

Abstract
Beclin1 has a key regulatory role in the initiation of autophagy and is a tumor suppressor. We have examined the interplay between viral or human Bcl-2-like proteins and UVRAG and their opposite effects on Beclin1. We show that Beclin1 forms a dimer in solution via its coiled-coil domain both in vivo and in vitro. Viral Bcl-2 binds independently to two sites on the Beclin1 dimer, one with high affinity and one with lower affinity, whereas human Bcl-xL binds both sites equally with relatively low affinity. UVRAG disrupts the Beclin1-dimer interface, forming a heterodimer with Beclin1, suggesting that this is how UVRAG causes its effects on Beclin1 to activate autophagy. Both Bcl-2-like proteins reduce the affinity of UVRAG for Beclin1 ∼4-fold, suggesting that they stabilize the Beclin1 dimer. Moreover, coimmunoprecipitation assays show that UVRAG substantially reduces Beclin1 dimerization in vivo. These data explain the concentration-dependent interplay between Bcl-2, UVRAG, and Beclin1, as both tumor suppressors, UVRAG and Beclin1, have single-copy mutations in human cancers. Furthermore, our data suggest that an alternative strategy for developing anti-cancer compounds would be to disrupt the Beclin1-dimer interface.
Autophagy is an intracellular vesicular pathway for the degradation of long-lived proteins and organelles (1, 2). The bulk cytoplasm is engulfed in vesicles that mature into autophagosomes and are then targeted to the lysosome. This process is well conserved from yeast to mammals, and many of the autophagy-related (atg) genes that are involved were originally identified in yeast (2). Many have mammalian homologues with conserved functions in autophagy. For example the mammalian protein Beclin1 is a homologue of yeast Atg6, and both form a lipid-kinase complex with class III phosphoinositide-3 kinase (PI(3)KC34; Vps34 in yeast) that mediates vesicle nucleation.
Autophagy is required during cell growth and development and is thought to be predominantly a pro-survival process. For example, it is induced in response to cellular stress such as starvation as well as during differentiation when large scale changes within cells are required. However, excessive autophagy also induces cell death by a morphologically distinct process to apoptosis, and increasing evidence suggests that autophagy can also function as a programmed-cell-death pathway that is distinct from apoptosis. For example, RNAi against atg5, atg7, or beclin1 prevents cell death in apoptosis-incompetent mouse cells (3, 4). This suggests that cells can die by an alternative Beclin1-dependent pathway when apoptosis is inhibited.
Beclin1 is monoallelically deleted in many sporadic human breast and ovarian cancers (5), and beclin1 gene transfer inhibits tumorigenicity, suggesting that reduced Beclin1 expression increases breast cancer progression (6). Heterozygous disruption of beclin1 in mice increases the formation of spontaneous tumors (7, 8), showing Beclin1 is a haplo-insufficient tumor suppressor.
The Bcl-2 family of proteins regulate apoptosis (9). The Bcl-2-like proteins, including Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1, inhibit apoptosis, whereas the other Bcl-2 subfamilies, the Bax-like proteins, including Bax and Bak, and the Bcl-2-homology domain 3 (BH3)-only proteins are largely pro-apoptotic. Bcl-2-like proteins induce Bax and Bak to rearrange to form transmembrane pores that initiate the later stages of apoptosis, whereas BH3-only proteins bind to Bcl-2-like proteins and block their inhibition of apoptosis. Bcl-2-like proteins including Bcl-xL and Bcl-2 bind to Beclin1 (10), and Beclin1 contains a single BH3 domain, indicating that it is also a member of the BH3-only family (10-12). Therefore, Beclin1 may sequester Bcl-2, which may be responsible for the tumor suppressor activity of Beclin1 (11). The γ-herpesviruses, such as Kaposi's sarcoma-associated herpesvirus (KSHV), encode a viral form of the cellular Bcl-2 oncogene (vBcl-2) which also bind to BH3-only proteins including Beclin1 to prevent apoptosis (13, 14).
As well as inhibiting apoptosis, Bcl-2 binding to Beclin1 also inhibits autophagy, as it blocks the lipid-kinase activity of the Beclin1-PI(3)KC3 complex and prevents vesicle initiation (14). Recently another tumor suppressor, UVRAG, has been shown to interact with Beclin1 and stimulate autophagy by activating the kinase activity of PI(3)KC3 (13). Therefore, Bcl-2 and UVRAG have opposing effects on the Beclin1-PI(3)KC3 complex. Here we have analyzed the reasons for these antagonistic effects at the molecular level by examining Beclin1 and its interacting partners. We show that the central coiled-coil domain of Beclin1 dimerizes both in vivo and in vitro. Bcl-xL and viral Bcl-2 bind to Beclin1 by different mechanisms, but both proteins appear to stabilize the Beclin1 dimer interface, whereas UVRAG disrupts the Beclin1 dimer. Therefore, Beclin1 acts as a monomer-dimer“switch” regulated by UVRAG and Bcl-2-like proteins.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The DNA sequences coding for Beclin1, residues 133-266 and 95-266, and KSHV Bcl-2 residues 1-146 were isolated by PCR amplification and cloned into the BamHI and XhoI restriction sites of pGEX6P-1 (Amersham Biosciences). Beclin1 residues 95-266 and KSHV Bcl-2 residues 1-146 were also cloned, respectively, into the BamHI and SalI and into the NdeI and XhoI restriction sites of pETDuet-1 (Novagen). UVRAG residues 189-275 and Beclin1 residues 133-266 were cloned, respectively, into the BamHI and SalI and into the NdeI and XhoI restriction sites of pETDuet-1. These pETDuet-1 constructs were subsequently modified to insert an N-terminal glutathione S-transferase fusion with a PreScission protease (Amersham Biosciences) cleavage site instead of the N-terminal His tag into the upstream gene. The double mutant variant of KSHV Bcl-2 (N67D,V117A) was created by two rounds of site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene). The individual proteins and complexes were expressed in the Escherichia coli strain BL21 (DE3) and purified from clarified crude cell extracts by cleaving from a glutathione affinity column using PreScission protease followed by ion-exchange and gel-filtration chromatography. Bcl-xL (residues 1-208, N52D, N66D) in pET29b was purified as described previously (11). The nucleotide sequence of each expression clone was verified by automated DNA sequencing. Protein concentrations were determined from the absorbance at 280 nm using molar extinction coefficients derived by summing the contributions from tyrosine and tryptophan residues. The 24-mer Beclin1-BH3 peptide (residues 107-129 and an additional N-terminal tyrosine to determine the concentration by UV spectroscopy) was purchased high performance liquid chromatography-purified (1st Base).
