evidence that human mtHsp70 exhibits limited solubility due to aggregation mediated by its ATPase domain and show that human Hep directly enhances chaperone solubility through interactions with this domain. In the absence of Hep, mtHsp70 was insoluble when expressed in Escherichia coli, as was its isolated ATPase domain and a chimera having this domain fused to the peptide-binding domain of HscA, a soluble monomeric chaperone. In contrast, these proteins all exhibited increased solubility when expressed in the presence of Hep. In vitro studies further revealed that purified Hep regulates the interaction of mtHsp70 with nucleotides. Full-length mtHsp70 exhibited slow intrinsic ATP hydrolysis activity (6.8 ± 0.2 × 10-4 s-1) at 25 °C, which was stimulated up to 49-fold by Hep. Hep also stimulated the activity of the isolated ATPase domain, albeit to a lower maximal extent (11.5-fold). In addition, gel-filtration studies showed that formation of chaperone-escort protein complexes inhibited mtHsp70 self-association, and they revealed that Hep binding to full-length mtHsp70 and its isolated ATPase domain is strongest in the absence of nucleotides. These findings provide evidence that metazoan escort proteins regulate the catalytic activity and solubility of their cognate chaperones, and they indicate that both forms of regulation arise from interactions with the mtHsp70 ATPase domain.
The hsp70 protein family is a ubiquitous class of proteins found in most cellular compartments that have evolved to participate in a range of cellular processes, including vesicular trafficking, Fe/S-cluster biogenesis, the stress response, protein folding, and protein translocation (for reviews, see Refs. 1–4). Members of this protein family contain two domains, an N-terminal ATPase domain and a C-terminal peptide-binding domain. Central to all hsp70 functions is their ability to bind polypeptide substrates reversibly and to use conformational changes driven by ATP binding and hydrolysis to regulate substrate affinity. ATP binding leads to a conformation that exhibits weaker substrate affinity and faster substrate exchange, and subsequent hydrolysis to ADP and inorganic phosphate results in a conformational state with stronger substrate affinity and slower exchange (5–10).
Mitochondria require hsp70 chaperones for the translocation of nuclear-encoded proteins (11–13), the synthesis of Fe/S-clusters (2, 14–16), and protein folding (17). In yeast, two chaperones (Ssc1 and Ssq1) contribute to these functions, whereas mammals have a single hsp70 isoform (designated mtHsp70, HspA9b, Grp75, and mortalin) that is predicted to fulfill these roles (18, 19). Ssc1 and Ssq1 both appear to require the presence of a specialized hsp70 escort protein Hep1 (also designated Zim17 and Tim15) to maintain their solubility and perform their functions. Saccharomyces cerevisiae containing an inactivated Hep1 exhibit a phenotype consistent with Ssc1 and Ssq1 depletion. This includes decreased import of nuclear-encoded proteins into the mitochondrial matrix, reduced activities of Fe/S proteins, and pleiotropic effects on mitochondrial morphology (20–23). These phenotypes are thought to arise, because yeast chaperones exhibit reduced solubility in the absence of Hep1.
Bacterial expression studies have provided evidence that eukaryotic escort proteins are sufficient to promote the solubility of their cognate hsp70 chaperones (23, 24). Coexpression of Ssc1 with Hep1 in Escherichia coli led to the production of soluble Ssc1, whereas expression of Ssc1 alone yielded insoluble chaperone (23). In addition, the Chlamydomonas reinhardtii chloroplast Hsp70B could only be produced as a soluble functional protein in bacteria when it was coexpressed with Hep2 (24). Currently, the nature of chaperone misfolding reactions and escort protein regulation of chaperone folding are unclear. Escort protein activity could arise from interactions with the Hsp70 peptide-binding domain, i.e. with Hep1 serving as a substrate for the chaperone. Alternatively, Hep1 escort activity could result from interactions with the Hsp70 ATPase domain. In addition, it is not clear if metazoan escort protein homologs can promote the solubility of their cognate chaperones similar to that observed in yeast and green algae, and it is not known whether escort proteins elicit effects on chaperone and nucleotide interactions.
To better understand escort protein regulation of chaperone folding and function, we have characterized the solubility of human mtHsp70, its isolated ATPase domain (designated 70ATPase), and human-bacterial chaperone chimeras. In addition, we have examined the effect of human Hep on the solubility of these proteins and characterized the effect of Hep on mtHsp70 and nucleotide interactions.
EXPERIMENTAL PROCEDURES
Materials—E. coli XL1-Blue and Rosetta 2 cells were from Stratagene and EMD Biosciences, respectively. Enzymes for DNA manipulation were obtained from Roche Applied Science, New England Biolabs, and Promega. Synthetic oligonucleotides were obtained from Fischer Scientific, and pET vectors were from EMD Biosciences. NuPAGE Novex 10% Bis-Tris2 gels from Invitrogen were used for all electrophoresis experiments. Bacterial growth media components were from BD Biosciences, and all other reagents were from Sigma-Aldrich.
Vectors—The gene encoding human mtHsp70 was amplified from an Invitrogen Ultimate ORF Human Clone (accession #BC0004788) using VENT DNA polymerase and cloned into pET21d(+) using NcoI and HindIII restriction sites to generate pHsp70, a vector that produces full-length mtHsp70. The mtHsp70 gene and a gene encoding the mtHsp70 ATPase domain (residues 47–440) were also cloned into pET21d(+) using NcoI and NotI to generate pHsp70-His and pATPase, respectively, vectors that produce mtHsp70 and its isolated ATPase domain with C-terminal His tags. All of these constructs produce mtHsp70 without its mitochondrial targeting sequence (25) but with an extra N-terminal methionine.
Gene fragments of human mtHsp70 and E. coli HscA were PCR-amplified from pHsp70 and E. coli genomic DNA, splicing by overlap extension was used to generate chimeric chaperone genes (26), and these full-length chimeras were cloned into pET21d(+) using NcoI and NotI restriction enzymes. The first chimera, designated A-70 (pA-70), contained the HscA ATPase domain (residues 1–391) and the mtHsp70 peptide-binding domain (PBD, residues 441–679). The second chimera, designated 70-A (p70-A) contained the mtHsp70 ATPase domain (residues 47–440) and the HscA PBD (residues 392–616).
A vector (pHep-EGFP) that produces human Hep fused to EGFP was generated by chemically synthesizing the predicted human Hep gene (accession #NM_001080849) and cloning it into pEGFP-N1 using BglII and HindIII restriction endonucleases. In addition, the gene fragment encoding Hep without its predicted mitochondrial targeting sequence (residues 1–49) was PCR-amplified and cloned into pET28b(+) and pET30a at NcoI and HindIII restriction sites. The pET28-derived vector (pHep) produces human Hep without an affinity tag, whereas the pET30-derived vector (pHis-Hep) produces human Hep with an N-terminal (His)6 tag. This tag can be removed with enterokinase to produce the predicted mitochondrial isoform of Hep with an Ala-Met at its N terminus. The S. cerevisiae hep1 gene was amplified from genomic DNA using PCR and cloned into pET28b(+) using NcoI and HindIII restriction sites to create pHep1, a vector that produces Hep1 (residues 48–174) without its mitochondrial targeting sequence (23).
