

Abstract
Many neurodegenerative diseases including Alzheimer, Parkinson, and polyglutamine (polyQ) diseases are thought to be caused by protein misfolding. The polyQ diseases, including Huntington disease and spinocerebellar ataxias (SCAs), are caused by abnormal expansions of the polyQ stretch in disease-causing proteins, which trigger misfolding of these proteins, resulting in their deposition as inclusion bodies in affected neurons. Although genetic expression of molecular chaperones has been shown to suppress polyQ protein misfolding and neurodegeneration, toward developing a therapy, it is ideal to induce endogenous molecular chaperones by chemical administration. In this study, we assessed the therapeutic effects of heat shock transcription factor 1 (HSF1)-activating compounds, which induce multiple molecular chaperones, on polyQ-induced neurodegeneration in vivo. We found that oral administration of 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) markedly suppresses compound eye degeneration and inclusion body formation in a Drosophila model of SCA. 17-AAG also dramatically rescued the lethality of the SCA model (74.1% rescue) and suppressed neurodegeneration in a Huntington disease model (46.3% rescue), indicating that 17-AAG is widely effective against various polyQ diseases. 17-AAG induced Hsp70, Hsp40, and Hsp90 expression in a dose-dependent manner, and the expression levels correlated with its therapeutic effects. Furthermore, knockdown of HSF1 abolished the induction of molecular chaperones and the therapeutic effect of 17-AAG, indicating that its therapeutic effects depend on HSF1 activation. Our study indicates that induction of multiple molecular chaperones by 17-AAG treatment is a promising therapeutic approach for a wide range of polyQ diseases and possibly other neurodegenerative diseases.
The accumulation and deposition of misfolded proteins in the brain has been recognized as a common molecular pathogenesis of various neurodegenerative diseases including Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis, and the polyglutamine (polyQ)4 diseases, and hence these diseases are called protein misfolding diseases (1). Indeed, most genetic mutations responsible for these diseases produce mutant proteins that are prone to be misfolded. These facts strongly indicate that protein misfolding commonly occurs as the initial step in the pathogenic cascade of neurodegenerative diseases, and hence protein misfolding is considered to be a common therapeutic target for these neurodegenerative diseases.
The polyQ diseases are a group of inherited neurodegenerative diseases including Huntington disease, various types of spinocerebellar ataxia (SCA1, 2, 6, 7, and 17 and SCA3/MJD), dentatorubral-pallidoluysian atrophy, and spinobulbar muscular atrophy, all of which are caused by expansions of the polyQ stretch to greater than 35-40 repeats in each disease-causing protein (2, 3). These expanded polyQ stretches cause misfolding of the disease-causing proteins, leading to their pathogenic interactions with themselves (aggregation) or other cellular proteins, resulting in their deposition and recruitment as inclusion bodies in affected neurons (1). The pathogenic interactions of misfolded polyQ proteins with other cellular proteins such as transcription factors, proteasome subunits, and cytoskeletal proteins have been reported to cause dysfunction of these proteins, eventually leading to neuronal dysfunction (4-6). Although therapeutic approaches against the dysfunction of each of these cellular proteins have been proposed to date, such as histone deacetylase inhibitors to improve transcriptional dysregulation (7), their therapeutic effects were limited because polyQ-induced neuronal dysfunction results from the dysfunction of multiple cellular proteins (2). In contrast, misfolding of the polyQ protein is likely the initial event in the pathogenic cascade, and hence suppression of protein misfolding is expected to inhibit a wide range of multiple downstream events, resulting in the most effective suppression of neuronal dysfunction.
Molecular chaperones are known to suppress protein misfolding by synergistically assisting misfolded proteins in the refolding process, as well as newly synthesized proteins in the folding process. For example, Hsp70 has been reported to act in concert with Hsp40 to suppress misfolding of various polyQ proteins in vitro, resulting in suppression of their aggregation (8, 9). In a Drosophila model of the polyQ diseases, co-expression of Hsp70 and Hsp40 has been reported to synergistically suppress misfolding of the polyQ protein, resulting in remarkable suppression of polyQ-induced neurodegeneration, although expression of Hsp70 or Hsp40 alone exhibits weaker suppression (10-12). Furthermore, genetic expression of Hsp70 or Hsp40 alone is known to cause cytotoxicity to unstressed cells, which is not observed when both chaperones are co-expressed (10, 13), suggesting that the balance of the amounts of molecular chaperones is important for their proper function (14). In addition, expression of other molecular chaperones such as Hsp90, Hsp105, and Hsp27 is also reported to suppress polyQ-induced cytotoxicity in cell culture models (15-17). Based on these results, expression of multiple molecular chaperones is expected to synergistically suppress polyQ-induced neurodegeneration and to have low cytotoxicity to unstressed cells.
Expression of molecular chaperones upon exposure to various types of cellular stress is known to be regulated by heat shock transcription factor 1 (HSF1). Under unstressed conditions, HSF1 is localized in the cytosol and is inactivated in a protein complex including Hsp90 (18). Upon exposure to stress, HSF1 dissociates from the Hsp90 protein complex, translocates into the nucleus, and binds to the heat shock element in the promoter region of various molecular chaperone genes to simultaneously induce their expression (19). Activation of HSF1 is regulated through various post-translational modifications such as phosphorylation, as well as alternative splicing (20, 21). In fact, HSF1 is phosphorylated in response to heat shock stress in HeLa cells and induces multiple molecular chaperones such as Hsp70 and Hsp40, which confer tolerance against heat shock stress on the cells (22, 23). Furthermore, expression of a constitutively active mutant of HSF1, which lacks the regulatory domain that binds to the Hsp90 protein complex, has been reported to suppress polyQ protein misfolding through induction of molecular chaperones (24). Therefore, HSF1 has been considered to be an attractive therapeutic target for the polyQ diseases.
Although genetic expression of molecular chaperones and a constitutively active mutant of HSF1 ameliorates polyQ-induced phenotypes in vivo (11, 12, 24), delivery of exogenous genes into the human brain is extremely limited. As a next step toward developing a therapy, it is ideal to pharmacologically induce endogenous molecular chaperones by administration of small chemical compounds, instead of to express exogenous genes. Interestingly, some compounds are known to activate HSF1 and to induce multiple endogenous molecular chaperones even under unstressed conditions (25). These include Hsp90 inhibitors, which dissociate HSF1 from the Hsp90 protein complex, and protein kinase C activators, which promote the phosphorylation of HSF1, both leading to activation of HSF1 (18, 26).
In this study, we investigated the therapeutic effects of administration of various HSF1-activating compounds on polyQ-induced neurodegeneration in vivo. We here show that oral administration of 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), a derivative of geldanamycin, markedly suppresses polyQ-induced neurodegeneration in Drosophila models of two polyQ diseases through induction of multiple molecular chaperones. Our study indicates the potential of HSF1-activating compounds as therapeutic candidates for a wide range of polyQ diseases as well as other neurodegenerative diseases.
EXPERIMENTAL PROCEDURES
Fly Stocks and Treatment—Flies were cultured and crossed under standard conditions at 25 °C. Transgenic fly lines bearing the UAS-MJDtr-Q27, UAS-MJDtr-Q78, UAS-Htt-Q128, or gmr-GAL4 transgene have been described previously (27, 28). The transgenic fly line bearing the elav-GAL4 transgene was obtained from the Bloomington Drosophila Stock Center. The MJDtr-Q27 and MJDtr-Q78 fly lines express a truncated form of the MJD protein with the indicated number of glutamines (MJDtr-Q27 and MJDtr-Q78 proteins, respectively), and the Htt-Q128 fly line expresses an N-terminal fragment of the huntingtin protein with 128 glutamines (Htt-Q128 protein), under the control of the GAL4-UAS system. For the MJDtr-Q78 flies, the W line showing weak and the S line showing strong phenotypes were used (27). The chemical compounds to be tested were dissolved in ethanol, further diluted in water, and then mixed with Instant Drosophila medium (Carolina Biological Supply Company, Burlington, NC). 17-AAG was purchased from Biomol Research Laboratories (Plymouth Meeting, PA); geldanamycin (GA) and radicicol (RA) were from Sigma-Aldrich; celastrol (CL) was from Merck; and sodium butyrate (SB) was from Nacalai Tesque (Kyoto, Japan). Geranylgeranylacetone (GGA) supplemented with 0.2% α-tocopherol was kindly provided by Eisai Co. (Tokyo, Japan). These compounds were used for flies at the following concentrations: 17-AAG, GA, and RA (50 nm, 500 nm, 5 μm, and 50 μm); CL (1 μm, 10 μm, and 100 μm); SB (10 mm); and GGA (1 nm, 10 nm, 100 nm, and 40 mm). These compounds at the above concentrations did not affect the viability and fertility of the MJDtr-Q78 flies. For heat shock treatment, crawling third instar larvae were incubated at 37 °C for 15 min and recovered at 25 °C for 1 h.