Sedimentation Velocity—Sedimentation velocity experiments were performed using a ProteomeLab XL-A analytical ultracentrifuge (Beckman Coulter) with 2-channel centerpieces in an An-60Ti rotor at 42,000 rpm and 20 °C. Radial scans at a single wavelength (typically 280 nm) were taken every 300 s. The solvent density and viscosity and the protein partial specific volume were calculated using the program SEDNTERP. Protein samples were dialyzed into a buffer containing 20 mm Tris, pH 7.8, 150 mm NaCl, and 0.5 mm Tris[2-carboxyethyphosphine] hydrochloride (TCEP). The data were fitted to the continuous size-distribution functions c(S) or c(M) using the program SEDFIT (15).
Sedimentation Equilibrium—Sedimentation equilibrium experiments were performed using a ProteomeLab XL-A analytical ultracentrifuge using 6-channel centerpieces in an An-60Ti rotor at 20 °C. Protein samples were dialyzed into 20 mm Tris, pH 7.8, 150 mm NaCl, and 0.5 mm TCEP. Samples were centrifuged for ∼24 h until they reached equilibrium and no further change was seen in the distribution. Radial scans were measured at 280, 250, and 230 nm. The data were fitted to an ideal single-species model as well as a rapid monomer-dimer equilibrium using the program HETEROANALYSIS. The vBcl-2-Beclin1 BH3-CCD complex was also analyzed in terms of a heteroassociation model consisting of two sequential binding events. Where possible, scans were also taken after centrifugation at 42,000 rpm to measure the residual absorbance for initial offset values.
Isothermal Titration Calorimetry—Isothermal titration calorimetry was performed using a VP-ITC microcalorimeter (MicroCal Inc.). Samples were dialyzed into 20 mm Tris, pH 7.8, 150 mm NaCl, and 0.5 mm TCEP. The cell was loaded with 10-20 μm Beclin1 sample, and the injection syringe was loaded with 200-400 μm UVRAG-(189-275) or Bcl-2-like protein. Typically titrations consisted of 28 injections of 10 μl, with 240-s equilibration between injections. The data were analyzed using Origin 7.0.
Analytical Size-exclusion Chromatography—A superdex 200 HR 10/30 column (Amersham Biosciences) was equilibrated with 20 mm Tris, pH 7.8, 150 mm NaCl, and 0.5 mm TCEP. Protein samples in the same buffer were injected onto the column and monitored at 210 nm.
Mammalian Cell Transfection and Imaging—Full-length Beclin1, Beclin1-(95-266), Beclin1-(133-266), and full-length Bcl-xL were cloned into pXJ-YPet. This vector consists of the YPet coding sequence (16) in place of the FLAG tag in pXJ-FLAG (17). Full-length of BCL-xL were cloned into pXJ-YPet, pXJ-FLAG, and pXJ-mCherry vectors. UVRAG-(189-275) was cloned into pXJ-FLAG vector. Full-length Beclin1 was cloned into a vector with the streptavidin binding peptide pXJ-FLAG-SBP vector.5
HeLa or COS-7 cells were plated on 18 × 18-mm glass coverslips at 30% cell density. Cells were incubated with a mixture of Optifect (Invitrogen) and plasmids according to the manufacturer's protocol for 4 h and cultured in fresh medium for 2 h before fixation. MitoTracker Red CMXRos (Invitrogen) was added at 1:10,000 from a 1 mm stock to cells maintained for 30 min at 37 °C and transferred to normal medium for 30 min at 37 °C before fixation. Images were captured on an Olympus Fluoview FV1000 laser-scanning confocal microscope using a NA1.45 60× oil lens.
Streptavidin Bead Affinity Pulldown Assay and Western Blotting—COS-7 cells in 60-mm NUNC culture dishes were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 8 h fresh medium was added, and cells were cultured overnight. Total cell lysate was collected in 500 μl of lysis buffer (25 mm HEPES, pH 7.3, 100 mm NaCl, 0.15 mm MgCl2, 0.2 mm EDTA, 5% glycerol, 1 mm dithiothreitol, 0.5% Triton X-100, 1× Complete (Roche Applied Science), 1 mm phenylmethylsulfonyl fluoride) and passed through a 26G1/2 needle (10-) before clarification (10 min at 13,000 rpm). This Triton-X-100-soluble lysate (total cell lysate, ∼500 μg) was incubated with 25 μl of streptavidin-agarose (Upstate) and washed with 200 μl of phosphate-buffered saline, 0.1% Triton X-100 (3×). Bound proteins were eluted in SDS sample buffer. YPet- or FLAG-tagged proteins were detected with anti-green fluorescent protein and anti-FLAG antibodies, respectively, by Western blotting on polyvinylidene difluoride.
RESULTS
The Beclin1 CCD Forms a Dimer in Solution—Beclin1 is a tumor suppressor (6) and contains a number of structural domains, including a BH3, a coiled-coil domain (CCD), and an evolutionarily conserved domain. The evolutionarily conserved domain binds to PI(3)KC3 (18), and the CCD binds to a similar CCD of UVRAG (13), whereas the BH3 domain binds to Bcl-2 (11, 12). It has previously been shown that UVRAG binding to Beclin1 increases PI(3)KC3 kinase activity (13) and that Bcl-2 reduces this kinase activity (14). Here we have sought to understand how Bcl-2 and UVRAG cause these effects at the molecular level. First, to characterize the interactions between Beclin1, Bcl-2, and UVRAG, we expressed the CCD of Beclin1 (residues 133-266; Beclin1 CCD) or both the BH3 and CCD domains (residues 95-266; Beclin1 BH3-CCD) in E. coli and purified them to near homogeneity. The final purification step involved a size-exclusion column and the Beclin1 CCD/BH3-CCD samples eluted at a much higher molecular mass than would be expected for a compact monomer or even dimer (>100 kDa; data not shown). Therefore, we further characterized both Beclin1 fragments by analytical ultracentrifugation (Fig. 1, Tables 1 and 2). Beclin1 CCD and Beclin1 BH3-CCD were both analyzed by sedimentation equilibrium over a wide concentration range. The data for both proteins fitted well to a single-species model, and both gave very similar weight-averaged molecular weights to the formula mass of a dimer showing that Beclin1 dimerizes via its CCD. No concentration dependence was seen over the concentration range used for either protein, indicating that the dimer interface is high affinity.