The gene encoding human mitochondrial Isu2 was PCR-amplified from an Invitrogen plasmid (accession #BM921073) and cloned into pET30a using NcoI and HindIII restriction sites to create pHis-Isu2, a vector that produces Isu2 (residues 37–167) without its mitochondrial targeting sequence (27) but with an N-terminal (His)6 tag. All cloning and plasmid amplification was performed using a strain of E. coli (XL1-Blue, Stratagene Inc.) that lacks a T7 RNA polymerase, and all constructs were sequence-verified.
Solubility Analysis—Protein solubility was analyzed using Rosetta 2 E. coli grown in Luria broth. Cells transformed with vectors for expressing the indicated proteins were grown at 37 °C, induced with 0.1 mm isopropylthio-β-d-galactoside (IPTG) at A600 ≈ 1, and grown for ≈18 h at 23 °C to allow expression. In experiments expressing yeast Hep1, cells were only allowed to grow for 6 h after induction. Cells were harvested by centrifugation, resuspended in HED buffer (10 mm HEPES, pH 7.5, 0.5 mm EDTA, and 1 mm DTT) containing 1 mm MgCl2, 300 μg/ml lysozyme, 2 units/ml DNase I, and frozen at -80 °C for ≥24 h. The lysed cells were thawed and centrifuged at 40,000 × g to separate the insoluble and soluble proteins. The insoluble fractions were resuspended in HED buffer, and protein concentrations in each fraction were determined using Bradford analysis prior to SDS-PAGE analysis.
Protein Expression and Purification—Cells transformed with pHis-Hep were grown at 37 °C, induced with 0.1 mm IPTG at A600 ≈ 1 and grown for ≈18 h at 23 °C to allow for expression. Harvested cells were resuspended in TND buffer (50 mm Tris, pH 8.0, 150 mm NaCl, and 1 mm DTT) containing 1 mm MgCl2, 300 μg/ml lysozyme, and 2 units/ml DNase I. After two freeze-thaw cycles at -80 °C, lysed cells were centrifuged at 40,000 × g to remove cell debris. Cleared lysate was applied to nickel-nitrilotriacetic acid resin (Qiagen), the column was washed with TND buffer containing 15 mm imidazole, and (His)6-tagged Hep1 was eluted using TND buffer containing 150 mm imidazole. After dialysis against 10 mm Tris, pH 8.0, the fusion protein (15 mg/ml) was treated with 2 units/ml enterokinase (Promega) for 72 h at room temperature to remove the His tag and applied to Histrap (GE Healthcare) column to remove the affinity tag. Cleaved Hep was concentrated and chromatographed in TND buffer using a S75 Superdex column (GE Healthcare). Fractions appearing homogeneous were pooled and concentrated to >15 mg/ml.
Rosetta 2 E. coli harboring pHsp70-His and pHep1 were grown and lysed using a similar protocol as described for Hep purification, except that TNED buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 0.5 mm EDTA, and 1 mm DTT) was used to resuspend cell pellets. After lysis, cells were centrifuged at 40,000 × g to remove cell debris, cleared lysate was applied to nickel-nitrilotriacetic acid resin, and the column was washed with 10 column volumes of TNED buffer containing 500 mm NaCl, 0.5% Triton X-100, and 10 mm imidazole. This latter step was required to remove yeast Hep1 that remained bound to mtHsp70 on the nitrilotriacetic acid column. Chaperone was eluted using TNED buffer containing 150 mm imidazole, fractions containing protein were combined, and ammonium sulfate was added to a final concentration of 50% saturation to precipitate mtHsp70. The precipitated protein was resuspended in TED buffer (50 mm Tris, pH 8.0, 0.5 mm EDTA, and 1 mm DTT), centrifuged at 40,000 × g, dialyzed against TED buffer, and applied to a Q-Sepharose column. Protein was eluted from this anion exchange column using a linear gradient from 0 to 400 mm NaCl in TED buffer. Fractions appearing homogenous were pooled, dialyzed against TED buffer, and concentrated to ∼15 mg/ml. The mtHsp70 ATPase domain was expressed in Rosetta 2 E. coli harboring pATPase and pHep and purified using a procedure similar to that described for full-length mtHsp70.
Gel-filtration Chromatography—Protein molecular masses were estimated by comparing their elution to monomeric standards of known molecular weight on Superdex 75 and 200 columns, respectively, using an AKTA fast-protein liquid chromatography system (GE Healthcare). For experiments examining mtHsp70 oligomerization, 20 μm chaperone was incubated for 30 min at 4 °C in HKMD buffer (50 mm HEPES, pH 7.5, 150 mm KCl, 10 mm MgCl2, and 1 mm DTT) containing and lacking 1 mm ADP or ATP. This sample was applied to a Superdex 200 column equilibrated in HKMD buffer and having levels of nucleotides (50 μm) that allow for spectroscopic detection of the eluted proteins. In experiments examining Hep binding to mtHsp70, equimolar mtHsp70 (or its ATPase domain), and Hep were incubated in HKMD buffer for 30 min at 4 °C to allow for complex formation. Nucleotides were then added to a final concentration of 1 mm, and the mixture was further incubated for 30 min prior to chromatographic separation in HKMD buffer containing 50 μm ADP or ATP. For analysis of the absence of nucleotide, samples were immediately chromatographed in HKMD buffer after the 30-min incubation. The standard curves shown were generated using the elution volumes for: amylase (200 kDa), alcohol dehydrogenase (158 kDa), bovine serum albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome C (12.4 kDa).
Cell Culture—HEK293 cells were cultivated in Dulbecco's Modification of Eagle's media (Fisher) supplemented with 10% bovine calf serum. The cells were incubated at 37 °C in a 90% humidified atmosphere containing 5% CO2. Before transfection (1 day), cells were seeded in 6-well plates and transfection was done at a cell confluency of 70% by using Fugene6 transfection reagent (Roche Applied Science) according to the manufacturer's protocol. Mitochondria were stained 48 h after transfection by incubating cells with 50 nm MitoTracker Red (Molecular Probes) in phosphate-buffered saline at 23 °C for 30 min. After washing with phosphate-buffered saline two times, live cells were imaged using LSM-510 (Zeiss) confocal fluorescence microscope. All images are nearly confocal (thin optical slice, <1 μm).
ATPase Measurements—The rate of ATP hydrolysis was monitored as previously described using the Invitrogen EnzChek phosphate detection kit (9, 28, 29). Assays were performed in HKMD buffer that contained 1 mm DTT. Enzymes and coupled assay reagents were incubated in a 0.5-ml reaction at 25 °C for 5 min prior to starting the reaction through addition of 1 mm ATP. First order rates were corrected for the degradation of the coupled enzyme substrate, 2-amino-6-mercaptro-7-methylpurine ribonucleoside, as well as the level of phosphate present in ATP. Reaction rates obtained for mtHsp70 and its isolated ATPase domain were directly proportional to enzyme concentrations. Curves shown represent a least-squares fit of the data to a hyperbolic saturation function.
Analytical Methods—Protein concentrations were determined spectrophotometrically. The extinction coefficients of mtHsp70 (ε280 = 19,600 m-1cm-1), the mtHsp70 ATPase domain (ε280 = 16,800 m-1cm-1), and Hep (ε280 = 9,800 m-1cm-1) were calculated using average absorptivities for tryptophan and tyrosine of 5,600 and 1,400 (m-cm)-1, respectively (30–32). All UV-visible absorbance measurements were performed using a Cary 50 spectrophotometer.