Microscopy, Histology, and Immunohistochemistry—To evaluate the therapeutic effects of HSF1-activating compounds on compound eye degeneration in the MJDtr-Q78S flies, light microscopic images of the compound eye morphologies of 1-day-old adult flies were taken using a stereoscopic microscope model SZX9 (Olympus, Tokyo, Japan). To assess the therapeutic effects of HSF1-activating compounds on photoreceptor degeneration in the Htt-Q128 flies, heads of 1-day-old Htt-Q128 flies treated with 17-AAG, GA, or RA (5 μm) were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde and embedded in Epon. Compound eyes were sectioned at 1 μm and stained with 0.5% toluidine blue. Microscopic images were taken using a fluorescence microscope model DMR (Leica Microsystems, Wetzlar, Germany) with a CCD camera model DC500 (Leica Microsystems), and the average number of rhabdomeres/ommatidium was calculated. The percentage of rescue of photoreceptor degeneration was calculated by dividing the difference in the number of rhabdomeres between the 17-AAG-treated and untreated Htt-Q128 flies by the decrease in the number of rhabdomeres in the untreated Htt-Q128 flies. The number of rhabdomeres/ommatidium was counted in 20 ommatidia/fly, and at least 100 ommatidia were assessed for each condition except for SB treatment (60 ommatidia). The data are expressed as the means ± S.E.
For immunohistochemical analysis, eye discs of the MJDtr-Q78W flies treated with 17-AAG (1.5 μm) were dissected from crawling third instar larvae and fixed in 4% paraformaldehyde. The eye discs were immunostained with a rat monoclonal anti-hemagglutinin (HA) antibody (clone 3F10; Roche Applied Science) at 1:100 dilution as the primary antibody to detect the MJDtr-Q78 protein and Alexa 546-conjugated anti-rat IgG antibody (Invitrogen) at 1:2000 dilution as the secondary antibody. The images were taken using a confocal laser scanning microscope model LSM510 (Carl Zeiss, Oberkochen, Germany). The anteroposterior width of the eye disc area with cells containing inclusion bodies of the MJDtr-Q78 protein (Wi; see Fig. 5F) and that of the area with cells containing the diffusely distributed MJDtr-Q78 protein (Wd; see Fig. 5F) were measured using National Institutes of Health Image software. The ratio of Wi/Wd in each eye disc was calculated to evaluate the degree of inclusion body formation. At least seven discs were analyzed for each treatment condition. The data are expressed as the means ± S.E.
FIGURE 5.
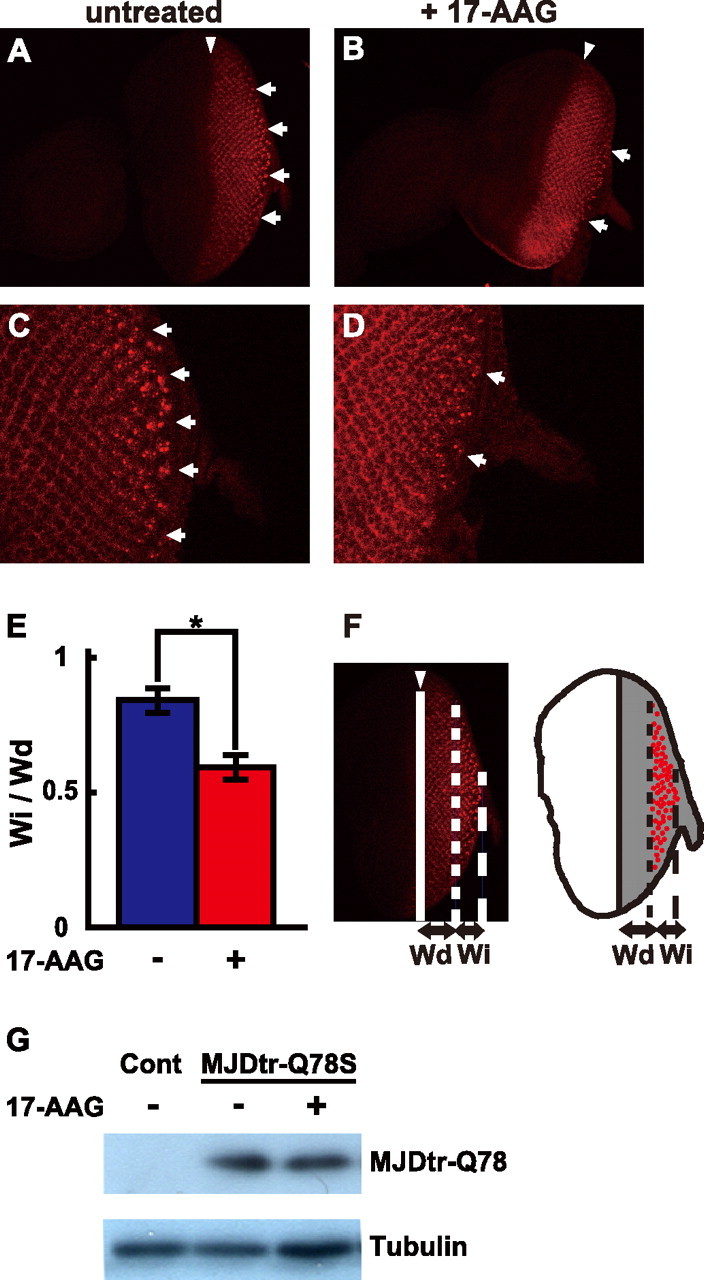
17-AAG suppresses inclusion body formation of the MJDtr-Q78 protein without affecting its expression level. A-D, confocal microscopic images of eye-antennal discs of third instar larvae of the MJDtr-Q78W flies treated with 17-AAG (1.5 μm), stained with an anti-HA antibody to detect the MJDtr-Q78 protein. Lower (×200, A, B) and higher (×630, C, D) magnification images are shown. Eye portions (posterior) are to the right, and antennal portions (anterior) to the left. The arrowheads indicate morphogenetic furrows. The 17-AAG-treated MJDtr-Q78W flies (B and D) have significantly fewer inclusion bodies (arrows) as compared with the untreated flies (A and C). E and F, quantitative analyses of the effects of 17-AAG on inclusion body formation. Schematic representation is shown of an eye disc, in which inclusion bodies (red) are formed in the area where the MJDtr-Q78 protein is expressed (gray in F, right). The ratio of the anteroposterior width of the area with cells containing inclusion bodies (Wi) to that of the area with cells containing the diffusely distributed MJDtr-Q78 protein (Wd) was calculated. The ratio of Wi/Wd was significantly decreased by 17-AAG treatment from 0.84 ± 0.04 to 0.59 ± 0.05 (E). The data are expressed as the means ± S.E. (n ≥ 7). *, p < 0.01 (Student's t test). Fly genotype is gmr-GAL4/+; +; UAS-MJDtr-Q78W/+. G, Western blot analyses of the MJDtr-Q78 protein in the MJDtr-Q78S flies treated with 17-AAG using an anti-HA antibody to detect the MJDtr-Q78 protein (upper panel) and an anti-tubulin antibody (lower panel). 17-AAG did not affect the expression level of the MJDtr-Q78 protein. Fly genotypes are gmr-GAL4/+, UAS-MJDtr-Q78S/+ (MJDtr-Q78S), or gmr-GAL4/+ (control, Cont).
Fly Survival Analyses—To examine the therapeutic effects of 17-AAG on the survival rate during development to adults of flies expressing the MJDtr-Q78 protein in the nervous system, we crossed flies homozygous for the UAS-MJDtr-Q78S transgene with flies bearing the elav-Gal4 driver transgene in trans to the balancer chromosome (CyO). 50% of the progeny are expected to bear both the elav-Gal4 driver and the UAS-MJDtr-Q78S transgenes (MJDtr-Q78S flies), which express the MJDtr-Q78 protein, whereas 50% of the progeny are expected to bear the UAS-MJDtr-Q78S transgene and CyO (control flies), which do not express the MJDtr-Q78 protein. The ratio of the MJDtr-Q78S flies to the control flies was calculated by dividing the number of emerging flies not bearing CyO (MJDtr-Q78S flies) by that of emerging flies bearing CyO (control flies) to evaluate the survival rate of the MJDtr-Q78S flies during their development to adults. The percentage of rescue of the lethality was calculated by dividing the difference in the survival rate between the 17-AAG-treated and untreated MJDtr-Q78S flies by the decrease in the survival rate of the untreated MJDtr-Q78S flies. At least 100 flies were scored for each treatment condition, and the experiments were repeated three times. The data are expressed as the means ± S.E.
