

Abstract
Reduced activity of CLK-1/MCLK1 (also known as COQ7), a mitochondrial enzyme that is necessary for ubiquinone biosynthesis, prolongs the lifespan of nematodes and mice by a mechanism that is distinct from that of the insulin signaling pathway. Here we show that 2-fold reduction of MCLK1 expression in mice reveals an additional function for the protein, as this level of reduction does not affect ubiquinone levels yet affects mitochondrial function substantially. Indeed, we observe that the phenotype of young Mclk1+/- mutants includes a severe reduction of mitochondrial electron transport, ATP synthesis, and total nicotinamide adenine dinucleotide (NADtot) pool size as well as an alteration in the activity of key enzymes of the tricarboxylic acid cycle. Surprisingly, we also find that Mclk1 heterozygosity leads to a dramatic increase in mitochondrial oxidative stress by a variety of measures. Furthermore, we find that the mitochondrial dysfunction is accompanied by a decrease in oxidative damage to cytosolic proteins as well as by a decrease in plasma isoprostanes, a systemic biomarker of oxidative stress and aging. We propose a mechanism for the conjunction of low ATP levels, high mitochondrial oxidative stress, and low non-mitochondrial oxidative damage in a long-lived mutant. Our model helps to clarify the relationship between energy metabolism and the aging process and suggests the need for a reformulation of the mitochondrial oxidative stress theory of aging.
Mutational inactivation of clk-1 in Caenorhabditis elegans (1) and partial inactivation of its orthologue Mclk1 in mice (2) prolong average and maximum lifespan in these organisms. Genetic evidence in nematodes indicates that the mechanism by which clk-1 prolongs lifespan is distinct from that of the insulin signaling pathway but overlaps with that of caloric restriction (3, 4), which makes the clk-1/Mclk1-dependent mechanism one of the very few molecularly defined and evolutionarily conserved pathways of animal aging that has been found to affect mammals (5-7). CLK-1/MCLK1 is a mitochondrial hydroxylase that is necessary for the biosynthesis of ubiquinone (coenzyme Q or UQ), a membrane antioxidant, lipid soluble enzymatic cofactor, and essential electron transporter of the mitochondrial respiratory chain (8, 9). In the absence of CLK-1/MCLK1, both worms and cultured mouse ES cells accumulate the biosynthetic intermediate demethoxyubiquinone (DMQ).2 Furthermore, no UQ can be detected in Mclk1-/- embryos, which start to die after embryonic day 8 (8). In contrast, clk-1 mutant worms, although they are equally unable to manufacture UQ9, survive thanks to their ability to absorb dietary (bacterial) UQ8 (endogenous UQ9 and bacterial UQ8 have different isoprenoid side chain lengths, as indicated by the subscript). These mutants, therefore, contain a mix of exogenous UQ8 and endogenous DMQ9. In such animals, Kayser et al. (10) discovered a reduction in electron transport from mitochondrial complex I but not from complex II, an observation that was interpreted as a difference in the way these complexes interact with the quinone pool. In contrast, in Mclk1-/- mouse ES cells, which contain DMQ9 but no detectable level of exogenous UQ, only electron transport from complex II is impaired (8).
Molecular and genetic analysis of different alleles of C. elegans clk-1 has previously suggested that CLK-1 might have other roles in mitochondrial function in addition to that of a DMQ hydroxylase. In particular, it was found that both the phenotypically severe allele clk-1(qm30), a partial deletion that does not produce any CLK-1 protein, and the phenotypically weaker allele clk-1(e2519), a Glu to Lys missense mutation that produces wild-type levels of a full-length mutant protein (11), are equally unable to sustain UQ biosynthesis (12). Furthermore, suppression of clk-1(e2519) by a tRNA missense suppressor produces only very small amounts of UQ9 but results in full phenotypic suppression (13). Together these observations suggest that at least part of the phenotype of clk-1 mutants is not because of the defect in UQ biosynthesis.
Previous analysis of Mclk1+/- mutants revealed that the level of MCLK1 protein was reduced ∼2-fold in these animals and that their lifespan was increased up to 30% compared with that of their Mclk1+/+ siblings in three distinct genetic backgrounds (2). It was also discovered that in the livers of very old Mclk1+/- mutants, analyzed shortly before natural death, there are areas of complete loss of expression of Mclk1, both mRNA and protein, through a phenomenon of loss-of-heterozygosity (2). We and others (14) have speculated that the presence of these Mclk1-/- clones might be responsible for the longevity of Mclk1+/- mutants. However, because Mclk1-/- clones have been found exclusively in very old animals, the notion of their importance for the extended lifespan of these animals implies that the extension is the result of a protective effect that takes place in old animals only and not the result of a reduction of the rate of aging throughout life.
In the present study we have tested various aspects of the phenotype of young Mclk1+/- mutants, with particular attention to mitochondria, to better understand the basis of the increased longevity of these animals. We demonstrate that the reduction of MCLK1 levels in young Mclk1+/- animals does not affect UQ levels yet profoundly alters the functions of the electron transport chain and the tricarboxylic acid (TCA) cycle while increasing mitochondrial oxidative stress. Additionally, we show that this early mitochondrial dysfunction leads to a reduction in the levels of cytosolic oxidative damage to proteins (protein carbonyls) as well as in a reduction in the levels of a systemic biomarker of aging (plasma isoprostanes), thus suggesting that the anti-aging effect triggered by low MCLK1 levels already occurs at a young age. These results provide a genetic model that will help to clarify the links between mitochondrial function and aging.
EXPERIMENTAL PROCEDURES
Animals—All the animals were housed in a pathogen-free facility at McGill University and were given a standard rodent diet and water ad libitum. Mice, 3-month-old Balb/c males, were anesthetized, sacrificed by cervical dislocation, and perfused with phosphate buffer. Tissues were then rapidly removed, rinsed, and placed in ice-cold mitochondrial isolation buffer or immediately frozen in liquid nitrogen. All procedures were approved by the McGill Animal Care and Ethics committees.
Identification of Quinones—The extraction of quinones as well as their quantification by HPLC were performed as we previously described (2). The total amount of quinone was normalized to the amount of protein.
Mitochondrial Oxygen Consumption and ATP Production—Unless otherwise stated, all the chemicals and reagents were purchased from Sigma. On the day of the experiment, fresh tissues were homogenized in 10 volumes (w/v) of the indicated homogenization buffer: 0.25 m sucrose, 10 mm Hepes buffer, pH 7.4, 1 mm EDTA (for liver and kidney); 100 mm sucrose, 10 mm EDTA, 100 mm Tris-HCl, pH 7.4, 46 mm KCl, 0.5% bovine serum albumin (BSA) (for muscle); 220 mm mannitol, 70 mm sucrose, 10 mm Hepes, pH 7.4, 1 mm EGTA, 0.04 mm BSA (for heart); 75 mm sucrose, 5 mm Hepes, pH 7.4, 1 mm EGTA (for brain). Mitochondria were isolated by standard differential centrifugation according to detailed procedures described else-where (15-18). The purity of the mitochondrial and cytosolic preparations obtained by following the published methods were checked by classic Western blotting experiments with antisera against mitochondrial porin (Calbiochem) and cytosolic α-tubulin. No porin and no α-tubulin could be detected in the cytosolic and the mitochondrial fractions respectively (Fig. 2A). Oxygen consumption of isolated mitochondria was measured polarographically with a Clark-type oxygen electrode connected to a suitable recorder (Yellow Springs instruments) in a 1.75-ml thermostatted water-jacketed closed chamber with magnetic stirring at 30 °C according to Schuh et al. (19) with some modifications. Briefly, the respiratory reaction medium consisted of 250 mm sucrose, 10 mm HEPES, pH 7.2, 20 mm KCl, 5 mm KH2PO4, and 2 mm MgCl2. Mitochondria were added to a final concentration of 0.5 mg protein/ml with external electron donor substrates consisting of either 5 mm glutamate or pyruvate (with 1 mm malate), 5 mm succinate (with 1 μm rotenone), or 0.15 mm N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD, with 2.5 mm ascorbate and 1 μm antimycin). Mitochondrial oxygen consumption in the presence of substrates and ADP (0.8 mm) is reported as state 3, whereas the state corresponding to the period after all added ADP has been converted into ATP is defined as State 4 respiration. State 4 respiration was also measured in the presence of 1.25 mg/ml oligomycin to eliminate the contribution of ATP cycling enzymes. The respiratory control ratio (state 3/state 4) of control animals was ∼7.5 with glutamate/malate and ∼4 with succinate plus rotenone, indicating intactness of the inner mitochondrial membrane. We also evaluated the coupling between ADP phosphorylation and oxygen consumption by calculating the adenosine diphosphate-to-oxygen ratio (ADP/O) from the trace graph (Fig. 2B), and the resulting data, ∼3.2 for glutamate/malate and ∼2 for succinate + rotenone, indicated that the control mitochondria were well coupled. The fully uncoupled rate of oxygen consumption was also measured after the addition of 1 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone, and no difference was observed between control and heterozygous animals (not shown). The quality of mitochondria preparation was confirmed by at least a 3-fold increase in respiration rate in the presence of the uncoupler. The quantification of ATP synthesis by isolated mitochondria was performed according to the method previously described by Drew and Leeuwenburgh (20) and using the ATP determination kit (Invitrogen) for quantitatively measuring ATP production. After the initial luminescence reading, ADP (200 μm) was added to the respiratory reaction medium containing freshly isolated mitochondria (0.5 mg/ml), and the increase in luminescence was monitored for 2 min at 30-s intervals on an infinite M200 plate reader (Tecan).