FIGURE 1.

Beclin1 CCD dimerizes in solution. Beclin1 CCD (A) and Beclin1 BH3-CCD (B) were analyzed by sedimentation equilibrium and fitted to an ideal single-species model. Representative fits for each protein are shown. C, Beclin1 CCD and Beclin1 BH3-CCD were also analyzed by sedimentation velocity and fitted to the c(S) and c(M) size-distribution functions. The c(M) distribution is shown for both proteins.
TABLE 1.
Summary of sedimentation equilibrium data
| Protein | Partial specific volume | Mr | Fitted Mr | Concentration range | Rotor speeds | Stoichiometry | Ka |
|---|---|---|---|---|---|---|---|
| μM | krpm | M−1 | |||||
| Beclin1 CCD | 0.7211 | 16,660 | 32,430 | 3-165 | 20, 23, 26 | Dimer | |
| Beclin1 BH3-CCD | 0.7213 | 20,720 | 41,810 | 2-145 | 16, 18, 20 | Dimer | |
| UVRAG | 0.7507 | 10,490 | 11,510 | 2-20 | 30, 32, 34 | Monomer | |
| vBcl-2 | 0.7431 | 16,620 | 17,370 | 1-22 | 20, 23, 26 | Monomer | |
| Beclin1 BH3-CCD + vBcl-2 | 0.7213, 0.7431 | 41,440, 16,200 | 0.3-15 | 14, 16, 18 | 2:2 | 5.55 × 107 | |
| 1.13 × 106 | |||||||
| Beclin1 BH3-CCD + Bcl-xL | 0.7199 | 41,440, 24,290 | 65,070 | 0.5-17 | 14, 17, 20 | 2:2 | |
| Beclin1 CCD + UVRAG CCD | 0.7326 | 10,490, 16,660 | 30,010 | 13-74 | 18, 21, 24 | 1:1 |
TABLE 2.
Summary of sedimentation velocity data
| Protein | Sapp (from c(S)) | S20,w | f/f0 | Mr | Mr (from c(M)) |
|---|---|---|---|---|---|
| Beclin1 CCD | 1.95 | 2.02 | 1.96 | 16660 | 34200 |
| Beclin1 BH3-CCD | 2.09 | 2.17 | 2.01 | 20720 | 40000 |
| Beclin1 BH3-CCD vBcl-2 | 3.24 | 3.36 | 1.63 | 73840 | 58800 |
| Beclin1 BH3-CCD Bcl-xL | 3.14 | 3.26 | 1.86 | 89990 | 64600 |
UVRAG CCD (residues 189-275) and KSHV Bcl-2 (excluding the transmembrane domain; hereafter referred to as vBcl-2) were also analyzed by sedimentation equilibrium. As expected, both proteins were monomeric in solution (Table 1). Bcl-xL, lacking the transmembrane domain, has previously been shown to be monomeric (19).
The Beclin1 proteins were also analyzed by sedimentation velocity to confirm that the proteins are dimeric and to get an indication of the molecular shape (Fig. 1C, Table 2). The data were fitted using the continuous distribution functions c(S) and c(M). These functions fit the data to a continuous distribution of sedimentation coefficients or weight-averaged molecular weights, respectively (15). This analysis showed that both Beclin1 proteins sedimented as single species with apparent molecular weights very close to that of a dimer. Surprisingly both proteins had very high frictional ratios of ∼2. Given that the Beclin1 CCD is largely α-helical in solution (13), this shows that both proteins are highly asymmetric.
vBcl-2 Binds to Two Distinct Sites on the Beclin1 Dimer—To test whether Beclin1 remains dimeric on binding to Bcl-2-family proteins, we analyzed the binding of vBcl-2 and Bcl-xL to the Beclin1 BH3-CCD dimer by isothermal titration calorimetry (ITC). These Bcl-2 proteins were used because of their high solubility and because both human Bcl-xL and vBcl-2 bind to Beclin1 and inhibit autophagy (14). Double mutant variants of both Bcl-2-like proteins were used because this increases their solubility (11, 20). The titration curve for vBcl-2 clearly shows biphasic binding, and therefore, the data were fitted to a two-site binding model (Fig. 2A, Table 3). One high affinity interaction occurred at a molar ratio of ∼0.5, i.e. 1 vBcl-2 per Beclin1 dimer, and the second interaction occurred at a molar ratio of ∼1, i.e. 2 vBcl-2 per Beclin1 dimer. Therefore, these data show that the Beclin1 dimer contains two distinct vBcl-2 binding sites, one with high affinity and one with lower affinity. The equilibrium dissociation constant (Kd) for the higher affinity site is 25 nm, whereas for the lower affinity site it is 4.8 μm.
FIGURE 2.

Two vBcl-2 molecules bind the Beclin1 dimer; one with high and one lower affinity. A, binding isotherm using ITC for vBcl-2 in the injection syringe titrated into Beclin1 BH3-CCD in the cell. The data were fitted to a two-site binding model (lower panel). B, a complex of vBcl-2-Beclin1 BH3-CCD was co-purified and analyzed by sedimentation equilibrium. The data were fitted to a heterodimer model in which vBcl-2 bound by two separate events to the Beclin1 dimer. A representative fit is shown. C, ITC titration for vBcl-2 in the syringe and Beclin1 BH3 peptide in the cell, and the data were fitted to a single-site model.
TABLE 3.