Western Blot Analyses—To assess the effect of 17-AAG on the expression level of the MJDtr-Q78 protein, 10 heads of the MJDtr-Q78S flies treated with 17-AAG (5 μm) were lysed in 100 μl of Laemmli sample buffer and then centrifuged at 17,000 × g for 10 min at 25 °C. The resultant supernatants were separated on a 10% polyacrylamide gel and then transferred onto Immobilon-P membranes (Millipore, Billerica, MA). The membranes were incubated overnight with either a rat monoclonal anti-HA antibody (clone 3F10; Roche Applied Science) at 1:2000 dilution to detect the MJDtr-Q78 protein or a sheep polyclonal anti-tubulin antibody (Cytoskeleton Inc., Denver, CO) at 1:4000 dilution. Horseradish peroxidase (HRP)-conjugated rabbit anti-rat Igs antibody (DakoCytomation, Glostrup, Denmark) or HRP-conjugated rabbit anti-sheep Igs antibody (DakoCytomation) were used at 1:5000 dilution as secondary antibodies. To detect the HSF1 protein in transfected Schneider line 2 (SL2) cell lysates, a mouse monoclonal anti-Myc antibody (clone 9E10; Invitrogen; 1:2000 dilution) and HRP-conjugated rabbit anti-mouse Igs antibody (DakoCytomation; 1:5000 dilution) were used. The HRP was detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
RT-PCR Analyses—Total RNA was purified from whole bodies or eye-antennal discs of crawling third instar larvae of the MJDtr-Q78S flies and control flies expressing the GAL4 activator protein alone using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions and was reverse transcribed with random hexamer primers. We performed PCR analyses of Hsp70, Hsp40, and Hsp90 mRNAs with the following primers: Hsp70-F, 5′-AGCCGTGCCAGGTTTG-3′ and Hsp70-R, 5′-CGTTCGCCCTCATACA-3′; Hsp40-F, 5′-CATAAAGCAGCCCGTGTAGC-3′ and Hsp40-R, 5′-AGATGTTGAGGCACCGATTC-3′; and Hsp90-F, 5′-CGATTAAGCGACCAGTCGAA-3′ and Hsp90-R, 5′-AAACGACAACTGCTCTTGAATG-3′. As an internal control, mRNA of rp49 (ribosomal protein 49), a housekeeping gene, was also analyzed with the primers rp49-F, 5′-AGCGCACCAAGCACTTCATCCGCCA-3′ and rp49-R, 5′-GCGCACGTTGTGCACCAGGAACTTC-3′. The RT-PCR products were separated on a 3% agarose gel and visualized by ethidium bromide staining. For precise quantification, real time quantitative PCR was performed with the above primers and Premix Ex Taq (Takara, Shiga, Japan) using ABI PRISM 7900 HT (Applied Biosystems, Foster City, CA). The relative abundance of each mRNA was calculated by normalizing to rp49 mRNA according to the manufacturer's instructions. To compare the expression levels of each mRNA, the ratio of each mRNA/rp49 mRNA of the 17-AAG-treated MJDtr-Q78S flies expressing the MJDtr-Q78 protein alone or co-expressing the HSF1 RNAi was divided by that of the untreated MJDtr-Q78S flies. The experiments were repeated at least three times. The data are expressed as the means ± S.E.
Vector Construction and Generation of Transgenic Fly Lines—For expression of the HSF1 protein, a DNA fragment coding for dHSFa, the major alternatively spliced isoform of Drosophila HSF1 (21), tagged with a c-Myc epitope was inserted into the pUAST vector, to generate the pUAS-HSF1 vector. For the HSF1 knockdown experiment, the pUAS-HSF1-RNAi vector harboring inverted repeats corresponding to the third to fifth exon of the HSF1 cDNA separated by the second intron of the HSF1 gene was constructed according to the strategy for splice activated RNAi. To establish HSF1 RNAi fly lines, the pUAS-HSF1-RNAi vector was injected into fly embryos by standard procedures.
Cell Culture and Transfection—SL2 cells, which were derived from late stage fly embryos, were cultured in Schneider's insect medium (Sigma-Aldrich) supplemented with 10% fetal calf serum at 25 °C. To evaluate the efficiency of RNAi-mediated HSF1 knockdown, SL2 cells were co-transfected with the pUAS-HSF1 vector along with either the pUAS-HSF1-RNAi or pUAST empty vector, as well as the pAct5C-GAL4 vector as a driver using Effectene transfection reagent (Qiagen) according to the manufacturer's instructions. The cells were harvested 48 h after transfection, and the cell lysates were subjected to Western blot analyses.
RESULTS
17-AAG Treatment Suppresses polyQ-induced Neurodegeneration in Drosophila—To evaluate the therapeutic effects of HSF1-activating compounds, which induce multiple endogenous molecular chaperones, on polyQ-induced neurodegeneration, we chose the following five compounds. GA, a benzo-quinone ansamycin anti-tumor antibiotic, specifically inhibits the activity of Hsp90, a negative regulator of HSF1, resulting in activation of HSF1 (18). 17-AAG, a derivative of GA, shares the property to inhibit Hsp90 but has less cytotoxicity than GA (29). RA, an antifungal macrolactone antibiotic, also inhibits the activity of Hsp90 (30). CL, a potent anti-inflammatory compound, activates HSF1 through unknown mechanisms (31). GGA, an anti-ulcer agent, activates protein kinase C, leading to the phosphorylation and activation of HSF1 (26). We employed Drosophila polyQ disease models to analyze the therapeutic effects of oral administration of the above compounds, because Drosophila melanogaster has been shown to be a useful in vivo model to study polyQ-induced neurodegeneration (27, 28). We administered the above HSF1-activating compounds by mixing them in culture food to the transgenic fly line MJDtr-Q78S, which expresses the MJDtr-Q78 protein in the eye under the gmr promoter, resulting in severe compound eye degeneration (27).
Treatment with the HSF1-activating compounds revealed that 17-AAG is most effective against polyQ-induced compound eye degeneration (Figs. 1 and 2). 17-AAG treatment at concentrations of 500 nm, 1.5 μm, and 5 μm strongly suppressed polyQ-induced compound eye degeneration (Fig. 2, C-E), whereas treatment at 50 nm showed a moderate therapeutic effect (Fig. 2B). However, 17-AAG treatment at 50 μm unexpectedly failed to improve compound eye degeneration in the MJDtr-Q78S flies (Fig. 2F), indicating that the therapeutic effect of 17-AAG is dose-dependent at least up to 5 μm. Treatment with GA at concentrations of 500 nm and 5 μm also showed modest suppression of polyQ-induced compound eye degeneration (Fig. 1C), whereas treatment at 50 nm and 50 μm did not (data not shown). Treatment with RA at concentrations ranging from 50 nm to 5 μm was also effective against polyQ-induced compound eye degeneration (Fig. 1D). However, treatment with either CL (1 to 100 μm) or GGA (10 μm to 40 mm) did not show any detectable changes (Fig. 1, E and F). Compared with the therapeutic effects of SB, a histone deacetylase inhibitor, which was previously reported to suppress polyQ-induced neurodegeneration (7), 17-AAG showed a remarkably greater improvement of polyQ-induced compound eye degeneration (Fig. 1, B and G).
FIGURE 1.

17-AAG treatment suppresses compound eye degeneration in the MJDtr-Q78S flies. Light microscopic images of the compound eye morphologies of 1-day-old MJDtr-Q78S flies treated with various HSF1-activating compounds. A, untreated MJDtr-Q78S flies show severe compound eye degeneration. B, notably, treatment with 17-AAG (1.5 μm), an HSF1-activating compound, strongly suppressed compound eye degeneration in the MJDtr-Q78S flies. C and D, treatment with GA (5 μm, C) or RA (5 μm, D) moderately suppressed compound eye degeneration. E and F, treatment with neither CL (100 μm, E) nor GGA (40 mm, F) showed any detectable changes in compound eye degeneration. G, treatment with SB (10 mm), a histone deacetylase inhibitor used as a positive control, weakly suppressed compound eye degeneration. Fly genotype is gmr-GAL4/+; UAS-MJDtr-Q78S/+.