FIGURE 2.
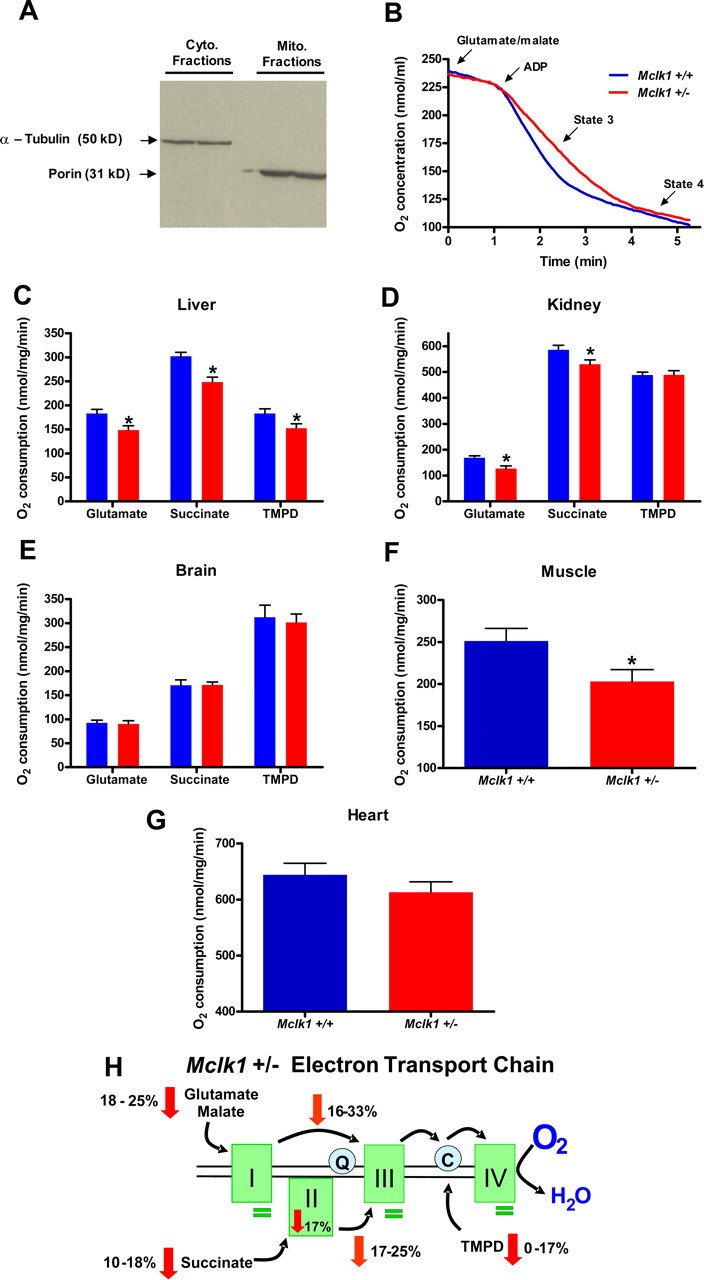
Reduction of MCLK1 levels alters mitochondrial function in young Mclk1+/- mice. A, purity of cytosolic and mitochondrial fractions was assessed by Western blotting with antisera against cytosolic (Cyto.) α-tubulin and mitochondrial (Mito.) porin. B, typical trace graph representing oxygen consumption by Mclk1+/+ and Mclk1+/- liver mitochondria after glutamate/malate addition in respiratory reaction medium. Oxygen consumption levels of isolated liver (C), kidney (D), and brain (E) mitochondria from young (3 months) males were measured with glutamate/malate, succinate, and TMPD as specific respiratory substrates. Results with Mclk1+/+ are represented with blue bars, and those with Mclk1+/- are represented with red bars. For muscle (F) and heart (G) mitochondria, respiration was measured with glutamate/malate only. Each bar in the graphs represents the mean ± S.E. of 12 Mclk1+/+ and 10 Mclk1+/- animals. The asterisk denotes statistical significance of the difference between Mclk1+/+ and Mclk1+/- animals, p < 0.05. H, schematic representation of the electron transport chain in affected tissues of Mclk1+/- mice. The rates of oxygen consumption by isolated mitochondria are decreased with complex I-, complex II-, and cytochrome c/complex IV-specific substrates (red arrows). The rates of electron transport between complexes I and III as well as between complexes II and III are reduced, as shown in orange arrows. Complex II activity is down-regulated (red arrow), whereas the activities of the other complexes are unaffected (green bars). See Table 1 for complete numerical results.
Quantitative Determination of Mitochondrial and Tissue ATP, ADP, and Total NAD Levels—Mitochondrial and tissue adenine nucleotides (ADP and ATP) levels were determined using an ATP determination kit (Invitrogen) as described else-where (21). Total nicotinamide adenine dinucleotide (NAD, NADH) levels were determined using the NAD+/NADH quantification kit (Biovision) according to the manufacturer's instructions. Standard curves generated with known amounts of purified ATP and NADH were used to calculate mitochondrial and tissue concentrations.
Electron Transport Chain Assays—Mitochondrial samples were diluted with 30 mm potassium phosphate, pH 7.4, to reach a concentration of about 1 mg/ml protein concentration and then subjected to three rounds of freezing-thawing. Activities of the respiratory chain enzymes and levels of electron transport were measured at 30 °C using a Beckman DU 640 spectro-photometer as described (22). All reactions were initiated by the addition of mitochondrial proteins, complex I (10 μg), complex II (15 μg), complex III (10 μg), complex IV (5 μg), complex I-II (5 μg), and complex II-III (5 μg). For complex II activity, activity was measured by following the decrease in absorbance due to the coupled reduction of 2,6-dichlorophenolindophenol at 600 with 750 nm as the reference wavelength. The reaction mixture containing 50 mm potassium phosphate, pH 7.4, 20 mm succinate, and 5 mm MgCl2 was preincubated with 15 μg of mitochondrial protein at 30 °C. After 10 min of incubation, antimycin (2 μg), rotenone (2 μg), KCN (2 mm), and 2,6-dichlorophenolindophenol (150 μm) were added. 2,6-Dichlorophenolindophenol can accept electrons from the complex II (23), the complex II-bound ubiquinone, or exogenous ubiquinone. Thus, the activity was first monitored for 1 min without the addition of exogenous ubiquinone. Subsequently, complex II activity was also measured with the addition of ubiquinone (Q1) (100 μm) and was monitored for 2 min. No difference was observed between both genotypes with the addition of exogenous Q1 to the reaction.
Assay of TCA Cycle Enzymes—Activities of TCA cycle enzymes (citrate synthase, aconitase, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase) in mitochondrial preparations were measured as previously described (24).