RESULTS
Domain Requirements for mtHsp70 Insolubility—Human mtHsp70 contains two domains, an N-terminal ATPase domain and a C-terminal peptide-binding domain, but the domain responsible for its low solubility upon expression in E. coli is not known. To investigate the relative contributions of these domains to mtHsp70 insolubility, we have created bacterial expression vectors for mtHsp70 and chimeras of mtHsp70 and HscA, a constitutively expressed bacterial hsp70 family member that is soluble and monomeric (28). The first chimera, designated 70-A, contains an mtHsp70 ATPase domain and an HscA PBD. The second chimera, designated A-70, contains an HscA ATPase domain and an mtHsp70 PBD. In addition, we created an expression vector for a truncation mutant of mtHsp70 that contains only the ATPase domain.
Fig. 1 shows the expression of mtHsp70, 70-A, A-70, and 70ATPase in Rosetta 2 E. coli grown in Luria broth medium. In all cases, IPTG-induced expression led to major bands with apparent molecular weights similar to those predicted. Analysis of the soluble and insoluble cell fractions indicated that mtHsp70 is predominantly within the insoluble fraction. In addition, the 70-A chimera and mtHsp70 ATPase domain were found within the insoluble fractions of the cells. The A-70 chimera, in contrast, was soluble under similar expression conditions. Taken together, these results suggest that the mtHsp70 ATPase domain contributes to the low solubility of full-length mtHsp70 under these assay conditions.
FIGURE 1.
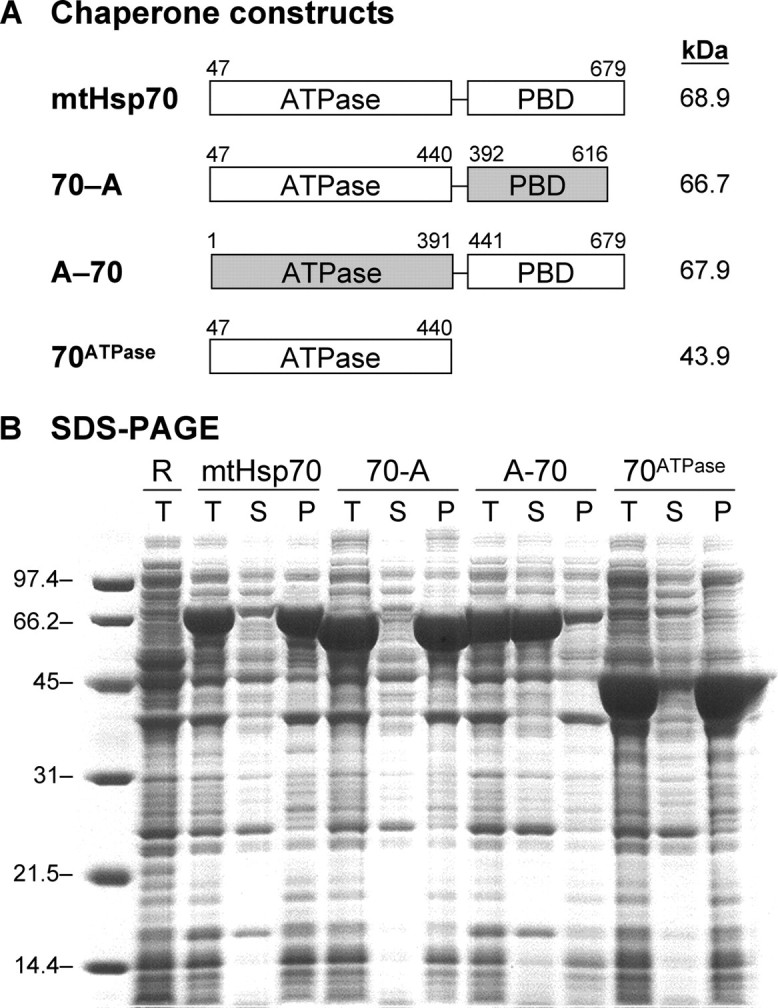
Solubility of mtHsp70, mtHsp70-HscA chimeras, and 70ATPase. A, domain composition of chaperone constructs. B, SDS-PAGE analysis of protein solubility upon expression in E. coli. Molecular weight markers (lane 1), Rosetta 2 E. coli lacking plasmids (lane 2), and cells harboring plasmids for expressing full-length mtHsp70 (pHsp70, lanes 3–5), a chimera having an mtHsp70 ATPase domain and a HscA peptide-binding domain (p70-A, lanes 6–8), a chimera having an HscA ATPase domain and a mtHsp70 PBD (pA-70, lanes 9–11), and the isolated mtHsp70 ATPase domain (pATPase, lanes 12–14). For each sample, 15 μg of total protein from whole cells (T) is shown as well as the soluble lysates (S) and pellets (P) derived from a sample that contained 15 μg of protein before fractionation.
Hep-EGFP Is Localized to Mitochondria—The N-terminal portion of Hep (residues 1–49) is predicted to constitute a mitochondrial localization signaling sequence (33), suggesting that this protein is translocated to mitochondria like the yeast escort protein Hep1 (20). To examine this, we created an expression vector that produced full-length Hep with EGFP fused to its C terminus and examined the localization of Hep-EGFP in HEK293 cells transiently transfected with this vector. Fig. 2 shows that the EGFP signal appears localized with that of MitoTracker Red, a dye that stains mitochondria by detecting the membrane potential.
FIGURE 2.

Mitochondrial localization of Hep. HEK293 cells transiently transfected with pHep-EGFP were stained with 50 nm MitoTracker Red for 30 min at 48 h after transfection. Live cells were imaged using a confocal microscope. A, EGFP channel representing Hep-EGFP fusion protein. B, bright field, C, Mito-Tracker Red channel representing mitochondria, and D, overlap of A, B, and C. The MitoTracker Red dye was excited with a 543 nm argon laser with emission collected through a 560 nm long-pass filter, and the EGFP was imaged using 488 excitation and emission detected through a 500 to 530 nm band pass. The scale bar represents 20 μm.
Hep Enhances mtHsp70 Solubility—Expression studies with yeast Ssc1 have shown that this chaperone can only be produced as a soluble recombinant protein in E. coli when it is coexpressed with yeast Hep1 (23), implicating escort proteins as sufficient to maintain mitochondrial chaperone solubility. To see if human Hep exhibits hsp70 escort activity, we have created a bacterial expression vector that produces the predicted mitochondrial isoform of Hep with a cleavable N-terminal His tag (33) and evaluated the escort activity of this recombinant protein.
Fig. 3A shows an SDS-PAGE of total protein from cells overexpressing Hep. A major band is observed within the soluble fraction that migrates with an apparent molecular mass that is heavier than that calculated for His-tagged Hep (18.6 kDa), indicating that Hep migrates slower than expected under these conditions. Previous studies analyzing yeast Hep1 migration on SDS-PAGE have found similar results (23), suggesting that abnormalities in SDS binding or protein conformation during electrophoresis are responsible for the slower than expected migration of these escort proteins. Fig. 3A also shows the effect of Hep on the solubility of mtHsp70, 70-A, and 70ATPase. In all three cases, more than half of the expressed chaperone was observed in the soluble cellular fraction. This can be contrasted with experiments performed in the absence of Hep, where we could not detect soluble mtHsp70, 70-A, or 70ATPase (see Fig. 1). These findings provide evidence that human Hep exhibits chaperone escort activity similar to that observed with yeast Hep1 (23). Furthermore, they suggest that Hep regulates chaperone solubility through interactions with the mtHsp70 ATPase domain.