FIGURE 2.

17-AAG suppresses polyQ-induced compound eye degeneration in a dose-dependent manner. Light microscopic images of the compound eye morphologies of 1-day-old MJDtr-Q78S flies treated with various concentrations of 17-AAG. A, untreated MJDtr-Q78S flies. B-F, 17-AAG treatment at concentrations of 500 nm (C), 1.5 μm (D), and 5 μm (E) strongly suppressed compound eye degeneration in the MJDtr-Q78S flies, whereas treatment at 50 nm (B) showed modest suppression. However, 17-AAG treatment at 50 μm (F) failed to improve compound eye degeneration in the MJDtr-Q78S flies. These data indicate that 17-AAG treatment up to 5 μm suppresses polyQ-induced neurodegeneration in a dose-dependent manner. Fly genotype is gmr-GAL4/+; UAS-MJDtr-Q78S/+.
To examine whether HSF1-activating compounds are widely effective against various polyQ diseases, we administered 17-AAG, GA, and RA to the transgenic fly line Htt-Q128, which expresses the Htt-Q128 protein in the eye under the gmr promoter, resulting in progressive photoreceptor degeneration (28). The loss of photoreceptor neurons in the Htt-Q128 flies can be quantitatively evaluated as a decrease in the number of rhabdomeres in each ommatidium (Fig. 3, B and G), which normally has seven rhabdomeres (Fig. 3A). We found that 17-AAG treatment markedly suppresses photoreceptor degeneration, resulting in an increase in the number of rhabdomeres/ommatidium from 4.6 ± 0.3 to 5.7 ± 0.1 in the Htt-Q128 flies (Fig. 3, C and G), corresponding to a 46.3 ± 5.5% rescue (Fig. 3H). Treatment with GA and RA modestly increased the number of rhabdomeres in the Htt-Q128 flies to 5.1 ± 0.2 (22.8 ± 8.3% rescue) and to 5.1 ± 0.2 (22.0 ± 8.5% rescue), respectively (Fig. 3, D, E, G, and H), which were similar levels to SB treatment (29.8 ± 10.5% rescue) (Fig. 3, F-H), although the suppression was not statistically significant. Among the HSF1-activating compounds tested, 17-AAG was the most effective against photoreceptor degeneration in the Htt-Q128 flies (Fig. 3H), consistent with their therapeutic effects in the MJDtr-Q78S flies (Fig. 1). These data indicate that 17-AAG widely suppresses eye degeneration induced by various polyQ proteins.
FIGURE 3.
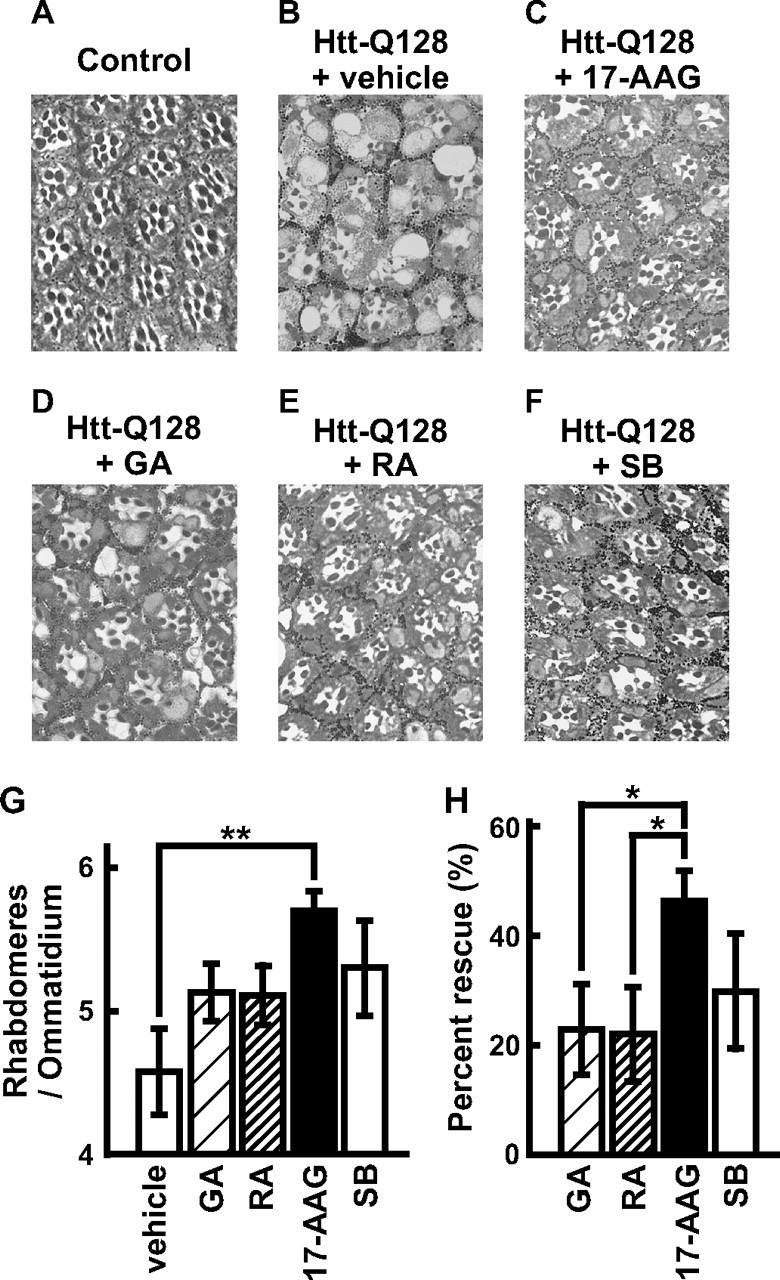
17-AAG suppresses degeneration of photoreceptor neurons in the Htt-Q128 flies. A-F, toluidine blue-stained sections of the eyes of 1-day-old Htt-Q128 flies treated with various HSF1-activating compounds (5 μm). Expression of the Htt-Q128 protein caused progressive photoreceptor degeneration, resulting in loss of rhabdomeres in each ommatidium (B), whereas flies expressing the GAL4 activator protein alone (Control) showed normal structures of ommatidia (A). Notably, 17-AAG remarkably suppressed photoreceptor degeneration in the Htt-Q128 flies (C). GA (D) and RA (E) moderately suppressed photoreceptor degeneration. SB, used as a positive control, also showed moderate suppression (F). Fly genotypes are gmr-GAL4/+; UAS-Htt-Q128/+ (B-F) or gmr-GAL4/+ (A). G, the average number of rhabdomeres/ommatidium in the Htt-Q128 flies treated with various HSF1-activating compounds. 17-AAG significantly suppressed photoreceptor degeneration, resulting in an increase in the number of rhabdomeres/ommatidium from 4.6 ± 0.3 to 5.7 ± 0.1. GA, RA, and SB modestly increased the number of rhabdomeres to 5.1 ± 0.2, to 5.1 ± 0.2, and to 5.3 ± 0.3, respectively. H, the percentage of rescue of photoreceptor degeneration in the Htt-Q128 flies by treatment with HSF1-activating compounds. 17-AAG most effectively rescued photoreceptor degeneration in the Htt-Q128 flies (46.3 ± 5.5%) as compared with GA, RA, and SB (22.8 ± 8.3, 22.0 ± 8.5, and 29.8 ± 10.5% respectively). The data are expressed as the means ± S.E. (n ≥ 3). *, p < 0.05; **, p < 0.01 (Student's t test).
We next examined whether 17-AAG suppresses polyQ-induced neurodegeneration broadly within the nervous system, because various regions of the nervous system are widely affected in human patients. Expression of the MJDtr-Q78 protein within the nervous system under the elav promoter significantly decreased the survival rate of flies during their development to adults, because of neurodegeneration (46.2 ± 14.4%; Fig. 4), whereas expression of the MJDtr-Q27 protein, which has a normal-length polyQ stretch, did not cause any significant changes (105.2 ± 12.6%). We found that 17-AAG treatment significantly increases the survival rate of the MJDtr-Q78S flies (86.0 ± 9.8%; Fig. 4), corresponding to a 74.1 ± 16.5% rescue of the lethality. These data indicate that 17-AAG is effective against polyQ-induced neurodegeneration broadly within the nervous system, and its efficiency is not limited to compound eye degeneration. We therefore conclude that 17-AAG, an HSF1-activating compound, is effective against neurodegeneration in Drosophila models of various polyQ diseases.