H2O2 Production—Hydrogen peroxide (H2O2) production from isolated liver mitochondria was measured fluorimetrically utilizing the Amplex Red™-horseradish peroxidase method (Invitrogen) as previously described (25) with some modifications. Briefly, the reaction medium contained 125 mm KCl, 2 mm MgCl2, 0.2 mm EGTA, 2 mm KH2PO4, 10 mm HEPES, pH 7.2, 5 units/ml horseradish peroxidase, 20 units/ml superoxide dismutase, and 1 μm Amplex Red. After mitochondrial addition, the oxidizable substrate consisting of 5 mm glutamate plus malate was added. Adenosine diphosphate (0.8 mm) was added a minute later followed by oligomycin (1.25 μg/ml). Detection of H2O2 production was measured as an increase in fluorescence of Amplex Red dye at 590 nm with the excitation wavelength set at 545 nm. The dye response was calibrated with the addition of known amounts of H2O2.
Enzymatic Antioxidant Assays—Mitochondrial and cytosolic Se-GPx activities were determined by the standard indirect method of NADPH oxidation using t-butyl hydroperoxide (26). Manganese-dependent and CuZn-superoxide dismutase activities were assessed using a commercially available kit (Cayman Chemical).
ROS Damage—Lipid peroxidation was determined by the indirect measurement of free aldehydes, malondialdehydes plus 4-hydroxyalkenals such as 4-hydroxy-2(E) nonenal, using a commercially available kit (LPO-586, OxisResearch). The level of protein carbonyl in liver tissues was determined with the Protein Carbonyl Assay kit (Cayman Chemical) according to manufacturer's instructions. Plasma free and esterified 8-isoprostanes were quantified with an 8-isoprostanes enzyme immunoassay kit (Cayman Chemical) according to the provided protocol. The plasma levels of 8-hydroxy-2-deoxyguanosine, a biomarker for oxidative damage to DNA, were determined using an enzyme-linked immunosorbent assay kit according to the manufacturer's protocol (Stressgen).
Statistical Analysis—Analyses were performed using an unpaired two-tailed Student's t test, and differences between the two genotypes (10-12 Mclk1+/+ and Mclk1+/- animals unless otherwise stated in the figure legends) were considered to be significant when p was <0.05.
RESULTS
Reduction in Mitochondrial Electron Transport between Mitochondrial Complexes Despite Normal Levels of Ubiquinone—We have shown previously that Mclk1+/- tissues exhibit the expected 2-fold decrease in Mclk1 mRNA and MCLK1 protein levels but that UQ9 levels were unaffected in tissue homogenates from such mice (2, 8). We have now extended these studies to mitochondrial extracts, which we also find to display no reduction in either UQ9 or UQ10 levels in either of the liver, kidney, or brain of the very animals used in the various studies described below (Fig. 1). This suggests that Mclk1 is in fact fully recessive for its function in UQ biosynthesis. However, as we have found that the heterozygous mice have an extended lifespan, and for other reasons out-lined in the introduction, there are grounds to believe that CLK-1/MCLK1 could also have an additional activity unrelated to UQ biosynthesis (13) but responsible for the phenotypes observed in Mclk1+/- heterozygous mutants.
FIGURE 1.

Ubiquinone levels are normal in Mclk1+/- mice. A, quantification of UQ9 levels in whole liver homogenates from 3-month-old Mclk1+/+ and Mclk1+/- Balb/c males was performed by HPLC analysis. B, UQ9 levels in isolated liver mitochondria from 3-month-old Mclk1+/+ and Mclk1+/- males. UQ10 is barely detectable in liver (not shown). C-F, UQ9 and UQ10 levels in kidney and brain mitochondria from 3-month-old Mclk1+/+ and Mclk1+/- Balb/c males. Each dot in the graphs represents an individual mouse.
As MCLK1 is a mitochondrial protein, we tested various aspects of mitochondrial function in young (3 months old) Balb/c Mclk1+/- males in five representative organs. We found that in liver (Fig. 2, B and C) and kidney (Fig. 2D), the rate of electron transport measured by oxygen consumption from intact isolated mitochondria is reduced with both complex I substrates (glutamate in combination with malate) and a complex II substrate (succinate) as well as with an artificial substrate (TMPD plus ascorbate) that donates electrons directly to cytochrome c and, thus, measures electron transport from cytochrome c to complex IV (27). Similar results were observed with Mclk1+/- males from the C57BL/6J and the 129SV/j X Balb/c genetic backgrounds (data not shown). We also tested electron transport between pairs of complexes (complex I to III and complex II to III) and found decreases in electron transport of a similar magnitude as for overall electron transport measured by oxygen consumption (Table 1; Fig. 2H). In addition, we tested oxygen consumption with glutamate in skeletal muscle and heart (Fig. 2, F and G). A clear decrease of oxygen consumption was observed in skeletal muscle similar to that seen in liver and kidney, and a similar trend was also observed in heart mitochondria. The reduction in electron transport in Mclk1+/- mutants in the absence of any reduction in UQ levels in these animals indicates that partial loss of MCLK1 affects electron transport independently of the function of the protein in UQ biosynthesis. This conclusion is also consistent with the finding of a reduction of electron transport from cytochrome c to complex IV, a step that does not require UQ.
TABLE 1.
Mitochondrial electron transport chain and TCA cycle-related measurements
ND, not determined.
|
Liver |
Kidney |
Brain |
||||
|---|---|---|---|---|---|---|
| Mclk1+/+ | Mclk1+/− | Mclk1+/+ | Mclk1+/− | Mclk1+/+ | Mclk1+/− | |
| nmol/mg/min | nmol/mg/min | nmol/mg/min | ||||
| Electron transport chain assays | ||||||
| C Ia | 144 ± 14b | 136 ± 8 | 211 ± 7 | 218 ± 7 | 182 ± 8 | 200 ± 9 |
| C II | 53 ± 4 | 41 ± 4c | 215 ± 5 | 192 ± 6c | 118 ± 10 | 113 ± 13 |
| C III | 261 ± 35 | 254 ± 19 | 262 ± 16 | 265 ± 15 | 748 ± 30 | 765 ± 30 |
| C IV | 207 ± 10 | 220 ± 16 | 213 ± 14 | 233 ± 17 | 736 ± 29 | 754 ± 21 |
| C I-III | 263 ± 33 | 178 ± 16c | 1007 ± 34 | 852 ± 23c | 467 ± 27 | 483 ± 26 |
|
C II-III
|
267 ± 28
|
201 ±
9c |
427 ± 17
|
355 ±
13c |
210 ± 13
|
234 ± 7
|
| TCA cycle enzymes | ||||||
| Citrate S. | 106 ± 8 | 95 ± 8 | ND | ND | ND | ND |
| Aconitase | 21.3 ± 1 | 16 ± 1.1c | ND | ND | ND | ND |
| α-KGDH | 10.9 ± 1.2 | 15.9 ± 1.4c | 56.2 ± 3.7 | 71.6 ± 3.7c | 39.7 ± 2 | 41.2 ± 4 |
| Iso. D. | 29.1 ± 5.3 | 33.1 ± 2.8 | ND | ND | ND | ND |
| Malate D. | 3570 ± 281 | 3261 ± 210 | ND | ND | ND | ND |
| GDH | 354 ± 77 | 290 ± 37 | 875 ± 31 | 892 ± 33 | 509 ± 42 | 561 ± 34 |
C I-IV, mitochondrial complexes; C I-III and C II-III, electron transport between complexes; Citrate S., citrate synthase; αKGDH, α-ketoglutrate dehydrogenase; Iso. D., isocitrate dehydrogenase; Malate D., malate dehydrogenase; GDH, glutamate dehydrogenase.
Data are the means ± S.E. of 10-15 samples.
Statistically significant at p < 0.05.
Using specific electron donors and acceptors we also tested the activities of complexes I through IV individually (Table 1 and Fig. 2H). In liver and kidney the activity of all complexes appeared essentially intact in the Mclk1+/- mutants. Only complex II is affected in Mclk1+/- mice but only when no soluble UQ1 is provided as a mediator to enhance electron transport to the oxidized acceptor (see“Experimental Procedures”). This suggests that there is a very mild defect in the mutant complex that can only be revealed under conditions in which electron transport by the complex cannot be efficient.
