

Abstract
20S RNA virus is a persistent positive strand RNA virus found in Saccharomyces cerevisiae. We previously observed that the virus generated in vivo from a launching vector possessed the correct RNA termini without extra sequences. Here we present evidence that the SKI1/XRN1 5′-exonuclease plays a major role in the elimination of the non-viral upstream sequences from the primary transcripts. The virus, once generated, however, is fairly unaffected by overexpression or deletion of SKI1/XRN1. By contrast, the copy number of the L-A double-stranded RNA virus in the same host is greatly increased by the deletion of SKI1/XRN1, and overexpression of the gene cured L-A virus from the cells at a high frequency. 20S RNA virus, unlike L-A virus, has a strong secondary structure at its 5′-end: the first four nucleotides are G, and they are buried at the bottom of a long stem structure, features known to inhibit the SKI1/XRN1 5′-exonuclease progression. Mutations that weakened the 5′-stem structure made 20S RNA virus vulnerable to SKI1/XRN1. These results, together with the data on L-A virus, indicate a strong anti-RNA virus activity of SKI1/XRN1. Given that 20S RNA virus resides and replicates in the cytoplasm without a protective capsid, our results suggest that the strong secondary structure at the 5′-end is crucial for the 20S RNA virus to evade the host SKI1/XRN1 defense.
Narnavirus 20S RNA is one of the simplest positive strand RNA viruses found in nature. The small genome (2514 nucleotides (nt)3) only encodes a single 91-kDa protein (p91), the RNA-dependent RNA polymerase (1–3). Despite its simplicity, the virus prospers and is widespread among laboratory strains of the yeast Saccharomyces cerevisiae as a persistent virus. All the information necessary to replicate and persist in the host is packed within its small RNA genome. The viral genome lacks a poly(A) tail at the 3′-end (3) and has perhaps no cap structure at the 5′-end, resembling an intermediate of mRNA degradation. The reading frame for p91 spans almost the entire length of the genome, and there are no genes for capsid proteins. Therefore, the RNA genome is not encapsidated into an intracellular virion structure (4, 5). It raises the question of how the virus avoids its RNA genome destruction by the enzymes involved in mRNA degradation pathways.
mRNA degradation in eukaryotes usually begins with the shortening of the poly(A) tail at the 3′-end, followed by decapping at the 5′-end by the Dcp1p-Dcp2p-decapping enzyme (6, 7). Numerous proteins (Lsm1p-7p, Pat1p, Dhh1p, etc.) are known to assist this reaction. Then, the decapped RNA is degraded by the XRN1 5′-exonuclease. Alternatively, deadenylated RNA is digested by a multiprotein complex with 3′-exo-nuclease activity called the exosome (8, 9). In yeast, the XRN1 5′-exonuclease plays a major role in mRNA degradation. Although xrn1 mutants exhibit pleiotropic phenotypes, common 5′-exonuclease motifs (motifs I–III) are identified in Xrn1p, and mutations of two critical residues in motif II (D206A, D208A) abolish the 5′-exonuclease activity rather specifically without affecting functions unrelated to RNA turnover (10). The enzyme digests the RNA processively in the 5′ to 3′ direction (11), and its progression is inhibited by a G tract or by a strong secondary structure on the RNA (12, 13). A genetic approach has identified several chromosomal SKI genes affecting the double-stranded RNA (dsRNA) virus L-A and its satellite RNA M (14, 15). Recessive mutants of these genes exhibit a super-killer phenotype due to the derepressed production of a toxin encoded on the satellite RNA. SKI1 and XRN1 have been assigned to the same gene (16), suggesting that a higher toxin production in this mutant is due to the stability of viral RNA that has neither 5′ cap structure nor a 3′ poly(A) tail.
Although the 20S RNA virus has no capsid protein, the viral genome does not exist as naked RNA in cells. Instead, it forms a ribonucleoprotein complex with p91 in a 1:1 stoichiometry and resides in the cytoplasm (5, 17). This raises the possibility that formation of complexes contributes to the stability of the 20S RNA genome in the cell. Recently, we have analyzed p91 in the complex and found that p91 interacts with 20S RNA at the 5′- and 3′-end regions and, to a lesser extent, at the central region as well (18). Mutations in the 5′ or 3′ cis sites reduced the formation of complexes to a basal level (10% compared with the full activity), and a combination of them did not further reduce the activity. This suggests that the interactions of p91 at both sites are coordinated and that the central cis site is responsible for the basal level of complex formation. Further study indicated that the 3′ cis site is identical to or overlaps with the 3′ cis signal for replication (Fig. 1A). Especially, the third and fourth nucleotides (both are C) from the 3′-end are essential for both replication and formation of complexes. By contrast, mutations at the 3′-terminal or penultimate position did not affect either activity. Furthermore, viruses recovered from the latter experiments had regained the wild-type nucleotide C by replacing the modified nucleotide at the terminal or penultimate position. This suggests that, although the 3′-terminal and penultimate nucleotides may be removed for repair, such nibbling would not go further inside because of p91 binding. In contrast to the 3′ cis site, the 5′ cis site for complex formation is located not at the very 5′-terminus but at the second stem-loop structure (nt 72–104) from the 5′-end (Fig. 1A) (18). The terminal region (nt 1–71) forms an intricate secondary structure, and the terminal nucleotide is buried at the bottom of a long stem (Fig. 1B). This stem also contains the initiation codon for p91. If p91 bound to the first stem structure, it is feasible that this binding might interfere with p91 translation from the initiation codon.