FIGURE 4.
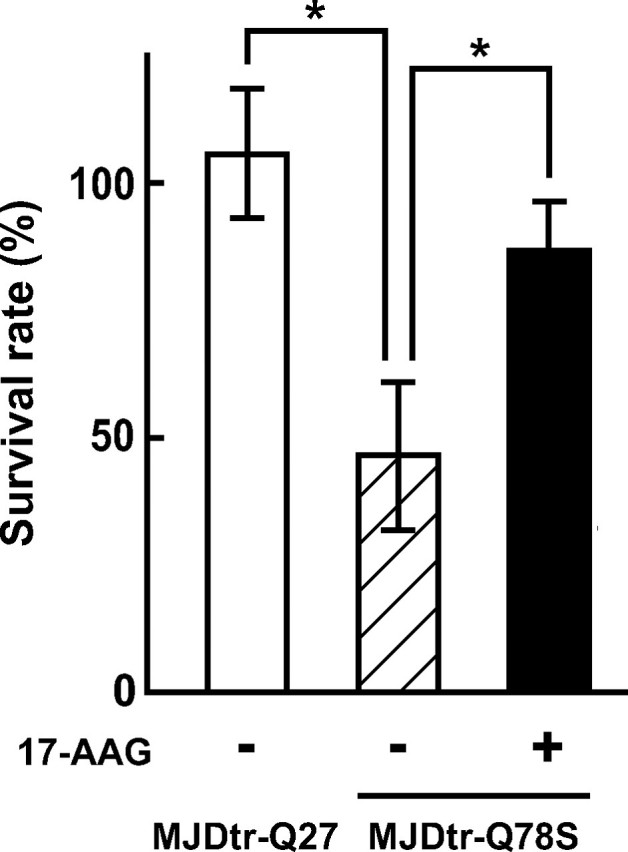
17-AAG increases the survival rate of the MJDtr-Q78S flies during their development to adults. The survival rates during the development to adults are shown of flies expressing the MJDtr-Q78 protein within the nervous system treated with 17-AAG. The ratio of the MJDtr-Q78S flies to the control flies was calculated by dividing the number of emerging flies not bearing CyO (MJDtr-Q78S flies) by that of emerging flies bearing CyO (control flies) to evaluate the survival rate of the MJDtr-Q78S flies. Expression of the MJDtr-Q78 protein significantly decreased the survival rate (46.2 ± 14.4%), whereas the MJDtr-Q27 protein did not cause any significant changes (105.2 ± 12.6%). Notably, 17-AAG treatment significantly increased the survival rate of the MJDtr-Q78S flies (86.0 ± 9.8%), corresponding to a 74.1 ± 16.5% rescue of the lethality. The data are expressed as the means ± S.E. (n = 3). *, p < 0.05 (Student's t test). Fly genotypes are elav-GAL4/UAS-MJDtr-Q78S (MJDtr-Q78S) or elav-GAL4/UAS-MJDtr-Q27 (MJDtr-Q27).
17-AAG Suppresses Inclusion Body Formation of the polyQ Protein without Affecting Its Expression Level—To evaluate the effect of 17-AAG on misfolding of the polyQ protein, we examined polyQ inclusion body formation upon 17-AAG treatment in a Drosophila polyQ disease model. Misfolded polyQ proteins are known to form aggregates and to eventually accumulate as inclusion bodies not only in human patients but also in Drosophila models (27, 28). Immunostaining of the eye discs of the MJDtr-Q78W fly larvae, which express the MJDtr-Q78 protein under the gmr promoter, revealed numerous inclusion bodies (Fig. 5A, arrows) in the region posterior to the morphogenetic furrow (arrowhead), as reported previously (27). We found that the MJDtr-Q78W flies treated with 17-AAG have significantly fewer inclusion bodies as compared with the untreated flies (Fig. 5, A and B, arrows). Upon higher magnification, the number and sizes of inclusion bodies in the MJDtr-Q78W flies were clearly reduced by 17-AAG treatment (Fig. 5, C and D), suggesting that 17-AAG suppresses polyQ inclusion body formation. For quantification of the effect of 17-AAG on inclusion body formation, we evaluated the anteroposterior width of the eye disc area with cells containing inclusion bodies (Fig. 5F, Wi). The ratio of the area with inclusions to the area with diffuse distribution of the MJDtr-Q78 protein (Wi/Wd) has been shown to gradually increase with disease progression (32), because terminal differentiation of cells and hence expression of the protein begin in the posterior end of the eye discs and spread toward the anterior. We found that 17-AAG treatment significantly decreases the Wi/Wd ratio in the eye discs of the MJDtr-Q78W flies from 0.84 ± 0.04 to 0.59 ± 0.05 (Fig. 5E). These data indicate that 17-AAG delays inclusion body formation of the MJDtr-Q78 protein. In addition, the intensity of the diffusely distributed MJDtr-Q78 protein in the 17-AAG-treated MJDtr-Q78W flies was similar to that in untreated flies (Fig. 5, A and B), suggesting that the delay in inclusion body formation by 17-AAG treatment is not due to a reduction in the expression level of the MJDtr-Q78 protein.
To further confirm that 17-AAG treatment does not affect the polyQ protein expression level, we evaluated MJDtr-Q78 protein expression by Western blot. We found that the expression level of the MJDtr-Q78 protein in the 17-AAG-treated MJDtr-Q78S flies was similar to that in untreated flies (Fig. 5G). These data suggest that 17-AAG suppresses polyQ inclusion body formation by suppression of protein misfolding rather than by the reduction of the polyQ protein expression level.
Therapeutic Effects of 17-AAG on polyQ-induced Neurodegeneration Correlate with the Induction Levels of Molecular Chaperones—To confirm whether the therapeutic effects of 17-AAG on polyQ-induced neurodegeneration depend on induction of molecular chaperones, we assessed their expression levels upon 17-AAG treatment. We examined the expression levels of Hsp70, Hsp40, and Hsp90 mRNAs in the 17-AAG-treated MJDtr-Q78S fly larvae by RT-PCR analyses, because these molecular chaperones are reported to suppress polyQ-induced cytotoxicity (11, 12, 16). We found that 17-AAG treatment up to 5 μm induces expression of Hsp70, Hsp40, and Hsp90 mRNAs in the MJDtr-Q78S flies, whereas treatment at 50 μm does not (Fig. 6A). For precise quantification of the increases in the mRNA levels of these Hsps, we performed real time quantitative RT-PCR analyses. We found that the expression levels of Hsp70 mRNA in the MJDtr-Q78S flies are significantly increased by 17-AAG treatment at concentrations ranging from 50 nm to 5 μm in a dose-dependent manner (50 nm, 4.53-fold; 500 nm, 29.04-fold; and 5 μm, 60.87-fold; Fig. 6B), although the levels of induction are not as robust as heat shock treatment (Fig. 6B, HS). However, 17-AAG treatment at 50 μm did not induce expression of Hsp70 mRNA at all (0.64-fold), which is consistent with the lack of a therapeutic effect on polyQ-induced neurodegeneration (Fig. 2F). We also found that 17-AAG treatment up to 5 μm induced expression of Hsp40 mRNA in a dose-dependent manner (50 nm, 1.41-fold; 500 nm, 1.53-fold; and 5 μm, 2.20-fold; Fig. 6C), whereas treatment at 50 μm did not. The expression levels of Hsp90 mRNA were also increased by 17-AAG treatment up to 5 μm in a dose-dependent manner (50 nm, 1.77-fold; 500 nm, 1.98-fold; and 5 μm, 2.42-fold; Fig. 6D) and were similar to that upon heat shock treatment (2.71-fold; Fig. 6D). 17-AAG treatment at 50 μm slightly induced expression of Hsp90 mRNA (1.47-fold). Taken together, we conclude that 17-AAG treatment up to 5 μm induces multiple molecular chaperones in a dose-dependent manner and that their expression levels correlate with the therapeutic effects of 17-AAG on polyQ-induced neurodegeneration.
FIGURE 6.