Interestingly, the defects in mitochondrial function that we describe were not observed in the brain (Fig. 2E). Brain mitochondrial function is known to be quite different from that of non-nervous tissue as is also apparent from our measurements of the wild type (Table 1). Further experiments will be necessary to determine why brain mitochondria are immune to a partial loss of MCLK1 function.
Reduction in the Rate of Mitochondrial ATP Synthesis, in Cellular ATP, and in Total Nicotinamide Adenine Dinucleotide Levels—The main function of mitochondrial electron transport is ATP production. We, therefore, determined the rate of ATP synthesis from liver mitochondria (Fig. 3A). We observed a marked decrease in the rate of ATP synthesis that correlated strongly with the rate of oxygen consumption in preparations from individual animals (Fig. 3B). We also measured ATP levels in the freshly isolated mitochondria at the beginning of the experiment (Fig. 3C) and observed a reduction in these levels. This suggests that ATP synthesis is reduced in vivo; that is, even when the mitochondria are not provided with saturating amounts of substrate, ADP, and oxygen, as is the case in vitro. We confirmed this observation by measuring ATP levels in samples of snap-frozen livers, kidneys, and hearts and observed strikingly lower ATP levels in all three organs (Fig. 3D). This decrease in ATP levels results in a lower ATP/ADP ratio in liver (Fig. 3E). Of note, the difference in absolute ATP levels between tissues and isolated mitochondria is likely attributable to a lost of ATP content that is known to occur during the isolation procedure (28). One crucial ATP-dependent cellular process that impinges on energy metabolism and many other processes is the biosynthesis of nicotinamide adenine dinucleotides (including NAD+ and NADH). We, therefore, determined the total NAD level in frozen liver and found it to be significantly lower in heterozygous mice (Fig. 3F). Thus, the altered mitochondrial function of Mclk1+/- likely affects the rate of a variety of energy-dependent cellular processes by reducing in vivo ATP levels as well as total NAD levels.
FIGURE 3.
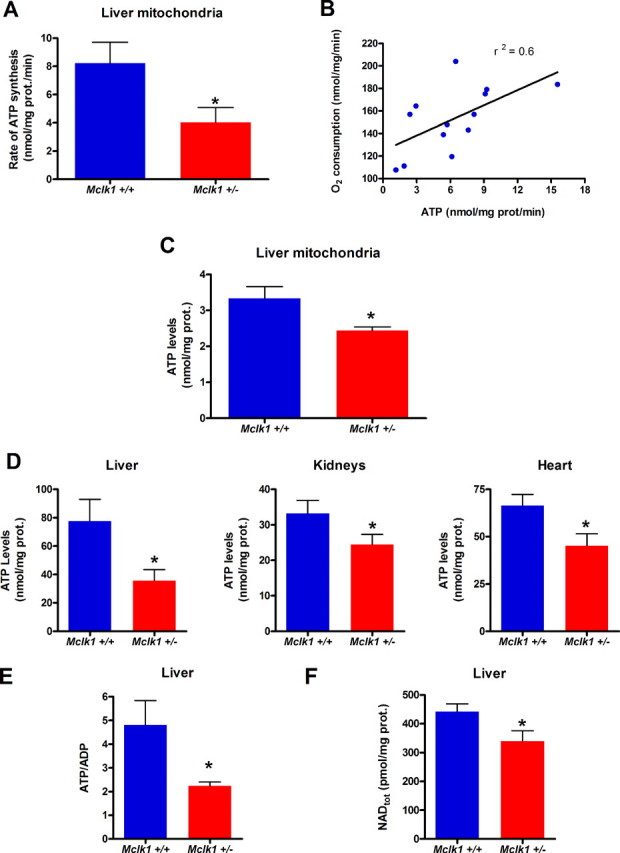
Mitochondrial ATP synthesis and cellular ATP and NAD levels are decreased in Mclk1+/- mutants. A, rates of ATP production by isolated mitochondria from liver of young (3 months) Balb/c males of both genotypes. B, correlation between the rates of ATP production and oxygen consumption by mitochondria isolated from individual animals. C, ATP levels in freshly isolated liver mitochondria from Mclk1+/+ and Mclk1+/- animals. D, cellular ATP levels measured in liver (*, p < 0.03), kidney and heart (*, p < 0.03) samples of both genotypes. E, ATP/ADP ratio measured in liver. F, total cellular NAD (NAD+, NADH) levels measured in liver. Each bar in the graphs represents the mean ± S.E. of 6 Mclk1+/+ and 7 Mclk1+/- animals. The asterisk denotes statistical significance of the difference between Mclk1+/+ and Mclk1+/- animals unless stated otherwise p < 0.05.
Altered Activities of the TCA Cycle Enzymes Aconitase and α-Ketoglutarate Dehydrogenase—Next, we tested the activities of the main enzymes of the TCA cycle in the Mclk1+/- mutants (Table 1 and Fig. 4F) and found that the activity of aconitase was severely reduced, whereas that of α-ketoglutarate dehydrogenase, a rate-limiting enzyme regulating the metabolic flux through the cycle, was increased. The other enzymes that were analyzed were unaffected. It is well known that aconitase is particularly susceptible to reaction with superoxide (29), and we believe that the reduction of aconitase is in fact because of an increase of oxidative stress in the mitochondria (see below). Interestingly, it has been predicted that in a situation in which aconitase is depleted due to oxidative stress the TCA cycle might function differently and use α-ketoglutarate dehydrogenase as a pivotal enzyme to produce NADH by way of the α-ketoglutarate produced from glutamate through transamination (30). Thus, the increased level of α-ketoglutarate dehydrogenase that we observe might represent a compensation for the loss of aconitase.
FIGURE 4.
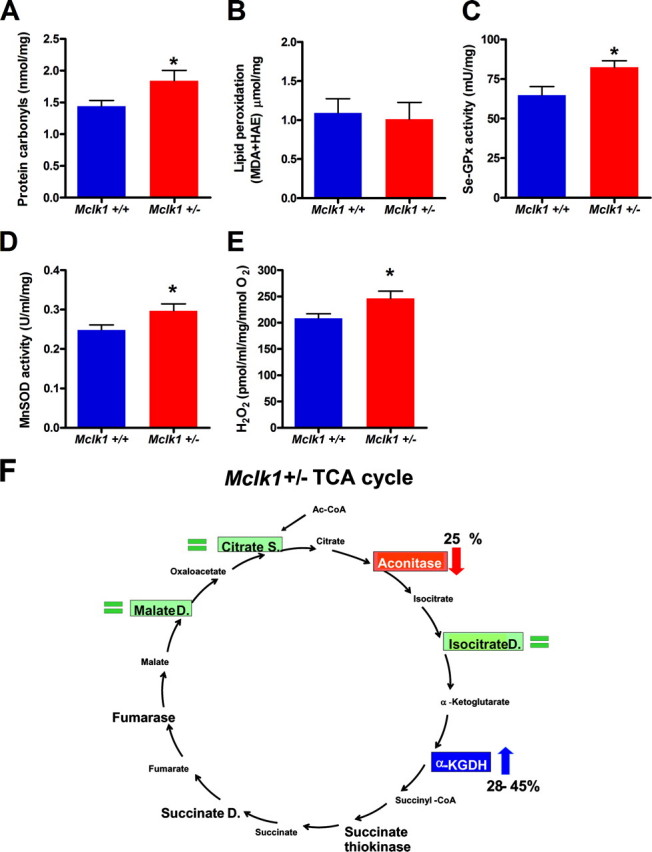
Young Mclk1+/- mice have higher mitochondrial oxidative stress and altered TCA cycle. Mitochondria of young Mclk1+/- mice display high oxidative stress as revealed by accumulation of protein carbonyls (A), higher levels, but not significantly, of lipid peroxidation (B), up-regulation of the major enzymatic antioxidant defenses, such as Se-GPx (C) and manganese-dependent superoxide dismutase (MnSOD; D), and increased ROS production per molecule of reduced O2 from isolated mitochondria (E). Each bar in the graphs represents the mean ± S.E. of 12 Mclk1+/+ and 10 Mclk1+/- animals. The asterisk denotes statistical significance of the difference between Mclk1+/+ and Mclk1+/- animals, p < 0.05. F, schematic representation of the effects of Mclk1 heterozygosity on key TCA cycle enzymes. Aconitase (in red) is partially inactivated, and α-ketoglutarate dehydrogenase (α-KGDH, in blue) is up-regulated in Mclk1+/- animals. Unaffected enzymes are showed in green.