FIGURE 3.
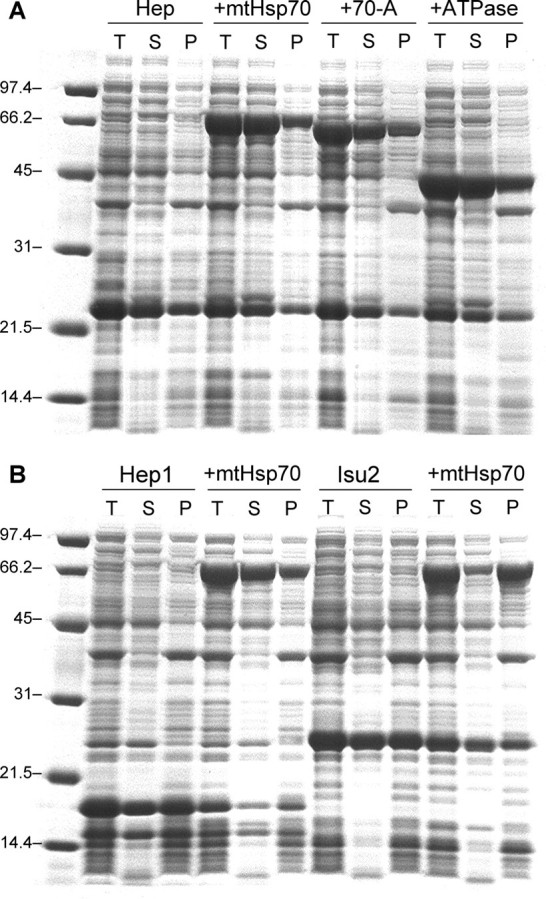
SDS-PAGE analysis of Hep escort activity. A, molecular weight markers (lane 1) and Rosetta 2 E. coli harboring a plasmid for expressing Hep alone (pHis-Hep, lanes 2–4) and with vectors for expressing mtHsp70 (pHsp70, lanes 5–7), the 70-A chimera (p70-A, lanes 8–10), and 70ATPase (pATPase, lanes 11–13). B, molecular weight markers (lane 1) and Rosetta 2 E. coli harboring plasmids for expressing yeast Hep1 (pHep1, lanes 2–4), Hep1 and mtHsp70 (pHep1 and pHsp70, lanes 5–7), Isu2 (pHis-Isu2, lanes 8–10), and Isu2 and mtHsp70 (pHis-Isu2 and pHsp70, lanes 11–13). For each sample, 15 μg of total protein from whole cells (T) is shown as well as the soluble lysates (S) and pellets (P) derived from a sample that contained the same amount of protein before fractionation.
Fig. 3B shows the effect of a non-cognate escort protein (S. cerevisiae Hep1) and a substrate (human Isu2) on the solubility of human mtHsp70 expressed in E. coli. IPTG-induced expression of both Hep1 and Isu2 led to major bands with apparent molecular weights slightly larger than those predicted for Isu2 (Mr = 19,200) and Hep1 (Mr = 14,600), as previously observed with yeast Hep1 (23) and a bacterial homolog of Isu2 (10). In addition, both proteins were predominantly in the soluble cell fractions. Although both proteins were expressed at similar levels, only the yeast escort protein Hep1 was able to promote human mtHsp70 to a similar extent as human Hep. In contrast, coexpression of mtHsp70 with Isu2 had little influence on chaperone solubility.
The slow migrations of Hep1 and Isu2 during electrophoresis are thought to arise from abnormalities in SDS binding or protein conformation, not retention of bound cofactors. Hep1 mutants having zinc-chelating cysteines mutated to serine exhibit similar migration during electrophoresis as native Hep1, even though these mutations abolish the solubility of recombinant Hep1 (data not shown). In addition, the Fe/S clusters coordinated by Isu2-type proteins are unstable in the presence of the oxygen levels where gel electrophoresis was performed (10).
Protein Expression and Purification—To obtain soluble mtHsp70 for purification, we coexpressed this chaperone with yeast Hep1 in E. coli. This approach was used because Hep1 promotes mtHsp70 solubility like human Hep, but it does not remain as strongly bound to the chaperone during chromatography, aiding in the separation of the escort protein from the chaperone during purification. In addition, the mtHsp70 ATPase domain was expressed in the presence of human Hep lacking a His tag. Hep lacking this affinity tag promotes 70ATPase and mtHsp70 solubility upon expression in E. coli, but it is expressed at lower levels than His-tagged Hep (data not shown). This lower expression aids in chromatographic separation of 70ATPase and Hep during purification.
Figs. 4A, 5A, and 6A show SDS-PAGE analysis of the final preparations of mtHsp70, Hep, and 70ATPase, respectively. In all cases, a single major band is obtained indicative of a high level of purity. In addition, we characterized the absorption spectrum of each protein. They exhibited spectra consistent with the known amino acid content of aromatic residues and showed no evidence for high levels of additional chromophoric groups, e.g. bound ADP/ATP that contributes significantly to absorption at 260 nm, as has been observed in purified preparations of other hsp70 chaperones like E. coli DnaK (34).
FIGURE 4.

Nucleotide effects on mtHsp70 oligomerization. A, SDS-PAGE analysis of purified mtHsp70: molecular weight markers (14.4, 21.5, 31, 45, 66.2, and 97.4 kDa) and 1, 3, and 10 μg of purified mtHsp70. B, elution profiles at 4 °C for 20 μm mtHsp70 (2 ml) chromatographed on a Superdex 200 column using HKMD buffer containing 50 μm ATP (top), 50 μm ADP (middle), and no nucleotide (bottom).
FIGURE 5.

Hep inhibits mtHsp70 oligomerization. A, SDS-PAGE analysis of purified mtHsp70: molecular weight markers (14.4, 21.5, 31, 45, 66.2, and 97.4 kDa) and 1, 3, and 10 μg of purified Hep. B, effect of mixing Hep and mtHsp70 on their elution at 4 °C from a Superdex 200 column chromatographed using HKMD buffer lacking nucleotides and containing ATP or ADP (50 μm). Samples (2 ml) introduced onto the column contained 20 μm Hep and mtHsp70.
FIGURE 6.

Hep binds to the mtHsp70 ATPase domain. A, SDS-PAGE analysis of purified mtHsp70 ATPase domain: molecular weight markers (14.4, 21.5, 31, 45, 66.2, and 97.4 kDa) and 1, 3, and 10 μg of purified 70ATPase. B, elution profiles at 4 °C for 70ATPase and Hep chromatographed alone and together on a Superdex 75 column using HKMD buffer lacking nucleotides and containing ATP or ADP (50 μm). Samples (2 ml) introduced onto the column contained 20 μm Hep and 70ATPase.