17-AAG induces expression of molecular chaperones in a dose-dependent manner. A, RT-PCR analyses of Hsp70, Hsp40, and Hsp90 mRNAs expressed in third instar larvae of the MJDtr-Q78S flies treated with 17-AAG (50 nm, 500 nm, 5 μm, or 50 μm). Flies expressing the GAL4 activator protein alone (Cont) exposed to heat shock (HS) were used as a positive control. The rp49 mRNA was also analyzed as an internal control. B-D, real time quantitative RT-PCR analyses of Hsp70, Hsp40, and Hsp90 mRNAs expressed in the MJDtr-Q78S fly larvae. Treatment with 17-AAG up to 5 μm induced expression of Hsp70 (B), Hsp40 (C), and Hsp90 (D) mRNAs in a dose-dependent manner (60.87-, 2.20-, and 2.42-fold at 5 μm, respectively). The results shown are from representative experiments. The data are expressed as the means ± S.E. (n = 3). Fly genotypes are gmr-GAL4/+, UAS-MJDtr-Q78S/+ (MJDtr-Q78S), or gmr-GAL4/+ (control).
Therapeutic Effects of 17-AAG on polyQ-induced Neurodegeneration Are Mediated by HSF1 Activation—We next determined whether the therapeutic effects of 17-AAG on polyQ-induced neurodegeneration through induction of multiple molecular chaperones are mediated by HSF1 activation. For this purpose, we examined whether RNAi-mediated knockdown of endogenous HSF1 affects the therapeutic effects of 17-AAG on the MJDtr-Q78S flies. We first designed a double-stranded RNA construct against HSF1 (HSF1 RNAi) and evaluated its efficiency of HSF1 knockdown in Drosophila SL2 cells. SL2 cells were co-transfected with expression vectors for the HSF1 protein and HSF1 RNAi, and the cell lysates were subjected to Western blot analysis. In the cells transfected with the HSF1 vector alone, we found that the HSF1 band appears as a doublet, probably because of phosphorylation, as reported previously (Fig. 7A, middle lane). In contrast, the HSF1 protein was hardly detected in the cells co-transfected with the HSF1 vector and the HSF1 RNAi vector (Fig. 7A, right lane), indicating that this HSF1 RNAi construct almost completely knocks down expression of the HSF1 protein. Accordingly, we established transgenic HSF1 RNAi flies expressing this HSF1 RNAi construct under the gmr promoter and confirmed that these flies do not show any detectable phenotypes in their compound eyes (data not shown). We then crossed these HSF1 RNAi flies with the MJDtr-Q78S flies to evaluate the effects of HSF1 knockdown on 17-AAG treatment. Notably, knockdown of endogenous HSF1 almost completely abolished the therapeutic effect of 17-AAG on compound eye degeneration in the MJDtr-Q78S flies (Fig. 7, D and E). The level of compound eye degeneration in the 17-AAG-treated MJDtr-Q78S flies co-expressing the HSF1 RNAi was similar to that in the untreated MJDtr-Q78S flies (Fig. 7, B and E). These data indicate that endogenous HSF1 is essential for the therapeutic effects of 17-AAG on polyQ-induced neurodegeneration. However, we cannot exclude the possibility that knockdown of HSF1 simply masks the therapeutic effects of 17-AAG independently of affecting the induction of molecular chaperones, because HSF1 knockdown itself slightly enhances polyQ-induced compound eye degeneration in the MJDtr-Q78S flies (Fig. 7C), consistent with a previous report (33).
FIGURE 7.
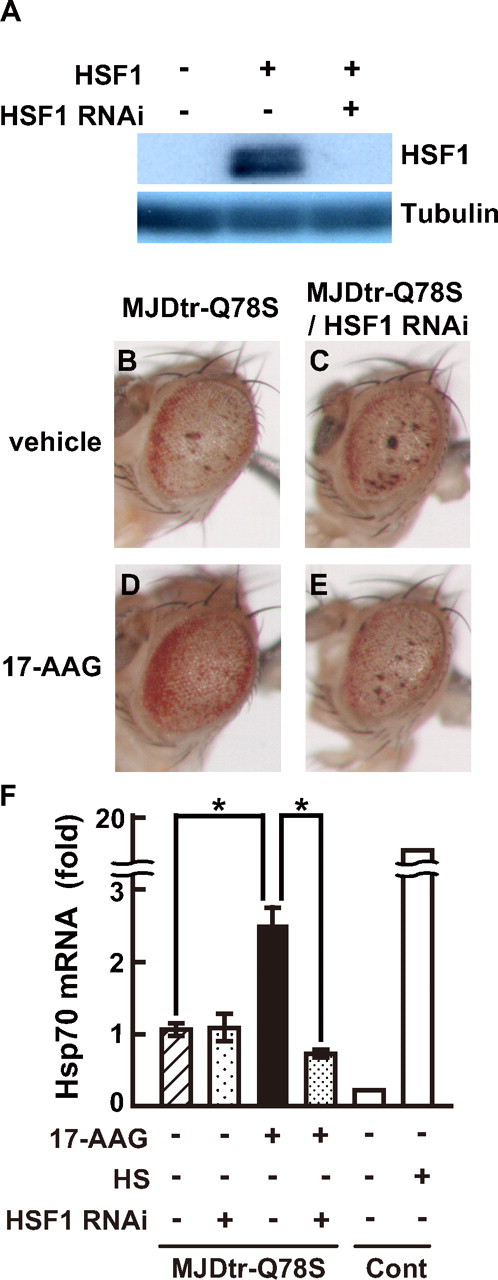
RNAi-mediated knockdown of HSF1 abolishes the therapeutic effect of 17-AAG on compound eye degeneration in the MJDtr-Q78S flies. A, Western blot analyses of the HSF1 protein expressed in SL2 cells co-transfected with expression vectors for the HSF1 protein and HSF1 RNAi, using the anti-Myc antibody to detect the HSF1 protein (upper panel) and the anti-tubulin antibody (lower panel). Co-expression of the HSF1 RNAi almost completely knocks down expression of the HSF1 protein. B-E, light microscopic images of the compound eye morphologies of the 17-AAG (5 μm)-treated MJDtr-Q78S flies expressing the MJDtr-Q78 protein alone (D) or co-expressing the HSF1 RNAi (E). Untreated MJDtr-Q78S flies expressing the MJDtr-Q78 protein alone (B) or co-expressing the HSF1 RNAi (C) are also shown. Co-expression of the HSF1 RNAi almost completely abolished the therapeutic effect of 17-AAG on compound eye degeneration in the MJDtr-Q78S flies. Fly genotypes are gmr-GAL4/+, UAS-MJDtr-Q78S/+, +/+ (B and D) or gmr-GAL4/+, UAS-MJDtr-Q78S/+, and UAS-HSF1-RNAi/+ (C and E). F, real time quantitative RT-PCR analyses of Hsp70 mRNA expressed in the eye-antennal discs of the 17-AAG (5 μm)-treated MJDtr-Q78S fly larvae co-expressing the HSF1 RNAi or expressing the MJDtr-Q78 protein alone. Flies expressing the GAL4 activator protein alone (control, Cont) exposed to heat shock (HS) were used as a positive control. Co-expression of the HSF1 RNAi decreased the expression level of Hsp70 mRNA in the 17-AAG-treated MJDtr-Q78S fly larvae from 2.37- to 0.68-fold. The experiments were repeated at least four times except for control flies. The results from representative experiments are shown for control flies. The data are expressed as the means ± S.E. (n ≥ 3). *, p < 0.01 (Student's t test). Fly genotypes are gmr-GAL4/+, UAS-MJDtr-Q78S/+ (MJDtr-Q78S), gmr-GAL4/+, UAS-MJDtr-Q78S/UAS-HSF1-RNAi (MJDtr-Q78S/HSF1 RNAi), or gmr-GAL4/+ (Cont).
To further confirm whether the therapeutic effects of 17-AAG depend on HSF1-mediated induction of molecular chaperones, we examined the expression level of Hsp70 mRNA in the 17-AAG-treated MJDtr-Q78S flies co-expressing the HSF1 RNAi. We performed quantitative RT-PCR analyses using eye-antennal discs of the fly larvae, because the HSF1 RNAi construct is expressed only in the eye discs under the gmr promoter. We confirmed that 17-AAG treatment significantly induces expression of Hsp70 mRNA in the eye-antennal discs of the MJDtr-Q78S fly larvae (2.37-fold; Fig. 7F) as was observed in their whole bodies (Fig. 6). Notably, knockdown of endogenous HSF1 dramatically decreased the expression level of Hsp70 mRNA in the 17-AAG-treated MJDtr-Q78S fly larvae (0.68-fold; Fig. 7F), which was similar to that in the untreated MJDtr-Q78S flies. These data indicate that endogenous HSF1 is required for induction of molecular chaperones by 17-AAG treatment, and we therefore conclude that the therapeutic effects of 17-AAG on polyQ-induced neurodegeneration depend on HSF1-mediated induction of molecular chaperones.