High Oxidative Stress in the Mitochondria of Young Mutants—The loss of aconitase activity, which is a commonly used biomarker of mitochondrial oxidative stress, suggested that Mclk1+/- mutants sustain high intramitochondrial oxidative stress. To investigate this possibility, we determined the level of oxidative damage to mitochondrial proteins and mitochondrial lipids. We found that the level of protein carbonylation was increased (Fig. 4A), although the level of lipid peroxidation remained unaffected (Fig. 4B). This apparent discrepancy between protein and lipid oxidative damage could be explained by the different sensitivities of these reactions to the exact ROS species present but also by the fact that proteins appear to be one of the primary targets of mitochondrial ROS (31, 32). Furthermore, it is well known that cells react to increased oxidative stress by increasing the activity of detoxifying enzymes (33, 34). Thus, we tested two of the principal components of this detoxification system in mitochondria, the glutathione peroxidases and the manganese-dependent superoxide dismutase. All activities were increased in Mclk1+/- mutants (Fig. 4, C and D). We also measured the extramitochondrial production of ROS by isolated mitochondria. Production of ROS by mitochondria is believed to be mostly due to electron transport through the electron transport chain (33, 35). Furthermore, ROS production increases sharply when the rate of electron transport is blocked, for example by treatment with inhibitors (36). We found no difference in the absolute level of ROS produced from Mclk1+/- mitochondria (data not shown). However, the level of ROS produced relative to oxygen consumption was increased (Fig. 4E). This suggests that electron transport is less efficient in Mclk1+/- mutants in terms of channeling electrons through the electron transport chain while preventing the loss of electrons to superoxide production. Thus, in Mclk1+/- mutants, aconitase and mitochondrial proteins in general sustain increased oxidative damage despite the up-regulation of detoxifying enzyme activities, and absolute levels of ROS production by mitochondria are at least as high as in the wild type. Taken together, these results suggest that there is a substantial increase in mitochondrial levels of ROS (Fig. 5H).
FIGURE 5.
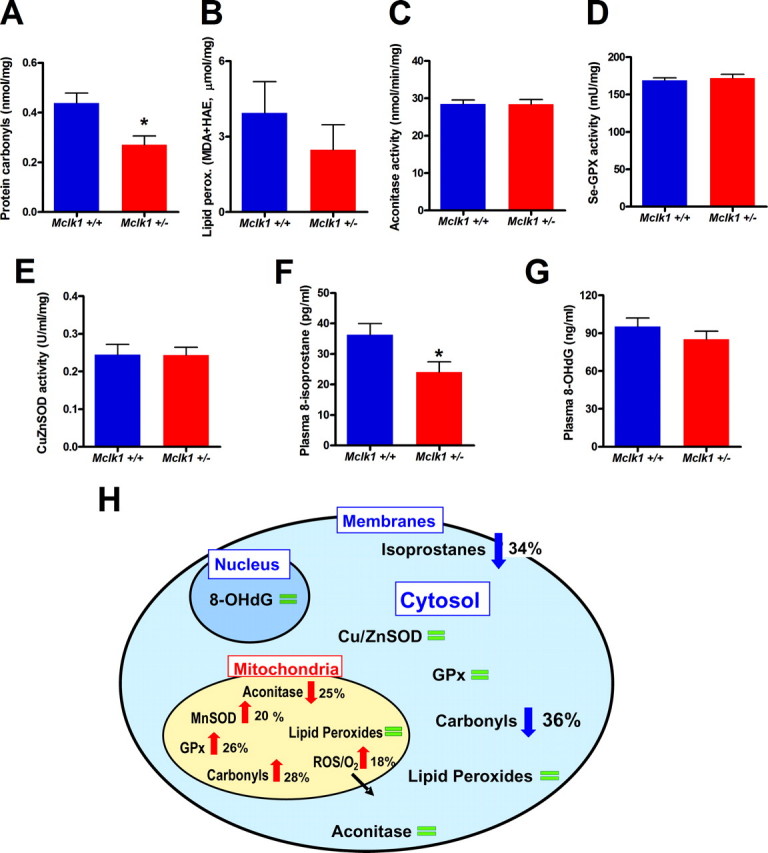
Normal or low cytoplasmic and systemic oxidative stress in young Mclk1+/- mutants. Analysis of cytoplasmic oxidative stress markers, including the levels of cytoplasmic protein carbonyls (A) (*, p < 0.01), lipid peroxidation (B), and aconitase activity (C) as well as glutathione peroxidase (GPx) and copper-zinc superoxide dismutase (Cu,ZnSOD) activities (D-E) revealed low or normal oxidative stress in non-mitochondrial compartments in Mclk1+/- mice. MDA, malondialdehyde; HAE, 4-hydroxy-alkenal. Furthermore, the measurement of the levels of plasmatic 8-hydroxy-2′-deoxyguanosine (8-OHdG) (F) and free and etherified isoprostanes (*, p < 0.03) (G) also indicate low or normal systemic oxidative stress in Mclk1+/- mutants. Each bar in the graphs represents the mean ± S.E. of 12 Mclk1+/+ and 10 Mclk1+/- animals. The asterisk denotes statistical significance of the difference between Mclk1+/+ and Mclk1+/- animals. H, schematic representation of the effects of Mclk1 heterozygosity on mitochondrial, cytosolic, and systemic oxidative stress. Changes that indicate the presence of increased oxidative stress are indicated with red arrows, whereas blue arrows indicate changes that indicate low oxidative stress and green bars indicate no difference between genotypes.
Normal or Low Cytoplasmic Oxidative Stress in Young Mutants—Cytoplasmic ROS damage parameters were also measured, but no indication of an increase in cytoplasmic ROS or ROS damage was found (Fig. 5, A-G). In fact, we observed a decrease in protein carbonylation (Fig. 5A). Cytoplasmic lipid peroxidation appeared reduced, but the effect did not reach significance (Fig. 5B). Aconitase activity was also normal (Fig. 5C). It is notable that, because CLK-1/MCLK1 is a mitochondrial protein, the reduced cytoplasmic protein carbonylation is likely a secondary effect of the increased oxidative stress or the decreased electron transport observed in the mitochondria. However, our analysis of detoxifying enzymes (Fig. 5, D and E), which revealed no change in cytoplasmic enzyme activities, suggests that increased protection from cytoplasmic ROS in reaction to the mitochondrial stress is not the mechanism that keeps cytoplasmic carbonylation low. A more likely mechanism is a reduction in the rate of cytoplasmic ROS-producing processes due to the decreased levels of ATP and total NAD in these animals (Figs. 2 and 6).
FIGURE 6.