FIGURE 1.
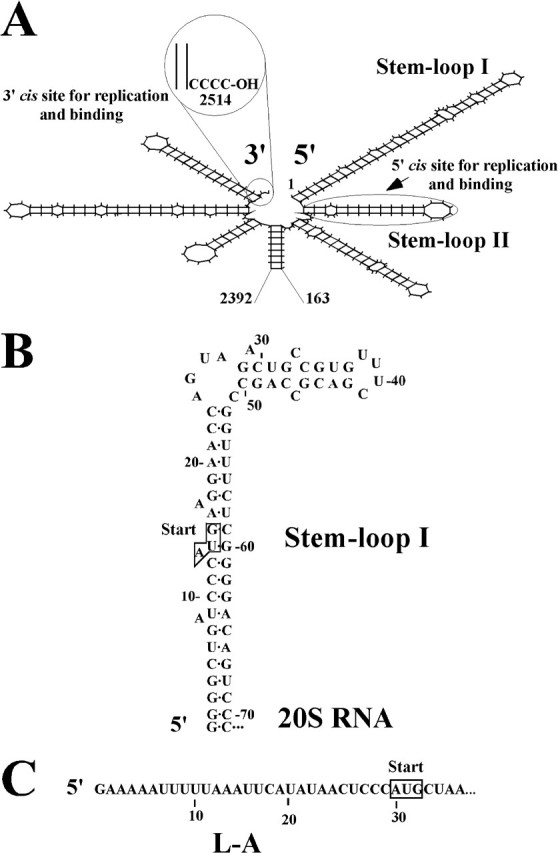
Terminal regions of the 20S RNA genome and the L-A positive strand. A, secondary structures in the 5′- and 3′-terminal regions of the 20S RNA genome. The 5′ stem-loops I and II and cis sites for replication and formation of complexes at the 5′- and 3′-terminal regions are indicated. Nucleotides are numbered from the 5′-end. B and C, nucleotide sequence of the 5′-terminal stem-loop I of the 20S RNA genome (B) and the 5′-terminal sequence of the L-A positive strand (C). The initiation codons (start) for p91 (B) and for the major coat protein of L-A virus (C) are indicated.
In this work, we show that the 20S RNA virus is quite resistant to the SKI1 5′-exonuclease because of the secondary structure at the 5′-end of the genome. Weakening the 5′-terminal stem structure made the virus susceptible to SKI1. By contrast, L-A virus has no strong secondary structure but an AU-rich sequence at the 5′-end of the positive strand (Fig. 1C) and can easily be eliminated from the cells by overexpression of SKI1. These results indicate that the SKI1 5′-exonuclease plays a critical role in defense against RNA viruses. During the course of reverse genetics using the in vivo 20S RNA virus launching system, we observed that the 5′-exonuclease activity of Ski1p plays a major role in removing non-viral upstream sequences from primary transcripts containing the 20S RNA genome. DHH1 was also found to be involved in this process. Our 20S RNA virus launching system thus provides a convenient method to analyze mRNA processing in vivo.
EXPERIMENTAL PROCEDURES
Strains and Media—20S RNA-negative strain 2928-4 (a ura3 trp1 his3, 20S RNA-o, 23S RNA-o, L-A-o) has been described previously (19). Standard strain BY4741 (a ura3Δ0, leu2Δ0 his3Δ1, met15Δ0 20S RNA-o, 23S RNA-o, L-A) and a haploid deletion series (BY4741 background) from the yeast knockout collection were a kind gift from Dr. J. L. Revuelta (Salamanca). For cytoplasmic mixing experiments (cytoduction), we used strain 1101 (α kar1-1, his4, L-A, M1, 20S RNA) (20). L-A-o derivatives of the strains were constructed during this work by overexpression of the SKI1 gene. Cells were grown in rich YPAD medium (1% yeast extract, 2% peptone, 0.04% adenine sulfate, and 2% glucose) or synthetic medium deprived of uracil or histidine, or both (21). Induction of 20S RNA virus under nitrogen starvation was performed as described previously (22).