DISCUSSION
Recently, many neurodegenerative diseases including Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis, and the polyQ diseases are thought to be caused by protein misfolding, and hence suppression of protein misfolding by molecular chaperones is considered to be a common therapeutic approach for these neurodegenerative diseases (14). In fact, genetic expression of Hsp70, a molecular chaperone, has been shown to suppress neurodegeneration in cell culture and animal models of Parkinson disease, amyotrophic lateral sclerosis, and the polyQ diseases (34-36). Furthermore, induction of multiple molecular chaperones including Hsp70 and Hsp40 by genetic expression of a constitutively active mutant of HSF1 has been shown to be more effective against amyotrophic lateral sclerosis and the polyQ diseases compared with expression of Hsp70 alone (24, 37). As a next step toward developing a therapy, we evaluated the therapeutic effects of pharmacological induction of multiple endogenous molecular chaperones by HSF1-activating compounds in Drosophila models of the polyQ diseases. In this study, we show that 17-AAG treatment successfully suppresses neurodegeneration through induction of Hsp70, Hsp40 and Hsp90 in a Drosophila model of SCA3/MJD, one of the polyQ diseases (Figs. 1, 2, 4, and 6). 17-AAG also significantly suppressed neurodegeneration in a model of Huntington disease, another polyQ disease (Fig. 3), indicating that 17-AAG is widely effective against various polyQ diseases. In addition, 17-AAG clearly reduced inclusion bodies composed of misfolded polyQ proteins (Fig. 5), consistent with previous reports showing that co-expression of Hsp70 together with Hsp40 synergistically suppresses polyQ inclusion body formation (8, 38). Moreover, induction of Hsp70 by GA treatment has been reported to suppress neurodegeneration in a Drosophila model of Parkinson disease (39). Induction of molecular chaperones by treatment with arimoclomol, which is also known to activate HSF1, has been reported to suppress neurodegeneration in a mouse model of amyotrophic lateral sclerosis (40). Therefore, treatment with HSF1-activating compounds to induce multiple endogenous molecular chaperones is a promising therapeutic approach for the polyQ diseases as well as other neurodegenerative diseases.
We show that 17-AAG is the most effective agent against polyQ-induced neurodegeneration in Drosophila among the HSF1-activating compounds we studied (Fig. 1). In addition to 17-AAG, GA, and RA also showed weaker suppression of polyQ-induced neurodegeneration, consistent with previous reports showing that GA and RA suppress polyQ protein misfolding in vitro (41). On the other hand, GGA was not effective against polyQ-induced neurodegeneration in this study, although it has been reported to mitigate the neurological phenotypes of a mouse model of spinobulbar muscular atrophy (42). GGA has recently been demonstrated to activate HSF1 by inhibiting the chaperone activity of Hsp70 (43), which may account for the lack of its therapeutic effect in our study. Importantly, 17-AAG has been shown to be less toxic than GA (29), which shows hepatotoxicity, and 17-AAG is currently being tested in clinical trials for the treatment of human cancer patients (44). In addition, intraperitoneal injection of 17-AAG into mice has been reported to induce multiple molecular chaperones in their spinal cord (45). Taken together, 17-AAG is the most promising therapeutic candidate for the polyQ diseases among the HSF1-activating compounds.
17-AAG has been reported to mitigate the neurological phenotypes of a mouse model of spinobulbar muscular atrophy by degradation of the mutant androgen receptor protein, an Hsp90 client protein, via its Hsp90 inhibitor activity, rather than by induction of molecular chaperones (45). In addition, Hsp90 inhibitors such as GA and RA have been reported to accelerate degradation of the mutant androgen receptor protein even in HSF1 knock-out cells (46), further supporting the possibility that the therapeutic effects of these Hsp90 inhibitors are not mediated by activation of HSF1 in the case of spinobulbar muscular atrophy models. Therefore, the effectiveness of Hsp90 inhibitors has been considered to be limited to spinobulbar muscular atrophy so far. In contrast, we show that knockdown of HSF1 abolished the induction of molecular chaperones and therapeutic effect of 17-AAG on polyQ-induced neurodegeneration in a Drosophila model of SCA3/MJD (Fig. 7), indicating that the therapeutic effect of 17-AAG depends on HSF1-mediated induction of molecular chaperones and not on degradation of the mutant MJD protein (Fig. 5G). Furthermore, we show that 17-AAG is also effective against neurodegeneration in a Drosophila model of Huntington disease (Fig. 3), consistent with a recent report showing that 17-AAG suppresses misfolding of the mutant huntingtin protein without its degradation (47). Therefore, we conclude that 17-AAG is effective against a wide range of polyQ diseases through HSF1-mediated induction of molecular chaperones.
In this study, we evaluated the therapeutic effects of HSF1-activating compounds on polyQ-induced neurodegeneration in vivo. We found that oral administration of 17-AAG markedly suppresses polyQ-induced neurodegeneration in Drosophila models of two polyQ diseases through HSF1-mediated induction of multiple molecular chaperones. Therefore, we conclude that 17-AAG is a promising therapeutic candidate for the polyQ diseases as well as other neurodegenerative diseases caused by protein misfolding.
Acknowledgments
We thank M. Sone, H. Matsushima, R. Sasaki, and C. Ito for technical assistance. We thank Eisai Co. for providing GGA. We also thank N. M. Bonini for providing us with the MJDtr-Q27, MJDtr-Q78W, and MJDtr-Q78S fly lines; J. T. Littleton for the Htt-Q128 fly line; and the Bloomington Drosophila Stock Center and Drosophila Genetic Resource Center at Kyoto Institute of Technology for the other fly lines.
This work was supported in part by Grants-in-Aid for Scientific Research on Priority Areas (to Y. N.; Advanced Brain Science Project, Research on Patho-mechanisms of Brain Disorders, Life of Proteins, and Protein Community) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by a Grant-in-Aid for the Research Committee for Ataxic Diseases (to Y. N.) from the Ministry of Health, Labor and Welfare, Japan; and by Grants-in-Aid for Scientific Research (B) (to Y. N.) and for Young Scientists (B) (to N. F.) from the Japan Society for the Promotion of Science. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: polyQ, polyglutamine; SCA, spinocerebellar ataxia; HSF1, heat shock transcription factor 1; 17-AAG, 17-(allylamino)-17-demethoxygeldanamycin; GA, geldanamycin; RA, radicicol; CL, celastrol; SB, sodium butyrate; GGA, geranylgeranylacetone; HA, hemagglutinin; SL2, Schneider line 2; HRP, horseradish peroxidase; RT, reverse transcription; RNAi, RNA interference.