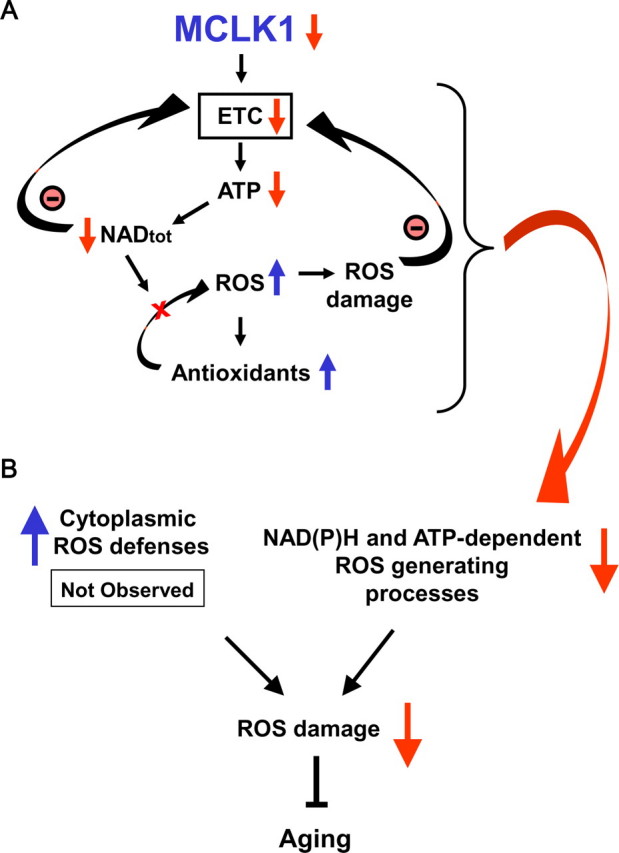
Links between the early mitochondrial dysfunction, the attenuation of systemic oxidative stress, and the long-lived phenotype of Mclk1+/- mice. A, reduction of MCLK1 levels in Mclk1+/- mutants reduces electron transport chain function. This results in a reduction in ATP levels, which is likely to have a negative effect on various ATP-dependent mitochondrial and cellular processes such as total nicotinamide adenine dinucleotide, NADtot, biosynthesis. Because the integrity of the NADtot (NAD+, NADH) pool is crucial for electron transport chain (ETC) function, the resulting reduction in NADtot levels will also negatively affect mitochondrial energy production, thus creating a vicious cycle. Additionally, the decrease in NADtot levels will compromise the ROS-detoxifying capacity of crucial mitochondrial NADPH-dependent antioxidants such as the glutathione peroxidases (GPx) that required reduced glutathione generated by glutathione reductase in an NADPH-dependent reaction. This situation leads to an increase in intramitochondrial ROS damage despite an up-regulation of antioxidant activities which can further contribute to reduction in electron transport chain function. B, the early mitochondrial dysfunction observed in Mclk1+/- mutant is paradoxically link to a decrease in the levels of cytosolic and systemic oxidative stress. This could be the result of an up-regulation of cytoplasmic antioxidant defenses in response to mitochondrial oxidative stress, but this was not observed. Thus, a more likely mechanism is a reduction in the rate of cytoplasmic ROS-producing oxidases due to the decreased levels of ATP and NADtot. Because it is well known that systemic oxidative stress is tightly linked to aging, our model could explain how a reduction in MCLK1 levels ultimately slows down the aging process.
Reduced Levels of Systemic Oxidative Stress Biomarkers—We carried out two tests for markers of systemic oxidative stress. We measured plasma levels of isoprostanes, which can be secreted into the plasma from multiple tissues as a result of damage to membrane lipids (37). These levels, which are known to increase sharply with age, were reduced in Mclk1 heterozygotes compared with Mclk1+/+ (Fig. 5F). As the majority of the oxidative metabolites of lipids that find their way into the plasma do not originate in the mitochondria but in cellular membranes, the reduction observed in young Mclk1+/- mutants are likely because of a decrease in cytoplasmic ROS-generating processes (Fig. 6). We also analyzed oxidative damage to DNA by measuring plasma levels of 8-hydroxy-2′-deoxyguanosine, a modified nucleoside base that is present in the plasma, excreted in the urine, and associated with aging (38). Although the levels of 8-hydroxy-2-deoxyguanosine appear slightly reduced in Mclk1+/- animals, we found no significant difference between genotypes (Fig. 5G).
DISCUSSION
Our study strongly suggests that CLK-1/MCLK1 determines lifespan in a ubiquinone-independent manner. Previously, we had found that UQ levels were reduced in the livers of very old, moribund, Mclk1+/- mutants but not in young animals due to the presence in the former of cells that had lost both copies of Mclk1 by loss-of-heterozygosity (2). At the time, the only documented phenotype of Mclk1+/- mutants was their increased lifespan, which could have been the result of the loss of Mclk1 in very old animals (14). The present findings, however, strongly imply that the well known function of CLK1/MCLK1 in UQ biosynthesis is not linked to the increased lifespan phenotype. Indeed, we find that Mclk1 heterozygosity affects the function of mitochondria in multiple organs in young animals in the absence of any effect on the levels of UQ (Fig. 1). Mitochondrial function appears very sensitive to the additional activity of MCLK1, as profound effects result from an only 2-fold reduction of wild-type MCLK1. This suggests that this haplo-insufficient function of MCLK1 might not be enzymatic in nature but, rather, involves a stoichiometric relation to other mitochondrial proteins. Our conclusion of a second function for CLK-1/MCLK1 is consistent with a recent study which indicates that exogenous CoQ10 is unable to restore wild-type ATP levels in dissociated cells from Mclk1-/- embryos cultured in vitro (39).
We have found by several measures that the reduction of MCLK1 levels alters the function of the electron transport chain and, thus, leads to early mitochondrial dysfunction. This defect results in a reduction of energy metabolism as revealed by a lower oxygen consumption rate and a substantial decrease in mitochondrial ATP production as well as in cellular ATP levels. Because ATP is the principal carrier of energy in cells, this reduction is likely to interfere with many important ATP-dependent cellular processes. This hypothesis was quickly confirmed by our finding that cellular levels of total NAD (NAD+, NADH), whose generation is dependent on the levels of ATP (40), were significantly decreased in Mclk1+/- mutants. It is well established that the NAD+/NADH pool plays crucial roles in mediating mitochondrial energy metabolism such as by donating electrons to the electron transport chain or by acting as a coenzyme for rate-limiting TCA cycle enzymes (41). Thus, the decrease in NAD and in ATP levels observed in Mclk1+/- mice likely results in a vicious cycle that depresses energy metabolism (Fig. 6A). Moreover, NAD as well as ATP are necessary for the generation of nicotinamide adenine dinucleotide phosphates (including NADP+ and NADPH), molecules involved in important cellular functions such as energy metabolism, signal transduction, and calcium homeostasis (42). Interestingly, NADPH is also the coenzyme for many enzymes participating in mitochondrial ROS detoxification, including glutathione reductases, thioredoxin reductases, and cytochrome P450 reductases (43). In fact, it was shown that a diminished NADPH concentration in mitochondria results in an elevated level of ROS damage and a significant reduction of the cellular ATP concentration (44). Therefore, the increase in mitochondrial oxidative stress that occurs in Mclk1+/- mice despite the up-regulation of the main antioxidants, especially glutathione peroxidases, could be attributable to a reduction in the formation of NADPH, and this phenomenon could further contribute to the vicious cycle leading to mitochondrial dysfunction (Fig. 6A).
The analysis of the mitochondrial phenotypes of Mclk1+/- mutants supports the notion that mitochondrial energy metabolism is crucial to aging. The electron transport chain in mitochondria is the site of ATP production and the main source of energy production in the cells of animals. Therefore, one expects mitochondrial function to be tightly linked to global energy production and utilization. We have found that mitochondrial function as well as ATP and total NAD levels are significantly reduced in young Mclk1+/- mutants and further discovered that this decrease is linked to lower levels of cytoplasmic oxidative damage and to a reduction in an age-associated systemic biomarkers of oxidative stress to membranes. As Mclk1+/- mutants are known to be long-lived, our data supports one aspect of the rate-of-living theory of aging that postulates the existence of a link between energy metabolism and aging (45, 46). Indeed, this theory proposes that what determines the lifespan of an organism is the rate at which it produces and uses energy at the cellular level. This theory was originally mostly supported by the observation that individuals belonging to species that have lower mass-specific metabolic rates tend to live longer than individual belonging to species with higher mass-specific metabolic rates. However, the inferences drawn from data about basal metabolic rates and its scaling in species of different body masses and life spans are now considered flawed by many (45, 46). On the other hand, our current as well as other findings (3, 47, 48) lead to the same conclusion, that is, the importance of energy metabolism, by way of a very different type of evidence.
Interestingly, our observations also suggest that the reduction of Mclk1 partially mimics some of the down-stream effects of caloric restriction, which include reduced metabolism (49) and reduced systemic oxidative damage (50). We also found that TCA cycle enzymes from liver mitochondria of Mclk1+/- mutants and mitochondrial antioxidants are regulated in part like those of calorie-restricted rodents (51, 52). Furthermore, given the evolutionary conservation of the action of clk-1/Mclk1 on lifespan, it is likely relevant that our previous work with C. elegans has shown that the effects of caloric restriction and clk-1 on lifespan are partially redundant (4). However, in contrast to what is observed in calorie-restricted animals (53), we observe an increase in mitochondrial oxidativestressinMclk1+/-mutants. This might indicate that caloric restriction and Mclk1 converge on the same downstream mechanisms but that the reduction in mitochondrial ROS levels observed in calorie-restricted animals might not be the mechanism by which caloric restriction extends life span.