Subsequent to purification, mtHsp70 and 70ATPase remained soluble in the absence of Hep and nucleotides when stored in Tris pH 8. In the case of the ATPase domain, the protein remained soluble at concentrations as high as 200 μm even when stored for 24 h at room temperature. However, the mtHsp70 ATPase domain could not be stably stored at high concentrations for a similar period of time in buffers having pH values (= 7.5) more closely resembling the physiological environment within E. coli where these proteins were overexpressed, unless it was incubated with equimolar Hep (data not shown).
Gel-filtration Analysis—Hsp70 binding to ATP results in a conformation change that leads to the formation of a tense state with reduced affinity for peptide substrates, and subsequent ATP hydrolysis to ADP results in formation of a relaxed state with increased substrate affinity (5–10). For some chaperones, like yeast mitochondrial Ssc1, these nucleotide-induced conformational changes influence the oligomeric state of the chaperone (23), suggesting that human mtHsp70 may also self-associate in a nucleotide-dependent manner. To evaluate this possibility, we have investigated mtHsp70 migration on a gel-filtration column in the presence and absence of ATP and ADP.
Fig. 4B shows elution profiles for mtHsp70 on a Superdex 200 size-exchange column. In the presence of 50 μm ATP, a single major peak was observed. A comparison of this peak elution volume to protein standards of known size indicates that mtHsp70 exhibits an apparent molecular mass (81 kDa) that is similar to the calculated mass for a mtHsp70 monomer. In the presence of ADP, two major peaks were observed at elution volumes with predicted molecular masses (86 and 168 kDa), consistent with the presence of mtHsp70 monomers and dimers. Nucleotide-free mtHsp70 also contained a mixture of monomeric and dimeric mtHsp70. Under these conditions, however, a larger fraction of the mtHsp70 eluted near the void volume (43.9 ml) of this column. This indicates that mtHsp70 forms higher order oligomers in the absence of nucleotides.
To evaluate whether Hep influences mtHsp70 oligomerization, we investigated whether Hep altered mtHsp70 elution on a Superdex 200 size-exclusion column. Fig. 5B compares the elution of Hep and a mixture of Hep and mtHsp70. In the absence of nucleotide, the protein mixture eluted as a single major peak. This can be contrasted with elution of mtHsp70 alone, which eluted as a mixture of monomers, dimers, and higher order oligomers (see Fig. 4B). A comparison of the elution volume for this peak to that of the standards indicates that the Hep-mtHsp70 complex exhibits an apparent molecular mass (124 kDa) that is less than that expected for a mtHsp70 dimer, suggesting that Hep promotes formation of mtHsp70 monomers. Hep alone eluted as a monodisperse peak at an apparent molecular mass (29 kDa) that is ∼2-fold greater than that calculated for a Hep monomer after removal of its His tag (13.6 kDa).
To determine whether Hep forms a stable complex with the mtHsp70 ATPase domain, we examined whether Hep altered 70ATPase elution from a Superdex 75 size-exclusion column. Fig. 6B shows a comparison of the peaks obtained to protein standards. When chromatographed alone, 70ATPase displayed an apparent molecular mass (∼54 kDa) similar to that calculated for a monomer (43.9 kDa). This indicates that this domain does not self-associate like the full-length protein (see Fig. 4B). In addition, an equimolar mixture of 70ATPase and Hep eluted at a single volume greater than that observed for 70ATPase or Hep alone. Under these conditions, little free Hep or 70ATPase was detected. This suggests that these proteins bind with a stoichiometry of 1:1, because non-stoichiometric binding for an equimolar mixture would have yielded two major peaks, one representing the 70ATPase that forms a complex with Hep and the other representing the remaining free 70ATPase. The finding that Hep migrates with an apparent molecular weight similar to that of a dimer further suggests that Hep must dissociate prior to binding to 70ATPase, or that Hep has an asymmetrical shape that causes it to migrate more rapidly during gel-filtration chromatography than a symmetrical monomer.
Nucleotide Effects on Hep Binding—The finding that Hep binds directly to the mtHsp70 ATPase domain suggested that nucleotide-induced conformational changes could influence the stability of the Hep-mtHsp70 complex. To address this, gel-filtration analyses of the escort-chaperone complexes were repeated in buffers containing ADP and ATP. Fig. 5B shows that the mtHsp70-Hep mixture eluted as two peaks in the presence of ATP. This can be contrasted with experiments performed in buffer lacking nucleotides, where mtHsp70 and Hep eluted together as a single major peak. In the presence of ATP, the faster peak eluted at a volume identical to that observed for mtHsp70-ATP chromatographed alone (see Fig. 4B), and the slower peak eluted at a volume identical to that observed when Hep was analyzed alone. In the presence of ADP, multiple peaks were also observed. Again, elution of these peaks occurred at volumes similar to those observed in experiments analyzing the migration of the individual proteins.
We also investigate whether nucleotides promoted dissociation of Hep from the mtHsp70 ATPase domain. Fig. 6B shows the effects of ADP and ATP on the elution of Hep and 70ATPase mixtures from a size-exclusion column. In an ATP-containing buffer, the Hep-70ATPase mixture eluted as two distinct peaks at volumes corresponding to the elution observed when experiments were performed with the individual proteins. This can be contrasted with experiments performed in the absence of nucleotides where the Hep-70ATPase complex eluted as a single peak at a smaller volume. In addition, gel-filtration analysis in a buffer containing ADP resulted in multiple elution peaks. However, the two peaks observed under these conditions were not as resolved as in the presence of ATP, suggesting that ADP-bound 70ATPase binds Hep stronger than ATP-bound protein.
ATPase Activity—To investigate whether Hep affects mtHsp70 and nucleotide interactions, we examined the effect of Hep on the ATPase activity of mtHsp70 and its isolated ATPase domain using a coupled enzyme assay for phosphate release (9, 28, 29). Fig. 7A shows the effect of 40 μm Hep on the rate of ATP hydrolysis catalyzed by 70ATPase (2 μm) in the presence of 1 mm ATP. Under these conditions, Hep increased the activity of 70ATPase ∼11-fold from 0.0054 to 0.062 mol of ATP hydrolyzed per mol of 70ATPase per second. Purified Hep alone exhibited a low background level of ATPase activity under these conditions (6 × 10-5 s-1).
FIGURE 7.