References
- 1.Ross, C. A., and Poirier, M. A. (2005) Nat. Rev. Mol. Cell Biol. 6 891-898 [DOI] [PubMed] [Google Scholar]
- 2.Gusella, J. F., and MacDonald, M. E. (2000) Nat. Rev. Neurosci. 1 109-115 [DOI] [PubMed] [Google Scholar]
- 3.Zoghbi, H. Y., and Orr, H. T. (2000) Annu. Rev. Neurosci. 23 217-247 [DOI] [PubMed] [Google Scholar]
- 4.Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001) Science 292 1552-1555 [DOI] [PubMed] [Google Scholar]
- 5.Nagai, Y., Onodera, O., Chun, J., Strittmatter, W. J., and Burke, J. R. (1999) Exp. Neurol. 155 195-203 [DOI] [PubMed] [Google Scholar]
- 6.Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., Wanker, E. E., Bates, G. P., Housman, D. E., and Thompson, L. M. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 6763-6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steffan, J. S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A., Apostol, B. L., Kazantsev, A., Schmidt, E., Zhu, Y. Z., Greenwald, M., Kurokawa, R., Housman, D. E., Jackson, G. R., Marsh, J. L., and Thompson, L. M. (2001) Nature 413 739-743 [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi, Y., Kume, A., Li, M., Doyu, M., Hata, M., Ohtsuka, K., and Sobue, G. (2000) J. Biol. Chem. 275 8772-8778 [DOI] [PubMed] [Google Scholar]
- 9.Muchowski, P. J., Schaffar, G., Sittler, A., Wanker, E. E., Hayer-Hartl, M. K., and Hartl, F. U. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 7841-7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan, H. Y., Warrick, J. M., Gray-Board, G. L., Paulson, H. L., and Bonini, N. M. (2000) Hum. Mol. Genet. 9 2811-2820 [DOI] [PubMed] [Google Scholar]
- 11.Kazemi-Esfarjani, P., and Benzer, S. (2000) Science 287 1837-1840 [DOI] [PubMed] [Google Scholar]
- 12.Warrick, J. M., Chan, H. Y., Gray-Board, G. L., Chai, Y., Paulson, H. L., and Bonini, N. M. (1999) Nat. Genet. 23 425-428 [DOI] [PubMed] [Google Scholar]
- 13.Rørth, P. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 12418-12422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muchowski, P. J., and Wacker, J. L. (2005) Nat. Rev. Neurosci. 6 11-22 [DOI] [PubMed] [Google Scholar]
- 15.Ishihara, K., Yamagishi, N., Saito, Y., Adachi, H., Kobayashi, Y., Sobue, G., Ohtsuka, K., and Hatayama, T. (2003) J. Biol. Chem. 278 25143-25150 [DOI] [PubMed] [Google Scholar]
- 16.Mitsui, K., Nakayama, H., Akagi, T., Nekooki, M., Ohtawa, K., Takio, K., Hashikawa, T., and Nukina, N. (2002) J. Neurosci. 22 9267-9277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyttenbach, A., Sauvageot, O., Carmichael, J., Diaz-Latoud, C., Arrigo, A. P., and Rubinsztein, D. C. (2002) Hum. Mol. Genet. 11 1137-1151 [DOI] [PubMed] [Google Scholar]
- 18.Zou, J., Guo, Y., Guettouche, T., Smith, D. F., and Voellmy, R. (1998) Cell 94 471-480 [DOI] [PubMed] [Google Scholar]
- 19.Baler, R., Dahl, G., and Voellmy, R. (1993) Mol. Cell Biol. 13 2486-2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu, B., Soncin, F., Price, B. D., Stevenson, M. A., and Calderwood, S. K. (1996) J. Biol. Chem. 271 30847-30857 [DOI] [PubMed] [Google Scholar]
- 21.Fujikake, N., Nagai, Y., Popiel, H. A., Kano, H., Yamaguchi, M., and Toda, T. (2005) FEBS Lett. 579 3842-3848 [DOI] [PubMed] [Google Scholar]
- 22.Holmberg, C. I., Illman, S. A., Kallio, M., Mikhailov, A., and Sistonen, L. (2000) Cell Stress Chaperones 5 219-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia, W., Vilaboa, N., Martin, J. L., Mestril, R., Guo, Y., and Voellmy, R. (1999) Cell Stress Chaperones 4 8-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimoto, M., Takaki, E., Hayashi, T., Kitaura, Y., Tanaka, Y., Inouye, S., and Nakai, A. (2005) J. Biol. Chem. 280 34908-34916 [DOI] [PubMed] [Google Scholar]
- 25.Westerheide, S. D., and Morimoto, R. I. (2005) J. Biol. Chem. 280 33097-33100 [DOI] [PubMed] [Google Scholar]
- 26.Yamanaka, K., Takahashi, N., Ooie, T., Kaneda, K., Yoshimatsu, H., and Saikawa, T. (2003) J. Mol. Cell Cardiol. 35 785-794 [DOI] [PubMed] [Google Scholar]
- 27.Warrick, J. M., Paulson, H. L., Gray-Board, G. L., Bui, Q. T., Fischbeck, K. H., Pittman, R. N., and Bonini, N. M. (1998) Cell 93 939-949 [DOI] [PubMed] [Google Scholar]
- 28.Lee, W. C., Yoshihara, M., and Littleton, J. T. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 3224-3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Page, J., Heath, J., Fulton, R., Yalkowsky, E., Tabibi, E., Tomaszewski, J., Smith, A., and Rodman, L. (1997) Proc. Am. Assoc. Cancer Res. 38 308 [Google Scholar]
- 30.Schulte, T. W., Akinaga, S., Soga, S., Sullivan, W., Stensgard, B., Toft, D., and Neckers, L. M. (1998) Cell Stress Chaperones 3 100-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westerheide, S. D., Bosman, J. D., Mbadugha, B. N., Kawahara, T. L., Matsumoto, G., Kim, S., Gu, W., Devlin, J. P., Silverman, R. B., and Morimoto, R. I. (2004) J. Biol. Chem. 279 56053-56060 [DOI] [PubMed] [Google Scholar]
- 32.Warrick, J. M., Morabito, L. M., Bilen, J., Gordesky-Gold, B., Faust, L. Z., Paulson, H. L., and Bonini, N. M. (2005) Mol. Cell 18 37-48 [DOI] [PubMed] [Google Scholar]
- 33.Nollen, E. A., Garcia, S. M., van Haaften, G., Kim, S., Chavez, A., Morimoto, R. I., and Plasterk, R. H. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 6403-6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auluck, P. K., Chan, H. Y., Trojanowski, J. Q., Lee, V. M., and Bonini, N. M. (2002) Science 295 865-868 [DOI] [PubMed] [Google Scholar]
- 35.Bruening, W., Roy, J., Giasson, B., Figlewicz, D. A., Mushynski, W. E., and Durham, H. D. (1999) J. Neurochem. 72 693-699 [DOI] [PubMed] [Google Scholar]
- 36.Cummings, C. J., Sun, Y., Opal, P., Antalffy, B., Mestril, R., Orr, H. T., Dillmann, W. H., and Zoghbi, H. Y. (2001) Hum. Mol. Genet. 10 1511-1518 [DOI] [PubMed] [Google Scholar]
- 37.Batulan, Z., Taylor, D. M., Aarons, R. J., Minotti, S., Doroudchi, M. M., Nalbantoglu, J., and Durham, H. D. (2006) Neurobiol. Dis. 24 213-225 [DOI] [PubMed] [Google Scholar]
- 38.Rujano, M. A., Kampinga, H. H., and Salomons, F. A. (2007) Exp. Cell Res. 313 3568-3578 [DOI] [PubMed] [Google Scholar]
- 39.Auluck, P. K., and Bonini, N. M. (2002) Nat. Med. 8 1185-1186 [DOI] [PubMed] [Google Scholar]
- 40.Kieran, D., Kalmar, B., Dick, J. R., Riddoch-Contreras, J., Burnstock, G., and Greensmith, L. (2004) Nat. Med. 10 402-405 [DOI] [PubMed] [Google Scholar]
- 41.Hay, D. G., Sathasivam, K., Tobaben, S., Stahl, B., Marber, M., Mestril, R., Mahal, A., Smith, D. L., Woodman, B., and Bates, G. P. (2004) Hum. Mol. Genet. 13 1389-1405 [DOI] [PubMed] [Google Scholar]
- 42.Katsuno, M., Sang, C., Adachi, H., Minamiyama, M., Waza, M., Tanaka, F., Doyu, M., and Sobue, G. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 16801-16806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otaka, M., Yamamoto, S., Ogasawara, K., Takaoka, Y., Noguchi, S., Miyazaki, T., Nakai, A., Odashima, M., Matsuhashi, T., Watanabe, S., and Itoh, H. (2006) Biochem. Biophys. Res. Commun. 353 399-404 [DOI] [PubMed] [Google Scholar]
- 44.Ronnen, E. A., Kondagunta, G. V., Ishill, N., Sweeney, S. M., Deluca, J. K., Schwartz, L., Bacik, J., and Motzer, R. J. (2006) Invest. New Drugs 24 543-546 [DOI] [PubMed] [Google Scholar]
- 45.Waza, M., Adachi, H., Katsuno, M., Minamiyama, M., Sang, C., Tanaka, F., Inukai, A., Doyu, M., and Sobue, G. (2005) Nat. Med. 11 1088-1095 [DOI] [PubMed] [Google Scholar]
- 46.Thomas, M., Harrell, J. M., Morishima, Y., Peng, H. M., Pratt, W. B., and Lieberman, A. P. (2006) Hum. Mol. Genet. 15 1876-1883 [DOI] [PubMed] [Google Scholar]
- 47.Herbst, M., and Wanker, E. E. (2007) Neurodegener. Dis. 4 254-260 [DOI] [PubMed] [Google Scholar]