Summary of isothermal titration calorimetry data
| Cell ligand | Injectant | n | Ka | ΔH | TΔS (293K) |
|---|---|---|---|---|---|
| M−1 | Kcal·mol−1 | Kcal·mol−1 | |||
| Beclin1 BH3-CCD | vBcl-2 | 0.545 ± 0.004 | 4.02 × 107 ± 6.8 × 106 | −1.242 × 104 ± 68 | −7.56 |
| 0.929 ± 0.02 | 2.08 × 105 ± 3.5 × 104 | −5.36 × 103 ± 276 | 6.04 | ||
| Beclin1 BH3 peptide | vBcl-2 | 0.964 ± 0.02 | 1.77 × 105 ± 1.0 × 104 | −7.10 × 103 ± 161 | −0.20 |
| Beclin1 BH3-CCD | Bcl-xL | 0.978 ± 0.004 | 9.08 × 105 ± 4.6 × 104 | −1.09 × 104 ± 63 | −9.82 |
| Beclin1 BH3 peptide | Bcl-xL | 0.952 ± 0.002 | 1.54 × 106 ± 3.0 × 104 | −1.32 × 104 ± 29 | −16.6 |
| Beclin1 BH3-CCD | UVRAG CCD | 1.02 ± 0.01 | 2.04 × 105 ± 1.2 × 104 | 1.12 × 104 ± 240 | 62.4 |
| Beclin1 BH3-CCD (2× UVRAG CCD) | vBcl-2 | 0.976 ± 0.017 | 4.59 × 105 ± 5.6 × 104 | −500 ± 118 | 8.83 |
| Beclin1 BH3-CCD (2× UVRAG CCD) | Bcl-xL | 0.954 ± 0.002 | 1.16 × 106 ± 2.2 × 104 | −1.14 × 104 ± 32 | −11.0 |
| Beclin1 BH3-CCD (2× vBcl-2) | UVRAG CCD | 1.06 ± 0.06 | 4.99 × 104 ± 8.4 × 103 | 1.58 × 104 ± 1680 | 75.5 |
| Beclin1 BH3-CCD (0.5× vBcl-2) | UVRAG CCD | 1.02 ± 0.02 | 1.85 × 105 ± 1.2 × 104 | 1.24 × 104 ± 312 | 66.3 |
| Beclin1 BH3-CCD (2× Bcl-xL) | UVRAG CCD | 1.0 | 4.66 × 104 ± 1.8 × 103 | 5.53 × 104 ± 1254 | 210 |
To confirm the existence of higher and lower affinity Bcl-2-binding sites, we co-expressed and purified a complex of vBcl-2 and Beclin1 BH3-CCD and analyzed it by sedimentation velocity and sedimentation equilibrium (Fig. 2B, Tables 1 and 2). The sedimentation equilibrium data were analyzed in terms of heteroassociation models with either a single vBcl-2 or two distinct vBcl-2 molecules binding to the Beclin1 dimer. The data could not be fitted to a single-association model but fitted very well to a model incorporating two separate binding events, and the dissociation constants were similar to those seen with the ITC data. Therefore, this confirms that Beclin1 binds two Bcl-2 molecules, one with high affinity and one with lower affinity.
To examine whether the high affinity Bcl-2-binding site on Beclin1 is created by dimerization of Beclin1, we measured binding between vBcl-2 and a Beclin1-BH3 peptide residues 107-129, which has been shown to be bound tightly in a hydrophobic groove created by helices α2-α5 of Beclin1 (11) (Fig. 2C). The data fitted to a single binding-site model with a very similar affinity to that seen for the lower affinity binding site on the Beclin1 dimer (Kd 5.6 μm). Therefore, dimerization is required to form the higher affinity vBcl-2 binding site on Beclin1.
Bcl-xL Binds to Beclin1 by a Different Mechanism—Bcl-xL bound very differently to Beclin1, and only a single binding event was detected by ITC (Fig. 3), with a Kd of 1.1 μm. Therefore, Bcl-xL binds to each BH3 domain of the Beclin1 dimer with a similar affinity. To rule out the possibility that Bcl-xL binds to monomeric Beclin1, a complex of Beclin1 and Bcl-xL was purified by gel filtration and analyzed by sedimentation equilibrium and sedimentation velocity (Tables 1 and 2). The equilibrium data globally fitted to a weight-averaged molecular mass of 65 kDa, and individual scans showed a concentration dependence and fitted to between 58 and 68 kDa. This is considerably greater than that for a Bcl-xL-Beclin1 heterodimer (Mr 45) showing that Beclin1 BH3-CCD remains dimeric and is capable of binding two Bcl-xL molecules. The velocity data were analyzed by the c(M) method, and the major species gave a similar molecular mass of ∼64 kDa.
FIGURE 3.

Bcl-xL binds to Beclin1 by a different mechanism from vBcl-2. ITC titrations with Bcl-xL in the syringe and Beclin1 BH3-CCD (A) or Beclin1 BH3 (B) peptide in the cell. The data were fitted to single-site models.
Binding between Bcl-xL and the Beclin1-BH3 peptide was also measured to examine the effect of Beclin1 dimerization on the Beclin1-Bcl-xL interaction (Fig. 3B, Table 3). Bcl-xL actually bound to the peptide with a higher affinity than to the Beclin1 dimer (Kd 0.65 versus 1.1 μm). This result shows that dimerization of Beclin1 is not required for Bcl-xL binding and suggests that Bcl-xL makes additional unfavorable interactions on binding to the Beclin1 dimer.
UVRAG Binds to Beclin1 and Disrupts the Dimer Interface—The interaction between Beclin1 BH3-CCD and UVRAG CCD was also characterized by ITC (Fig. 4A). The titration curve for this interaction was fitted to a single-site model, and binding occurred at a molar ratio of 1, showing that Beclin1 and UVRAG bind with a 1:1 stoichiometry (Kd 4.9 μm). This interaction was highly endothermic and, therefore, was driven entirely by entropy at 20 °C (TΔS, 62 kcal·mol-1; Table 3). One possible explanation for this entropy-driven binding is that UVRAG disrupts an ordered Beclin1-dimer interface. To test this possibility we analyzed Beclin1 CCD, UVRAG CCD, and a mixture of the two by size-exclusion chromatography (Fig. 4B). A peak corresponding to a lower molecular weight than the Beclin1 CCD dimer is seen when the two proteins are mixed that is not present when either is analyzed alone, indicating that the two proteins form a complex of an intermediate molecular weight. Only a small amount of the complex was detected, presumably due to the low affinity of the interaction and the fact that both proteins have very different shapes (data not shown). Nevertheless this result was reproducible and also occurred for Beclin1 BH3-CCD mixed with UVRAG. To confirm this finding, we also analyzed an equimolar mixture of Beclin1 CCD and UVRAG CCD by sedimentation equilibrium (Table 1). All the individual equilibria fitted to mass-averaged molecular masses of 26-33 kDa and globally fitted to 30 kDa, less than the Beclin1-dimer molecular mass of 33 kDa. Given that the molecular mass of 2:1 or 2:2 Beclin1·UVRAG complexes would be 44 or 54 kDa, respectively, this shows that the sample consists of an equilibrium between a Beclin1 homodimer and a Beclin1-UVRAG heterodimer, suggesting that UVRAG may disrupt the Beclin1-dimer interface.