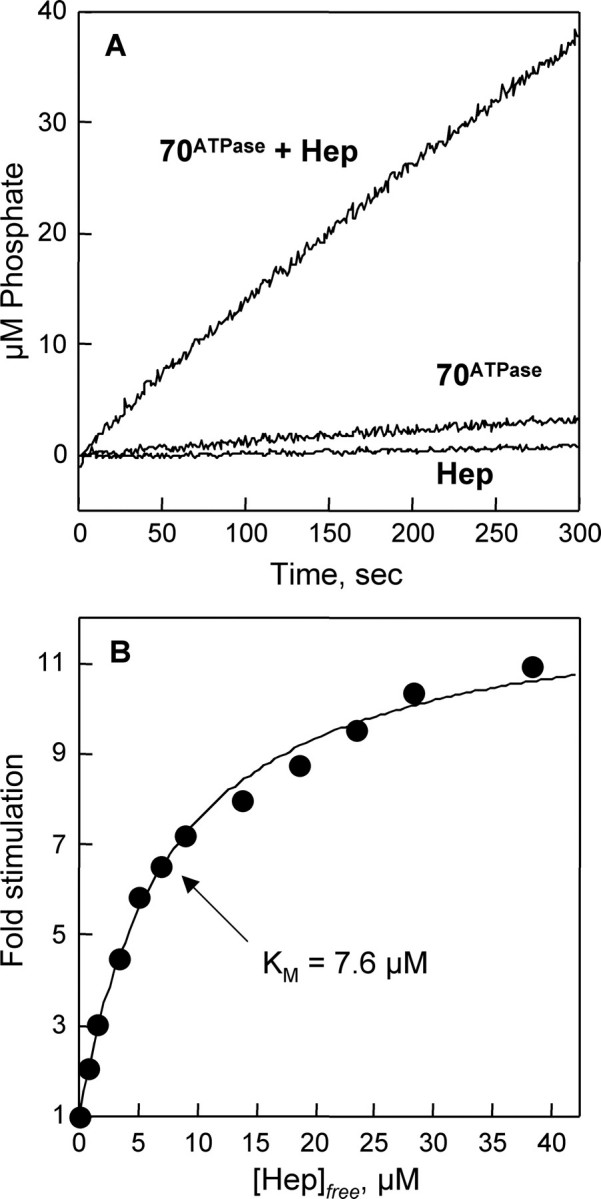
Effect of Hep on the ATPase activity of 70ATPase. A, time course of ATP hydrolysis at 25 °C in reactions containing 2 μm 70ATPase, a mixture of 2 μm 70ATPase and 40 μm Hep, and 50 μm Hep. B, increase in the basal ATPase rate of 70ATPase (2 μm) at 25 °C in the presence of 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, and 40 μm Hep. All experiments were performed in HKMD buffer that contained 1 mm ATP. The concentration of free Hep was calculated assuming 1:1 binding stoichiometry with 70ATPase using the equation, [Hep]free = [Hep]total - Δv·[E]/Δvmax, where [Hep]total is the concentration of Hep added to the reaction, [E] is the concentration of 70ATPase, Δv is the observed change in rate, and Δvmax is the maximal rate obtained by extrapolating to infinite Hep concentration. The data are fit to a hyperbolic saturation function assuming a maximal stimulation for 70ATPase of 11.5-fold with half-maximal stimulation at 7.6 μm. Hep (50 μm) yielded a turnover number (0.00006 s-1) that was ∼100-fold lower than the basal rate measured for 70ATPase (0.0054 s-1).
To determine the affinity of the mtHsp70 ATPase domain for Hep, ATPase measurements were carried out over a range of concentrations of Hep. Fig. 7B shows the increase in ATPase activity relative to the rate in the absence of Hep as a function of Hep concentration. Assuming a 1:1 stoichiometry for the binding of Hep to 70ATPase, as predicted from size-exclusion chromatography measurements (see Fig. 6B) and observed during calorimetric analysis of Hep and 70ATPase binding (data not shown), a hyperbolic saturation curve was obtained when the data were corrected for the amount of Hep bound. Extrapolation to saturating levels of Hep indicates a maximal stimulation of 11.5-fold with half-maximal stimulation at 7.6 μm Hep.
Fig. 8A shows the ATPase activity of full-length mtHsp70 (10 μm) in the absence and presence of 40 μm Hep. The basal activity of mtHsp70 (6.8 ± 0.2 × 10-4 mol ATP hydrolyzed per mol per mtHsp70 per second) was less than that of the isolated ATPase domain, as observed with other hsp70 family members (9). In addition, Hep (40 μm) elicited a greater stimulation (∼20-fold) of full-length mtHsp70 under these conditions. Furthermore, biphasic ATP hydrolysis kinetics was observed when reactions were started by adding ATP to mixtures of mtHsp70 and Hep that had been preincubated for 5 min. This can be contrasted with ATP hydrolysis by 70ATPase in the presence of Hep, which was linear over the time course of the assay. Reactions involving full-length mtHsp70 yielded linear rates when mtHsp70 was incubated with ATP prior to Hep addition (data not shown).
FIGURE 8.

Effect of Hep on the ATPase activity of mtHsp70. A, time course of ATP hydrolysis at 25 °C in reactions containing 10 μm mtHsp70 and a mixture of 10 μm mtHsp70 and 40 μm Hep. B, the effect of increasing Hep on the ATPase rate of mtHsp70 (10 μm) observed during the fast kinetic phase (0–30 s). C, the effect of increasing Hep on the ATPase rate of mtHsp70 (10 μm) observed after reactions had proceeded for 150 s. Reactions were performed in HKMD buffer containing 10 μm mtHsp70, and 0, 5, 10, 15, 20, 25, 30, 40, 50, 60, 75, and 100 μm Hep. All reaction components were mixed and incubated at 25 °C for 5 min prior to starting reactions through the addition of ATP to a final concentration of 1 mm. The concentration of free Hep was calculated assuming a 1:1 binding stoichiometry as described in Fig. 8, and the data in B and C were fit to a hyperbolic saturation functions assuming a maximal stimulation for the faster kinetic phase of 49-fold with half-maximal stimulation at 5 μm, and a maximal stimulation for the slower kinetic phase of 27.6-fold with half-maximal stimulation at 26 μm.
To examine whether mtHsp70 exhibits a similar affinity for Hep as 70ATPase, we examined the effect of Hep concentration on mtHsp70 ATPase activity. Fig. 8B shows the increase in the rate of ATP hydrolysis during the first 30 s of the reactions relative to the rate in the absence of Hep as a function of Hep concentration. In all reactions containing Hep, the fast phase exhibited linear rates over this time course. Extrapolation to saturating levels of Hep indicates a maximal stimulation of mtHsp70 activity (∼49-fold) that is greater than that observed with the ATPase domain alone. However, the concentration of Hep required for half-maximal stimulation (5 μm Hep) is similar to that observed with 70ATPase. Fig. 8C shows the effect of Hep on mtHsp70 activity after the reaction was allowed to proceed for 2 min. During this time, Hep stimulated mtHsp70 activity to a lower maximal extent (27.6-fold), and the concentration of Hep required for half-maximal stimulation was ∼5-fold greater (∼26 μm) than that observed when analyzing the initial rates. In all reactions, ATP hydrolysis rates were linear between 2 and 5 min.
DISCUSSION
In earlier studies, escort proteins from yeast mitochondria (Hep1) and algae chloroplasts (Hep2) were shown to maintain their cognate chaperones in an active conformation by preventing chaperone aggregation (20–22, 24). However, the domains responsible for aggregation and escort protein binding were not established. Our results herein provide evidence that the N-terminal ATPase domain of human mtHsp70 is responsible for its reduced solubility. Like full-length mtHsp70, a truncation mutant containing only the ATPase domain was insoluble when overexpressed in E. coli, as was the 70-A chimera containing the human mtHsp70 ATPase domain fused to the C-terminal peptide-binding domain of E. coli HscA, a monomeric hsp70-family member that has evolved to regulate Fe/S-cluster biosynthesis reactions (3, 28). In contrast, a chimera having the mtHsp70 PBD fused to an HscA ATPase domain was completely soluble upon overexpression in E. coli. These findings are consistent with studies performed by Craig and coworkers (2), which showed that an mtHsp70 truncation mutant having only the C-terminal PBD can be readily expressed as a soluble protein in the absence of Hep.