Our results also suggest that mitochondrial oxidative stress is not the link between energy generation and aging. Indeed, a plausible mechanism for the link between energy metabolism and lifespan is the generation of ROS as byproducts of mitochondrial function (46, 54). Mitochondrial ROS can damage mitochondria, and there is a well documented gradual loss of mitochondrial function with chronological age (55, 56). This loss, by impairing adequate cellular energy production, could be the cause of the dysfunction of senescent cells and organisms. Based on these findings, the mitochondrial oxidative stress theory of aging states that production of ROS during cellular respiration is responsible for the damage associated with the aging process and is a key factor affecting species longevity (57). Experimental evidence in C. elegans supports part of this model as it indicates that reduction in mitochondrial electron transport can dramatically prolong worm life span (47, 58). However, this effect in worms is likely not mediated by ROS (48, 58-60). Our findings with Mclk1 also appear incompatible with the mitochondrial oxidative stress theory of aging. Indeed, we have found that young Mclk1+/- mutant mitochondria sustain high oxidative stress, yet their altered function ultimately results in slow aging. The significance of our findings is that, as the Mclk1+/- mutants live long and differ minimally from their Mclk1+/+ siblings at the molecular level (reduced dosage of one naturally encoded gene), it is difficult to conceive of a mechanism by which the same minimal change could both decrease the rate of aging and increase mitochondrial oxidative stress if the latter is the cause of aging.
It is important to note that despite the above considerations, our results remain in line with the overwhelming number of studies which have shown that increased oxidative stress is tightly correlated with the aged phenotype (61). Indeed, this correlation is maintained in our experiments as we find that the phenotype of Mclk1+/- mutants is characterized by a low level of non-mitochondrial oxidative damage. In fact, most past observations as well as our current observations are consistent with the notion that increased oxidative stress is a consequence of aging that strongly impinges on the aged phenotype.
Our findings should help to redefine the link between mitochondrial ROS production, non-mitochondrial oxidative stress, and lifespan. In fact, our results are consistent with other studies which have found that early deregulation of mitochondrial ROS production can be associated with increased lifespan. Indeed, elevated ROS production has been measured in isolated mitochondria from long-lived C. elegans daf-2 mutants, and it has been shown that the rates of mitochondrial H2O2 production tended to increase with mean survival in different Drosophila lines (62, 63). Another recent study indicates that glucose restriction prolongs C. elegans lifespan by inducing mitochondrial ROS production (64). In mammals, it was shown that various tissues of the long-lived naked-mole rats exhibit high oxidative damage, that knock-out mice heterozygous for the mitochondrial manganese-dependent superoxide dismutase showed the expected increases in mitochondrial oxidative stress without any effects on lifespan, and that reduced levels of the antioxidant enzyme GPX4 increases mouse lifespan (65-67). One possibility to explain these striking results is that the increased mitochondrial oxidative stress induces compensatory mechanisms that reduce oxidative stress in other cellular compartments such as in the cytosol, nucleus, and membrane systems and ultimately contribute to the long lifespan phenotype. However, it remains unclear what these compensatory mechanisms would be as we could observe no obvious increase in the activity of cytosolic detoxifying enzymes. Alternatively, slow cellular metabolism due to low ATP synthesis and low ATP levels could result in lower activity of cytoplasmic energy-dependent processes that produce ROS and lead to oxidative stress (Fig. 6B). In addition, low levels of total NAD should negatively affect the activity of extra-mitochondrial NADH/NADPH oxidases, which are known to be major generators of ROS (68, 69) and, thus, also contribute to reduce the accumulation of systemic oxidative damage that is likely involved in the increased lifespan phenotype of the Mclk1+/- mice (Fig. 6B).
Acknowledgments
We thankÈve Bigras and Aurélie Masurel for help with animal colony maintenance and genotyping. We thank Zaruhi Stepanyan for preliminary results on aconitase activity. We thank Robyn Branicky for reading the manuscript.
This study was funded in part by a grant from the National Science and Engineering Research Council of Canada. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: DMQ, demethoxyubiquinone; TCA, tricarboxylic acid; HPLC, high performance liquid chromatography; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine; ROS, reactive oxygen species.
References
- 1.Wong, A., Boutis, P., and Hekimi, S. (1995) Genetics 139 1247-1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu, X., Jiang, N., Hughes, B., Bigras, E., Shoubridge, E., and Hekimi, S. (2005) Genes Dev. 19 2424-2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakowski, B., and Hekimi, S. (1996) Science 272 1010-1013 [DOI] [PubMed] [Google Scholar]
- 4.Lakowski, B., and Hekimi, S. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 13091-13096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenyon, C. (2005) Cell 120 449-460 [DOI] [PubMed] [Google Scholar]
- 6.Hekimi, S. (2006) Nat. Genet. 38 985-991 [DOI] [PubMed] [Google Scholar]
- 7.Taguchi, A., Wartschow, L. M., and White, M. F. (2007) Science 317 369-372 [DOI] [PubMed] [Google Scholar]
- 8.Levavasseur, F., Miyadera, H., Sirois, J., Tremblay, M. L., Kita, K., Shoubridge, E., and Hekimi, S. (2001) J. Biol. Chem. 276 46160-46164 [DOI] [PubMed] [Google Scholar]
- 9.Nakai, D., Yuasa, S., Takahashi, M., Shimizu, T., Asaumi, S., Isono, K., Takao, T., Suzuki, Y., Kuroyanagi, H., Hirokawa, K., Koseki, H., and Shirsawa, T. (2001) Biochem. Biophys. Res. Commun. 289 463-471 [DOI] [PubMed] [Google Scholar]
- 10.Kayser, E. B., Sedensky, M. M., Morgan, P. G., and Hoppel, C. L. (2004) J. Biol. Chem. 279 54479-54486 [DOI] [PubMed] [Google Scholar]
- 11.Hihi, A. K., Kebir, H., and Hekimi, S. (2003) J. Biol. Chem. 278 41013-41018 [DOI] [PubMed] [Google Scholar]
- 12.Jonassen, T., Larsen, P. L., and Clarke, C. F. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 421-426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Branicky, R., Nguyen, P. A., and Hekimi, S. (2006) Mol. Cell. Biol. 26 3976-3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilaniu, H., Durieux, J., and Dillin, A. (2005) Genes Dev. 19 2399-2406 [DOI] [PubMed] [Google Scholar]
- 15.Kwong, L. K., and Sohal, R. S. (1998) Arch. Biochem. Biophys. 350 118-126 [DOI] [PubMed] [Google Scholar]