FIGURE 4.
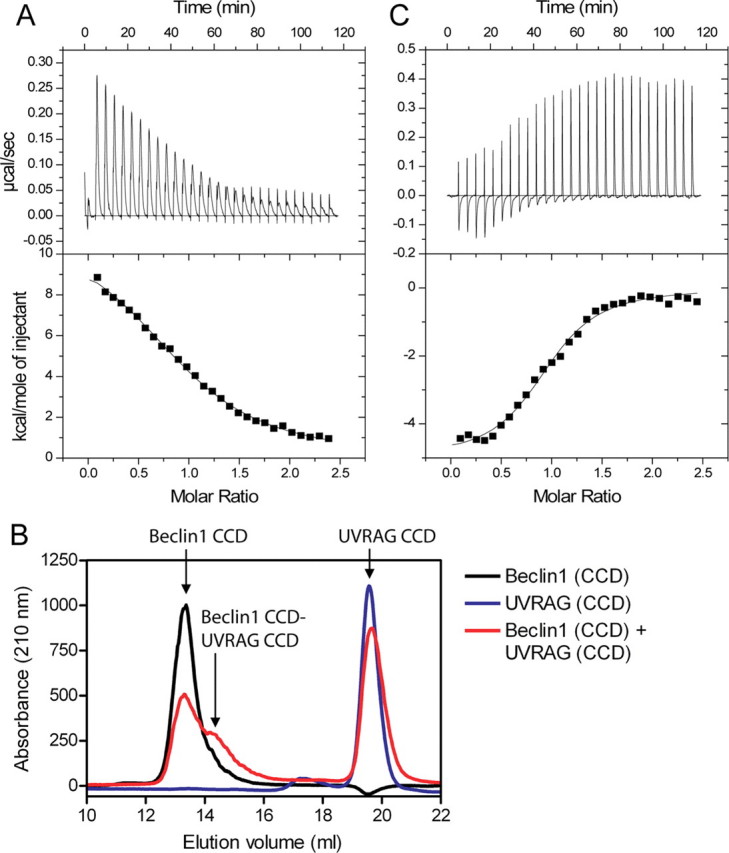
UVRAG disrupts the Beclin1 dimer interface. A, ITC titration for UVRAG CCD in the injection syringe and Beclin1 BH3-CCD in the cell. B, analytical gel-filtration profiles, measured at 210 nm for Beclin1 CCD, UVRAG CCD, or a mixture of the two. C, ITC titration for vBcl-2 in the injection syringe and Beclin1 BH3-CCD in the cell in the presence of a 2-fold excess of UVRAG CCD. The ITC data were fitted to a single-site model.
To further confirm that UVRAG really disrupts the Beclin1-dimer interface, binding between vBcl-2 and Beclin1 BH3-CCD was measured in the presence of a 2-fold molar excess of UVRAG (Fig. 4C). If UVRAG disrupts the Beclin1 dimer interface, one would expect that the high affinity interaction between vBcl-2 and Beclin1 would be removed. This binding isotherm was substantially different from that seen in the absence of UVRAG (compare Fig. 2A with Fig. 4C), and a single binding event occurred at a molar ratio of 1 showing that UVRAG does indeed bind a Beclin1 monomer. The interaction fitted to a Kd of 2.2 μm, i.e. much more similar to the lower affinity site seen in Fig. 2A. A similar experiment performed for Bcl-xL binding to Beclin1 BH3-CCD in the presence of a 2-fold excess of UVRAG CCD (Table 3) showed that UVRAG had little effect on the interaction between Bcl-xL and Beclin1 BH3-CCD.
Bcl-2-like Proteins Reduce the Affinity of UVRAG for Beclin1—To examine the effect of the Bcl-2-like proteins on the UVRAG-Beclin1 interaction, we performed further ITC titrations between UVRAG and Beclin1 BH3-CCD in the presence of a 2-fold molar excess of vBcl-2 or Bcl-xL (Fig. 5; Table 3). The titrations both show that UVRAG still binds to Beclin1 but with a reduced affinity. The Kd is reduced from 4.9 to ∼20 μm in both cases. Therefore, both vBcl-2 and Bcl-xL reduce the affinity of UVRAG for Beclin1 to a similar extent.
FIGURE 5.

Bcl-xL and Bcl-2 have a similar effect on the affinity of UVRAG for Beclin1. ITC titrations for UVRAG CCD in the syringe and Beclin1 BH3-CCD in the cell in the presence of a 2-fold excess of vBcl-2 (A) or Bcl-xL (B). The data were fitted to a single-site model and in the presence of Bcl-xL. n was held constant at 1.
The entropy driving both interactions is increased with respect to the Beclin1-UVRAG interaction alone, consistent with UVRAG breaking additional interactions on binding to the Bcl-2-xL-Beclin1 complex. This is particularly apparent for UVRAG binding to the Bcl-xL-Beclin1 complex (TΔS, 210 kcal·mol-1). It is not clear why vBcl-2 and Bcl-xL affect the affinity of UVRAG for Beclin1 by the same amount given the different manner of vBcl-2 and Bcl-xL binding to Beclin1. Therefore, we tested whether occupation of the high affinity site on Beclin1 alone could affect the affinity of the UVRAG-Beclin1 interaction. To do this we measured binding between UVRAG CCD and Beclin1 BH3-CCD in the presence of a 0.5 molar ratio of vBcl-2 (i.e. 1 vBcl-2 per Beclin1 dimer), but the affinity and energetics of the UVRAG-Beclin1 interaction were unaffected (Table 3). This indicates that both vBcl-2 molecules need to be bound to reduce the affinity of UVRAG for Beclin1.