Our findings also provide the first direct evidence that escort proteins promote chaperone solubility through interactions with the ATPase domain. Hep enhanced the solubility of full-length mtHsp70, a truncation mutant containing only the ATPase domain, and the 70-A chimera that contained a human mtHsp70 ATPase domain. In contrast, mtHsp70 solubility was not dramatically altered by coexpression with Isu2. In previous studies, Isu2-type proteins have been shown to serve as substrates for mitochondrial hsp70 family members (9, 35, 36). In addition, a recent study showed that the C-terminal peptide-binding domain of mtHsp70 binds to a polypeptide harboring a LPPVK motif, the minimal portion of Isu2-type proteins that is required for chaperone binding (2, 37). The low chaperone escort activity elicited by Isu2 suggests that efficient suppression of mtHsp70 aggregation requires a protein that interacts with its N-terminal ATPase domain.
To our surprise, we found that S. cerevisiae Hep1 could promote the solubility of human mtHsp70. The ability of yeast Hep1 to cross-react with human mtHsp70 suggests that mammalian and yeast escort protein homologs have similar topology (38), even though they display only 25% amino acid sequence identity. This also suggests that these escort protein homologs use a similar mechanism to bind their cognate chaperones, and it implicates a role for residues conserved among these homologs in mediating chaperone interactions. Support for this comes from a recent study evaluating the solubility and escort activity of yeast Hep1 mutants having residues conserved in the human homolog mutated, including His-107 and Asp-111 (38). Yeast Hep1 variants harboring these mutations all exhibited parent-like solubility when produced in yeast and E. coli, suggesting that these residues are not required for proper folding. In addition, Hep1 variants containing the D111A mutation (or a H107A mutation in combination with R106A) were impaired in their ability to promote the solubility of Ssc1 overexpressed in E. coli, and they could not support the growth of Δhep1 S. cerevisiae (38).
Gel-filtration studies evaluating chaperone and escort protein binding revealed that mtHsp70-Hep and 70ATPase-Hep complexes are most stable in the absence of nucleotides. These measurements also revealed that Hep inhibits the self-association of mtHsp70 in the absence of nucleotides. This self-association is thought to arise at least in part from mtHsp70-mtHsp70 interactions involving the ATPase and substrate-binding domains of different chaperone molecules, because 70ATPase did not self-associate in buffers lacking nucleotides. The former mirrors findings from a study examining the effect of nucleotides and Hep on the oligomerization of the yeast mitochondrial chaperone Ssc1 (23). In the absence of nucleotide, glutaraldehyde cross-linking treatment generated Ssc1 that migrated on SDS-PAGE with a molecular weight consistent with the presence of four or more proteins per complex. However, in the presence of Hep1 (or ADP and ATP), a majority of the Ssc1 was monomeric or dimeric, indicating that Hep1, ADP, and ATP inhibit the formation of higher order Ssc1 oligomers.
Purified mtHsp70 exhibited slow intrinsic ATPase activity (6.8 ± 0.2 × 10-4 s-1) characteristic of hsp70 family members (39), and this activity was enhanced by Hep. This implicates a role for Hep-type escort proteins in regulating chaperone and nucleotide interactions. This stimulation is predicted to arise at least in part from Hep binding to the ATPase domain of mtHsp70, because Hep was capable of stimulating the ATP hydrolysis activity of the isolated ATPase domain (up to 11.5-fold) and full-length mtHsp70 (up to 49-fold). Hep activation of mtHsp70 activity could arise from Hep stimulating the rate of ATP hydrolysis, similar to that reported for J-type auxiliary cochaperones (40, 41). Alternatively, Hep could enhance the rate of nucleotide exchange, similar to GrpE-type cochaperones (42). Both J and GrpE-type family members are present in the mammalian mitochondria (43–45), suggesting that Hep may cooperate or compete with these proteins in regulating mtHsp70 ATPase activity.
Full-length mtHsp70 exhibited linear ATP hydrolysis kinetics in the absence of Hep. In contrast, biphasic kinetics was observed in reactions involving Hep and mtHsp70. These two linear kinetic phases were stimulated to different extents by Hep. In the case of the fast phase, the rate of ATP hydrolysis was enhanced up to 49-fold with half-maximal stimulation occurring at 5 μm Hep. In contrast, the rate of ATP hydrolysis during the slower phase was only stimulated up to 27.6-fold with a higher concentration (26 μm) of Hep required for half-maximal stimulation. The stronger apparent affinity for the fast kinetic phase suggests that mtHsp70 and Hep form a complex in the absence of nucleotides that has a distinct conformational state from ATP-bound mtHsp70. In addition, the higher stimulation observed for the fast kinetic phase suggests that Hep directly stimulates the rate of ATP hydrolysis ∼49-fold during the first round of substrate turnover, and it implicates a subsequent ATPase reaction cycle step as rate-limiting during subsequent rounds of ATP hydrolysis. Additional studies are needed to establish the exact role that Hep plays in regulating the individual steps of the mtHsp70 ATPase reaction cycle.
The ability to produce high levels of functional, recombinant human mtHsp70 will facilitate future studies examining how the activities of this chaperone differ from Ssc1 and Ssq1, the current model systems for mitochondrial chaperone function. Not only will it enable studies that directly examine binding and regulation by auxiliary cochaperones and substrates, but it will allow studies that investigate the structural features of mtHsp70 that mediate binding to Hep. Human mtHsp70 is predicted to recognize a structured motif within Hep (38) using its ATPase domain, and it seems likely that auxiliary cochaperones (43–45), protein substrates (27), and post-translational modifications (46) could influence this interaction.
Acknowledgments
We thank Ryan M. McGuire and Robert M. Raphael for help with confocal imaging and Aruna Mahendravada for technical support.
This work was supported by the Robert A. Welch Foundation (Grant C-1614 to J. J. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Bis-Tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; Hep, hsp70 escort protein; mtHsp70, mitochondrial hsp70; PBD, peptide-binding domain; 70ATPase, mtHsp70 ATPase domain; 70-A, mtHsp70-HscA chimera having mtHsp70 ATPase domain; A-70, HscA-mtHsp70 chimera having HscA ATPase domain; DTT, dithiothreitol; IPTG, isopropylthio-β-d-galactoside; EGFP, enhanced green fluorescent protein.