- 16.Starkov, A. A., Fiskum, G., Chinopoulos, C., Lorenzo, B. J., Browne, S. E., Patel, M. S., and Beal, M. F. (2004) J. Neurosci. 24 7779-7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellard, J. P., McCudden, C. R., Tanega, C., James, K. A., Ratkovic, S., Staples, J. F., and Wagner, G. F. (2007) Mol. Cell. Endocrinol. 264 90-101 [DOI] [PubMed] [Google Scholar]
- 18.Zini, R., Berdeaux, A., and Morin, D. (2007) Free Radic. Res. 41 1159-1166 [DOI] [PubMed] [Google Scholar]
- 19.Schuh, R. A., Kristian, T., Gupta, R. K., Flaws, J. A., and Fiskum, G. (2005) Toxicol. Sci. 88 495-504 [DOI] [PubMed] [Google Scholar]
- 20.Drew, B., and Leeuwenburgh, C. (2003) Am. J. Physiol. Regul. Integr. Comp. Physiol. 285 1259-1267 [DOI] [PubMed] [Google Scholar]
- 21.Sone, H., Sasaki, Y., Komai, M., Toyomizu, M., Kagawa, Y., and Furukawa, Y. (2004) Biochem. Biophys. Res. Commun. 314 824-829 [DOI] [PubMed] [Google Scholar]
- 22.Kwong, L. K., and Sohal, R. S. (2000) Arch. Biochem. Biophys. 373 16-22 [DOI] [PubMed] [Google Scholar]
- 23.Yu, L., and Yu, C. A. (1982) J. Biol. Chem. 257 2016-2021 [PubMed] [Google Scholar]
- 24.Nulton-Persson, A. C., and Szweda, L. I. (2001) J. Biol. Chem. 276 23357-23361 [DOI] [PubMed] [Google Scholar]
- 25.Muller, F. L., Liu, Y., and Van Remmen, H. (2004) J. Biol. Chem. 279 49064-49073 [DOI] [PubMed] [Google Scholar]
- 26.Flohe, L., and Gunzler, W. A. (1984) Methods Enzymol. 105 114-121 [DOI] [PubMed] [Google Scholar]
- 27.Crinson, M., and Nicholls, P. (1992) Biochem. Cell Biol. 70 301-308 [DOI] [PubMed] [Google Scholar]
- 28.Austin, J., and Aprille, J. R. (1984) J. Biol. Chem. 259 154-160 [PubMed] [Google Scholar]
- 29.Gardner, P. R., Raineri, I., Epstein, L. B., and White, C. W. (1995) J. Biol. Chem. 270 13399-13405 [DOI] [PubMed] [Google Scholar]
- 30.Tretter, L., and Adam-Vizi, V. (2005) Philos. Trans. R. Soc. Lond. B. Biol. Sci. 360 2335-2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowaltowski, A. J., and Vercesi, A. E. (1999) Free Radic. Biol. Med. 26 463-471 [DOI] [PubMed] [Google Scholar]
- 32.Temple, M. D., Perrone, G. G., and Dawes, I. W. (2005) Trends Cell Biol. 15 319-326 [DOI] [PubMed] [Google Scholar]
- 33.Cadenas, E., Boveris, A., Ragan, C. I., and Stoppani, A. O. (1977) Arch. Biochem. Biophys. 180 248-257 [DOI] [PubMed] [Google Scholar]
- 34.Franco, A. A., Odom, R. S., and Rando, T. A. (1999) Free Radic. Biol. Med. 27 1122-1132 [DOI] [PubMed] [Google Scholar]
- 35.Turrens, J. F., Alexandre, A., and Lehninger, A. L. (1985) Arch. Biochem. Biophys. 237 408-414 [DOI] [PubMed] [Google Scholar]
- 36.Turrens, J. F., and Boveris, A. (1980) Biochem. J. 191 421-427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrow, J. D. (2000) Drug Metab. Rev. 32 377-385 [DOI] [PubMed] [Google Scholar]
- 38.Chiou, C. C., Chang, P. Y., Chan, E. C., Wu, T. L., Tsao, K. C., and Wu, J. T. (2003) Clin. Chim. Acta 334 87-94 [DOI] [PubMed] [Google Scholar]
- 39.Takahashi, M., Shimizu, T., Moriizumi, E., and Shirasawa, T. (2008) Mech. Ageing Dev. 129 291-298 [DOI] [PubMed] [Google Scholar]
- 40.Magni, G., Amici, A., Emanuelli, M., Raffaelli, N., and Ruggieri, S. (1999) Adv. Enzymol. Relat. Areas Mol. Biol. 73 135-182 [DOI] [PubMed] [Google Scholar]
- 41.Berger, F., Ramirez-Hernandez, M. H., and Ziegler, M. (2004) Trends Biochem. Sci. 29 111-118 [DOI] [PubMed] [Google Scholar]
- 42.Lerner, F., Niere, M., Ludwig, A., and Ziegler, M. (2001) Biochem. Biophys. Res. Commun. 288 69-74 [DOI] [PubMed] [Google Scholar]
- 43.Ying, W. (2008) Antioxid. Redox Signal. 10 179-206 [DOI] [PubMed] [Google Scholar]
- 44.Jo, S. H., Son, M. K., Koh, H. J., Lee, S. M., Song, I. H., Kim, Y. O., Lee, Y. S., Jeong, K. S., Kim, W. B., Park, J. W., Song, B. J., and Huh, T. L. (2001) J. Biol. Chem. 276 16168-16176 [DOI] [PubMed] [Google Scholar]
- 45.Pearl, R. (1922) The Biology of Death, J. B. Lippincott, Philadelphia
- 46.Speakman, J. R. (2005) J. Exp. Biol. 208 1717-1730 [DOI] [PubMed] [Google Scholar]
- 47.Feng, J., Bussiere, F., and Hekimi, S. (2001) Dev. Cell 1 633-644 [DOI] [PubMed] [Google Scholar]
- 48.Yang, W., Li, J., and Hekimi, S. (2007) Genetics 177 2063-2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarter, R. J., and McGee, J. R. (1989) Am. J. Physiol. 257 E175-E179 [DOI] [PubMed] [Google Scholar]
- 50.Lopez-Torres, M., Gredilla, R., Sanz, A., and Barja, G. (2002) Free Radic. Biol. Med. 32 882-889 [DOI] [PubMed] [Google Scholar]
- 51.Hagopian, K., Ramsey, J. J., and Weindruch, R. (2004) Exp. Gerontol. 39 1145-1154 [DOI] [PubMed] [Google Scholar]
- 52.Sreekumar, R., Unnikrishnan, J., Fu, A., Nygren, J., Short, K. R., Schimke, J., Barazzoni, R., and Nair, K. S. (2002) Am. J. Physiol. 283 E38-E43 [DOI] [PubMed] [Google Scholar]
- 53.Sohal, R. S., Ku, H. H., Agarwal, S., Forster, M. J., and Lal, H. (1994) Mech. Ageing Dev. 74 121-133 [DOI] [PubMed] [Google Scholar]
- 54.Finkel, T., and Holbrook, N. J. (2000) Nature 408 239-247 [DOI] [PubMed] [Google Scholar]
- 55.Cadenas, E., and Davies, K. J. (2000) Free Radic. Biol. Med. 29 222-230 [DOI] [PubMed] [Google Scholar]
- 56.Shigenaga, M. K., Hagen, T. M., and Ames, B. N. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 10771-10778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harman, D. (1972) J. Am. Geriatr. Soc. 20 145-147 [DOI] [PubMed] [Google Scholar]
- 58.Hekimi, S., and Guarente, L. (2003) Science 299 1351-1354 [DOI] [PubMed] [Google Scholar]
- 59.Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., Kamath, R. S., Ahringer, J., and Kenyon, C. (2002) Science 298 2398-2401 [DOI] [PubMed] [Google Scholar]
- 60.Lee, S. S., Lee, R. Y., Fraser, A. G., Kamath, R. S., Ahringer, J., and Ruvkun, G. (2003) Nat. Genet. 33 40-48 [DOI] [PubMed] [Google Scholar]
- 61.Sohal, R. S. (2002) Free Radic. Biol. Med. 33 573-574 [DOI] [PubMed] [Google Scholar]
- 62.Ballard, J. W., Katewa, S. D., Melvin, R. G., and Chan, G. (2007) Ann. N. Y. Acad. Sci. 1114 93-106 [DOI] [PubMed] [Google Scholar]
- 63.Brys, K., Vanfleteren, J. R., and Braeckman, B. P. (2007) Exp. Gerontol. 42 845-851 [DOI] [PubMed] [Google Scholar]
- 64.Schulz, T. J., Zarse, K., Voigt, A., Urban, N., Birringer, M., and Ristow, M. (2007) Cell Metab. 6 280-293 [DOI] [PubMed] [Google Scholar]
- 65.Andziak, B., O'Connor, T. P., Qi, W., DeWaal, E. M., Pierce, A., Chaudhuri, A. R., Van Remmen, H., and Buffenstein, R. (2006) Aging Cell 5 463-471 [DOI] [PubMed] [Google Scholar]
- 66.Ran, Q., Liang, H., Ikeno, Y., Qi, W., Prolla, T. A., Roberts, L. J., Jr., Wolf, N., VanRemmen, H., and Richardson, A. (2007) J. Gerontol. 62 932-942 [DOI] [PubMed] [Google Scholar]
- 67.Williams, M. D., Van Remmen, H., Conrad, C. C., Huang, T. T., Epstein, C. J., and Richardson, A. (1998) J. Biol. Chem. 273 28510-28515 [DOI] [PubMed] [Google Scholar]
- 68.Bedard, K., and Krause, K. H. (2007) Physiol. Rev. 87 245-313 [DOI] [PubMed] [Google Scholar]
- 69.Maia, L., Vala, A., and Mira, L. (2005) Free Radic. Res. 39 979-986 [DOI] [PubMed] [Google Scholar]