Beclin1 Co-localizes with Bcl-xL in the Mitochondria and Dimerizes via Its CCD Domain in Vivo—To determine whether Beclin1 binds to Bcl-xL in vivo, we expressed full-length Beclin1 as a fusion with YPet in HeLa cells. Beclin1 localized mainly to the cytoplasm with no obvious specific subcellular features (Fig. 6A, a-c (21)). When Beclin1 was co-expressed with Bcl-xL, then both proteins were predominantly co-localized at the mitochondria (Fig. 6A, d-f) agreeing with previous data showing that Beclin1 interacts with Bcl-xL in vivo (12, 14). Localization to the mitochondria was confirmed by fluorescent Mito-Tracker staining of HeLa cells expressing Bcl-xL. Beclin1 BH3-CCD co-localized with Bcl-xL, but Beclin1 CCD, which lacks the BH3 domain, did not (Fig. 6B), showing that the Beclin1 interaction with Bcl-xL is dependent on the BH3 domain in vivo.
FIGURE 6.
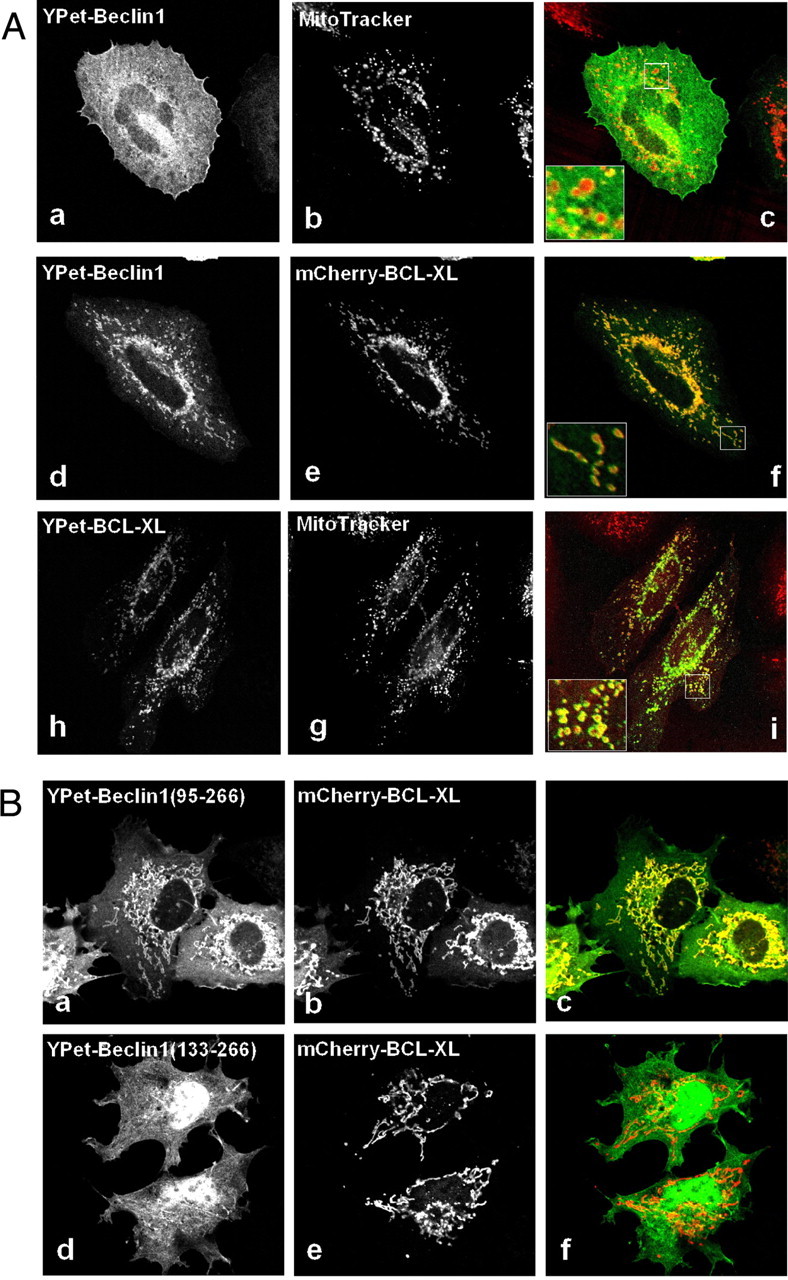
Beclin1 co-localization with Bcl-xL in mitochondria is dependent on its BH3 domain. A, HeLa cells were transfected with YPet-Beclin1 (a-c), YPet-Beclin1 and mCherry-Bcl-xL (d-f), or YPet-Bcl-xL (h-i) and fixed after 6 h. Mitochondrial co-localization was assessed using MitoTracker (b and g). Panels c, f, and i are merged images of a and b, d and e, and g and h, respectively. The insets show an enlarged area (×3.4). B, representative images of COS-7 cells expressing YPet-Beclin1-(95-266) and mCherry-Bcl-xL (a-c) or YPet-Beclin1-(133-266) and mCherry-Bcl-xL (d-f). Panels c and f are merged images of a and b and or d and e, respectively.
To show that Beclin1 homodimerizes in vivo via its CCD domain, we performed streptavidin pulldown experiments, with FLAG-SBP-Beclin1, which contains a streptavidin-binding peptide (SBP, Fig. 7A). YPet-Beclin1, YPet-Beclin1 BH3-CCD, YPet-Beclin1 CCD, and YPet-Bcl-xL all interacted with FLAG-SBP-Beclin1, showing that Beclin1 dimerizes in vivo via its CCD domain. YPet alone did not interact with FLAG-SBP-Beclin1, showing that the interaction is specific.
FIGURE 7.