References
- 1.Mayer, M. P., and Bukau, B. (2005) Cell Mol. Life Sci. 62 670-684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schilke, B., Williams, B., Knieszner, H., Pukszta, S., D'Silva, P., Craig, E. A., and Marszalek, J. (2006) Curr. Biol. 16 1660-1665 [DOI] [PubMed] [Google Scholar]
- 3.Vickery, L. E., and Cupp-Vickery, J. R. (2007) Crit. Rev. Biochem. Mol. Biol. 42 95-111 [DOI] [PubMed] [Google Scholar]
- 4.Young, J. C., Agashe, V. R., Siegers, K., and Hartl, F. U. (2004) Nat. Rev. Mol. Cell. Biol. 5 781-791 [DOI] [PubMed] [Google Scholar]
- 5.Palleros, D. R., Reid, K. L., Shi, L., Welch, W. J., and Fink, A. L. (1993) Nature 365 664-666 [DOI] [PubMed] [Google Scholar]
- 6.Schmid, D., Baici, A., Gehring, H., and Christen, P. (1994) Science 263 971-973 [DOI] [PubMed] [Google Scholar]
- 7.McCarty, J. S., Buchberger, A., Reinstein, J., and Bukau, B. (1995) J. Mol. Biol. 249 126-137 [DOI] [PubMed] [Google Scholar]
- 8.Takeda, S., and McKay, D. B. (1996) Biochemistry 35 4636-4644 [DOI] [PubMed] [Google Scholar]
- 9.Silberg, J. J., Hoff, K. G., Tapley, T. L., and Vickery, L. E. (2001) J. Biol. Chem. 276 1696-1700 [DOI] [PubMed] [Google Scholar]
- 10.Hoff, K. G., Silberg, J. J., and Vickery, L. E. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 7790-7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matouschek, A., Pfanner, N., and Voos, W. (2000) EMBO Rep. 1 404-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neupert, W., and Brunner, M. (2002) Nat. Rev. Mol. Cell. Biol. 3 555-565 [DOI] [PubMed] [Google Scholar]
- 13.Schneider, H. C., Berthold, J., Bauer, M. F., Dietmeier, K., Guiard, B., Brunner, M., and Neupert, W. (1994) Nature 371 768-774 [DOI] [PubMed] [Google Scholar]
- 14.Dutkiewicz, R., Marszalek, J., Schilke, B., Craig, E. A., Lill, R., and Muhlenhoff, U. (2006) J. Biol. Chem. 281 7801-7808 [DOI] [PubMed] [Google Scholar]
- 15.Muhlenhoff, U., Gerber, J., Richhardt, N., and Lill, R. (2003) EMBO J. 22 4815-4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muhlenhoff, U., Richhardt, N., Gerber, J., and Lill, R. (2002) J. Biol. Chem. 277 29810-29816 [DOI] [PubMed] [Google Scholar]
- 17.Kang, P. J., Ostermann, J., Shilling, J., Neupert, W., Craig, E. A., and Pfanner, N. (1990) Nature 348 137-143 [DOI] [PubMed] [Google Scholar]
- 18.Kaul, S. C., Deocaris, C. C., and Wadhwa, R. (2007) Exp. Gerontol. 42 263-274 [DOI] [PubMed] [Google Scholar]
- 19.Daugaard, M., Rohde, M., and Jaattela, M. (2007) FEBS Lett. 581 3702-3710 [DOI] [PubMed] [Google Scholar]
- 20.Burri, L., Vascotto, K., Fredersdorf, S., Tiedt, R., Hall, M. N., and Lithgow, T. (2004) J. Biol. Chem. 279 50243-50249 [DOI] [PubMed] [Google Scholar]
- 21.Sanjuan Szklarz, L. K., Guiard, B., Rissler, M., Wiedemann, N., Kozjak, V., van der Laan, M., Lohaus, C., Marcus, K., Meyer, H. E., Chacinska, A., Pfanner, N., and Meisinger, C. (2005) J. Mol. Biol. 351 206-218 [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto, H., Momose, T., Yatsukawa, Y., Ohshima, C., Ishikawa, D., Sato, T., Tamura, Y., Ohwa, Y., and Endo, T. (2005) FEBS Lett. 579 507-511 [DOI] [PubMed] [Google Scholar]
- 23.Sichting, M., Mokranjac, D., Azem, A., Neupert, W., and Hell, K. (2005) EMBO J. 24 1046-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willmund, F., Hinnenberger, M., Nick, S., Schulz-Raffelt, M., Muhlhaus, T., and Schroda, M. (2008) J. Biol. Chem. 283 16363-16373 [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharyya, T., Karnezis, A. N., Murphy, S. P., Hoang, T., Freeman, B. C., Phillips, B., and Morimoto, R. I. (1995) J. Biol. Chem. 270 1705-1710 [DOI] [PubMed] [Google Scholar]
- 26.Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J. K., and Pease, L. R. (1989) Gene (Amst.) 77 61-68 [DOI] [PubMed] [Google Scholar]
- 27.Tong, W. H., and Rouault, T. (2000) EMBO J. 19 5692-5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vickery, L. E., Silberg, J. J., and Ta, D. T. (1997) Protein Sci. 6 1047-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silberg, J. J., Hoff, K. G., and Vickery, L. E. (1998) J. Bacteriol. 180 6617-6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gill, S. C., and von Hippel, P. H. (1989) Anal. Biochem. 182 319-326 [DOI] [PubMed] [Google Scholar]
- 31.Mach, H., Middaugh, C. R., and Lewis, R. V. (1992) Anal. Biochem. 200 74-80 [DOI] [PubMed] [Google Scholar]
- 32.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995) Protein Sci. 4 2411-2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claros, M. G., and Vincens, P. (1996) Eur. J. Biochem. 241 779-786 [DOI] [PubMed] [Google Scholar]
- 34.Russell, R., Jordan, R., and McMacken, R. (1998) Biochemistry 37 596-607 [DOI] [PubMed] [Google Scholar]
- 35.Cupp-Vickery, J. R., Peterson, J. C., Ta, D. T., and Vickery, L. E. (2004) J. Mol. Biol. 342 1265-1278 [DOI] [PubMed] [Google Scholar]
- 36.Dutkiewicz, R., Schilke, B., Knieszner, H., Walter, W., Craig, E. A., and Marszalek, J. (2003) J. Biol. Chem. 278 29719-29727 [DOI] [PubMed] [Google Scholar]
- 37.Hoff, K. G., Ta, D. T., Tapley, T. L., Silberg, J. J., and Vickery, L. E. (2002) J. Biol. Chem. 277 27353-27359 [DOI] [PubMed] [Google Scholar]
- 38.Momose, T., Ohshima, C., Maeda, M., and Endo, T. (2007) EMBO Rep. 8 664-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silberg, J. J., and Vickery, L. E. (2000) J. Biol. Chem. 275 7779-7786 [DOI] [PubMed] [Google Scholar]
- 40.Misselwitz, B., Staeck, O., and Rapoport, T. A. (1998) Mol. Cell 2 593-603 [DOI] [PubMed] [Google Scholar]
- 41.Silberg, J. J., Tapley, T. L., Hoff, K. G., and Vickery, L. E. (2004) J. Biol. Chem. 279 53924-53931 [DOI] [PubMed] [Google Scholar]
- 42.Liberek, K., Marszalek, J., Ang, D., Georgopoulos, C., and Zylicz, M. (1991) Proc. Natl. Acad. Sci. U. S. A. 88 2874-2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borges, J. C., Fischer, H., Craievich, A. F., Hansen, L. D., and Ramos, C. H. (2003) J. Biol. Chem. 278 35337-35344 [DOI] [PubMed] [Google Scholar]
- 44.Oliveira, C. L., Borges, J. C., Torriani, I. L., and Ramos, C. H. (2006) Arch. Biochem. Biophys. 449 77-86 [DOI] [PubMed] [Google Scholar]
- 45.Qiu, X. B., Shao, Y. M., Miao, S., and Wang, L. (2006) Cell Mol. Life Sci. 63 2560-2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanai, M., Ma, Z., Izumi, H., Kim, S. H., Mattison, C. P., Winey, M., and Fukasawa, K. (2007) Genes Cells 12 797-810 [DOI] [PubMed] [Google Scholar]