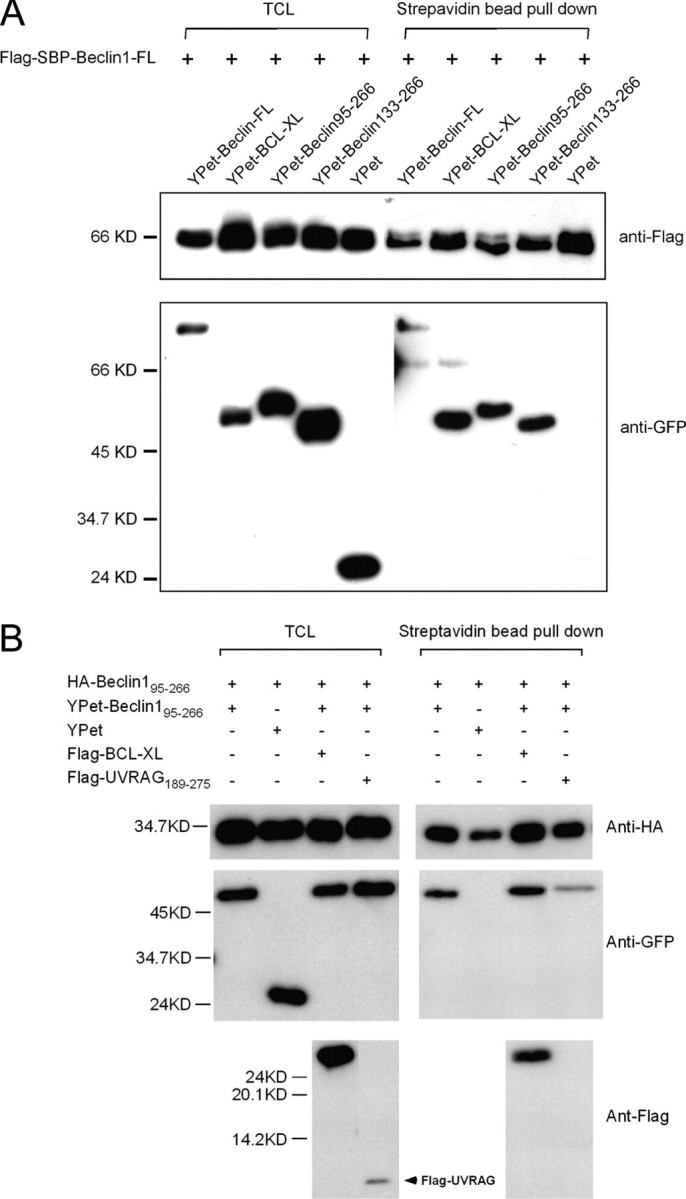
Beclin1 dimerizes via its CCD domain and this interaction is inhibited by UVRAG in vivo. A, COS7 total cell lysate (TCL, 400 μg) co-expressing FLAG-SBP-Beclin1 and YPet-fusion proteins as indicated were collected on 25 μl of streptavidin-agarose beads, and after washing, 10% of material was assessed by Western blotting for bound YPet proteins. Total cell lysate (5%) was used for comparison. GFP, green fluorescent protein. B, COS7 cells expressing hemagglutinin (HA)-Beclin1 BH3-CCD, YPet-Beclin1 BH3-CCD, and FLAG constructs as indicated were collected on 25 μl of streptavidin-agarose beads and processed as for A.
UVRAG Reduces Dimerization of Beclin1—To examine the effects of UVRAG and Bcl-xL on the homodimerization of Beclin1 in vivo, Beclin1 BH3-CCD (residues 95-266) and UVRAG CCD (residues 189-275) were used since the co-expressed full-length proteins were essentially Triton X-100-insoluble (data not shown). Beclin1 dimerization was assessed by the ability of the SBP-Beclin1 to bring down YPet-Beclin1. As shown in Fig. 7B, the dimerization of Beclin1 was unaffected by co-transfection of Bcl-xL but reduced significantly by UVRAG CCD co-expression. These results support the in vitro biophysical data, and together they provide strong evidence that UVRAG blocks Beclin1 dimerization.
DISCUSSION
It has previously been shown that UVRAG and Bcl-2-like proteins have antagonistic effects on the activation of autophagy. UVRAG binds to the CCD of Beclin1 and activates PI(3)KC3 leading to the initiation of autophagy (13), whereas Bcl-2-like proteins inhibit Beclin1-dependent activation of PI(3)KC3 activity and autophagy (14). In this work we have identified how these effects are conferred through Beclin1 by characterizing Beclin1 and its interactions with UVRAG and Bcl-2-like proteins in vitro and in vivo. Our data show that Beclin1 forms a high affinity dimer and that this dimerization can be regulated by the Bcl-2-like proteins Bcl-xL and vBcl-2 or by UVRAG. UVRAG disrupts the Beclin1-dimer, forming a heterodimer with Beclin1. Because UVRAG binding to Beclin1 activates PI(3)KC3 activity (13), it is very likely that monomerization of Beclin1 activates the kinase, possibly by allowing the kinase to bind to Beclin1. The Beclin1 dimer appears to be stabilized by the Bcl-2-like proteins, presumably by protein-protein interactions, but the molecular mechanism of this stabilization remains elusive. One possibility is that Bcl-xL forms a transient domain-swapped dimer on binding to Beclin1, thereby stabilizing the Beclin1 dimer. A domain-swapped dimer of Bcl-xL involving swapping of α-helix 1 at the N terminus has been observed in the recent crystal structure of Bcl-xL bound to the BH3 domain of Beclin1 (11), but such a domain-swapped dimer has not been shown to occur in solution (11, 24). It may occur transiently when bound to dimeric BH3-only proteins, with a relatively low activation barrier such that its formation is reversible.
Bcl-xL has also been shown to form a different domain-swapped dimer in solution, involving swapping the C-terminal α-helices 7 and 8 (22, 23). Bcl-xL is predominantly monomeric in vitro, and dimerization is promoted by heat and alkaline pH. The function of this dimer formation remains unclear, and dimers and higher order oligomers have been suggested to mediate pore formation in membranes during apoptosis (23). The energy barrier for forming the C-terminal domain-swapped dimer is relatively high, and dimerization is inhibited by the BH3 domain from BID (22). Therefore, it seems unlikely that this form of swapping occurs when bound to the Beclin1 dimer.
The effects of Bcl-xL and UVRAG on Beclin1 are modest, probably so that the reaction is reversible. For example the equilibrium dissociation constant for the interaction between UVRAG and Beclin1 is increased from ∼5 to ∼20 μm in the presence of Bcl-xL. This indicates that the change in Beclin1 monomer/dimerization is likely to be very sensitive to the local concentrations of Bcl-2-like proteins and UVRAG, as neither prevents the other from binding. An increase in UVRAG will push the equilibrium toward Beclin1 monomers, increasing the kinase activity and autophagy, whereas an increase in the Bcl-2-like protein pushes the equilibrium toward Beclin1 dimer and inhibition of autophagy. Therefore, we suggest that Beclin1 acts as a monomer-dimer switch to control autophagy. This would explain why both UVRAG and Beclin1 are haplo-insufficient tumor suppressors, i.e. loss of a single allele of each is found in human cancers (5, 7, 8, 25). Further research will be required to identify how this Beclin1 switch transmits its effects to PI(3)KC3. The PI(3)KC3 binding site on Beclin1 includes the CCD domain of Beclin1 as well as the evolutionarily conserved domain (13). It may be that Beclin1 must be monomeric for PI(3)KC3 to stably interact with the CCD domain and form an active complex to initiate autophagy.
It is not clear whether this concentration-dependent switch mechanism is used by other BH3 proteins to regulate apoptosis, as there is little indication of other BH3-only proteins forming homodimers. However, a single-allele knock-out of another BH3 protein, Bim, is counteracted by a Bcl-2 deficiency (26), showing that the relative levels of Bcl-2-like and BH3-only proteins is crucial in deciding whether programmed cell death occurs.
The interplay between Bcl-2-like proteins and UVRAG also suggests novel approaches for the development of anti-cancer agents. An important consequence of this mechanism is that compounds that disrupt the Beclin1-dimer interface or stabilize the interaction with UVRAG would be expected to activate autophagy and autophagic cell death. It will be important in future research to demonstrate that monomerization of Beclin1 alone can activate autophagy and autophagic cell death and thereby demonstrate that this is a bona fide anti-cancer target.
Acknowledgments
We thank Yigong Shi and Adam Oberstein for providing the BclxL construct, and Peter Erb and Isabelle Widmer for the gift of KSHV Bcl-2 cDNA.
This work was supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research) (to H. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PI(3)KC3, class III phosphoinositide-3 kinase; CCD, coiled-coil domain; BH, Bcl-2 homology; ITC, isothermal titration calorimetry; KSHV, Kaposi's sarcoma-associated herpesvirus; TCEP, Tris[2-carboxyethyphosphine] hydrochloride; SBP, streptavidin-binding peptide.
Z.-S. Zhao, unpublished information.
References
- 1.Levine, B., and Yuan, J. (2005) J. Clin. Investig. 115 2679-2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine, B., and Klionsky, D. J. (2004) Dev. Cell 6 463-477 [DOI] [PubMed] [Google Scholar]
- 3.Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., and Tsujimoto, Y. (2004) Nat. Cell Biol. 6 1221-1228 [DOI] [PubMed] [Google Scholar]
- 4.Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., Baehrecke, E. H., and Lenardo, M. J. (2004) Science 304 1500-1502 [DOI] [PubMed] [Google Scholar]
- 5.Aita, V. M., Liang, X. H., Murty, V. V., Pincus, D. L., Yu, W., Cayanis, E., Kalachikov, S., Gilliam, T. C., and Levine, B. (1999) Genomics 59 59-65 [DOI] [PubMed] [Google Scholar]
- 6.Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., and Levine, B. (1999) Nature 402 672-676 [DOI] [PubMed] [Google Scholar]
- 7.Yue, Z., Jin, S., Yang, C., Levine, A. J., and Heintz, N. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 15077-15082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., Rosen, J., Eskelinen, E. L., Mizushima, N., Ohsumi, Y., Cattoretti, G., and Levine, B. (2003) J. Clin. Investig. 112 1809-1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cory, S., and Adams, J. M. (2002) Nat. Rev. Cancer 2 647-656 [DOI] [PubMed] [Google Scholar]
- 10.Liang, X. H., Kleeman, L. K., Jiang, H. H., Gordon, G., Goldman, J. E., Berry, G., Herman, B., and Levine, B. (1998) J. Virol. 72 8586-8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oberstein, A., Jeffrey, P. D., and Shi, Y. (2007) J. Biol. Chem. 282 13123-13132 [DOI] [PubMed] [Google Scholar]
- 12.Maiuri, M. C., Le Toumelin, G., Criollo, A., Rain, J. C., Gautier, F., Juin, P., Tasdemir, E., Pierron, G., Troulinaki, K., Tavernarakis, N., Hickman, J. A., Geneste, O., and Kroemer, G. (2007) EMBO J. 26 2527-2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang, C., Feng, P., Ku, B., Dotan, I., Canaani, D., Oh, B. H., and Jung, J. U. (2006) Nat. Cell Biol. 8 688-699 [DOI] [PubMed] [Google Scholar]
- 14.Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., Packer, M., Schneider, M. D., and Levine, B. (2005) Cell 122 927-939 [DOI] [PubMed] [Google Scholar]
- 15.Schuck, P. (2000) Biophys. J. 78 1606-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen, A. W., and Daugherty, P. S. (2005) Nat. Biotechnol. 23 355-360 [DOI] [PubMed] [Google Scholar]
- 17.Manser, E., Huang, H. Y., Loo, T. H., Chen, X. Q., Dong, J. M., Leung, T., and Lim, L. (1997) Mol. Cell. Biol. 17 1129-1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuya, N., Yu, J., Byfield, M., Pattingre, S., and Levine, B. (2005) Autophagy 1 46-52 [DOI] [PubMed] [Google Scholar]
- 19.Thuduppathy, G. R., and Hill, R. B. (2006) Protein Sci. 15 248-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang, Q., Petros, A. M., Virgin, H. W., Fesik, S. W., and Olejniczak, E. T. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3428-3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang, X. H., Yu, J., Brown, K., and Levine, B. (2001) Cancer Res. 61 3443-3449 [PubMed] [Google Scholar]
- 22.Denisov, A. Y., Sprules, T., Fraser, J., Kozlov, G., and Gehring, K. (2007) Biochemistry 46 734-740 [DOI] [PubMed] [Google Scholar]
- 23.O'Neill, J. W., Manion, M. K., Maguire, B., and Hockenbery, D. M. (2006) J. Mol. Biol. 356 367-381 [DOI] [PubMed] [Google Scholar]
- 24.Feng, W., Huang, S., Wu, H., and Zhang, M. (2007) J. Mol. Biol. 372 223-235 [DOI] [PubMed] [Google Scholar]
- 25.Ionov, Y., Nowak, N., Perucho, M., Markowitz, S., and Cowell, J. K. (2004) Oncogene 23 639-645 [DOI] [PubMed] [Google Scholar]
- 26.Bouillet, P., Cory, S., Zhang, L. C., Strasser, A., and Adams, J. M. (2001) Dev. Cell 1 645-653 [DOI] [PubMed] [Google Scholar]