Genetic Methods—Standard methods for yeast genetics were used (23). Cytoduction was carried out using a strain carrying the karyogamy-deficient kar1-1 mutation (24).
Pull-down Assay—Immunoprecipitation of ribonucleoprotein complexes with anti-p91 antiserum, RNA extraction, slotblotting, and detection by Northern hybridization were described previously (18). Titration of 20S RNA transcripts in complex with p91 or in the lysates is shown in supplemental Fig. S1. Hybridization with the [γ-32P]ATP-labeled oligodeoxy probe RE365, that was complementary to nt 1–22 of the 20S RNA genome, was done as described (25). The oligonucleotide RE368 (5′-CCTCATAAAACTGATACGAGCTTCTGCTATCC-3′) was used to detect 25S rRNA.
In Vitro Mutagenesis—Site-directed in vitro mutagenesis was done as described (26). Mutations were confirmed by DNA sequencing.
Northern Hybridization—Cells were broken with glass beads as described previously (18). Phenol-extracted lysates were separated in a native agarose gel, denatured in the gel, blotted, and hybridized with 32P-labeled RNA probes as described (27). To analyze SKI1 transcripts, RNA samples were separated in a formaldehyde-containing denaturing gel (28) instead of using a native agarose gel. 32P-labeled probes to detect 20S RNA and L-A positive strand genomes were made by T3 run-off transcription using SmaI-digested pALI18 (complementary to the full sequence of 20S RNA) and PvuII-digested pRE687 (complementary to nt 1323–1786 of L-A), respectively. The SKI1 specific probe was T3 run-off transcripts from SpeI-digested pRE908 that were complementary to the second half of the SKI1 gene (nt 2240–4587, numbered from the AUG initiation codon).
Launching Plasmids—pRE763 is a derivative of the 20S RNA launching vector pRE740 previously described (19). pRE763 has a 1.1-kb fragment containing the URA3 gene inserted into the unique EcoRV site in the TRP1 marker of pRE740. The rest of the launching plasmids were made from pRE763 by site-directed in vitro mutagenesis. The changes introduced into the vector or 20S RNA sequence are indicated in Table 1.
TABLE 1.
Plasmids
|
A. Launching plasmids (2 μm derivatives) | ||||
|---|---|---|---|---|
| Name | Upstream | 20S RNA | Ribozyme | Marker |
| pRE763 | 47 nt | WT | Active | URA3 |
| pLOR38 | 19 nt | WT | Active | URA3 |
| pLOR35 | 9 nt | WT | Active | URA3 |
| pRE940 | 9 nt | G69 | Active | URA3 |
| pRE941 | 9 nt | 68GG69 | Active | URA3 |
| pRE948 | 9 nt | G69 | Inactive | URA3 |
| pRE949 | 9 nt | 68GG69 | Inactive | URA3 |
|
pRE950
|
9 nt
|
WT
|
Inactive
|
URA3 |
|
B. Plasmids Expressing SKI1 | ||||
|
Name |
Promoter |
Copy number |
SKI1 |
Marker |
| pRS313 | - | Centromeric | - | HIS3 |
| pRE908 | SKI1 | Centromeric | SKI1 | HIS3 |
| pRE921 | SKI1 | Centromeric | ski1a | HIS3 |
| pRE910 | - | Multicopy | - | HIS3 |
| pRE914 | PGK1 | Multicopy | SKI1 | HIS3 |
| pRE1066 | PGK1 | Multicopy | ski1 | HIS3 |
ski1-D206A, D208A.
Cloning of SKI1—The SKI1 gene was amplified by PCR from strain 2928-4 with oligos RE298 (5′-AATTAGGATCCAATCCAAACATTGTGCCCAC-3′) and RE299 (5′-AATTAGGATCCGGTTTGCTAAGCAAAATGAG-3′). The DNA fragment (5.5 kb) contains the SKI1 open reading frame flanked by a 0.5-kb upstream sequence with the SKI1 promoter and a 0.25-kb downstream sequence with polyadenylation and transcription termination signals. The PCR fragment was cloned into the unique BamH1 site of the pRS313 centromeric vector (29), resulting in plasmid pRE908. The cloned sequence of the SKI1 gene was confirmed by DNA sequencing. The SKI1 overexpression vector pRE914 was constructed as follows: A 4.7-kb DNA fragment containing the SKI1 coding sequence was amplified by PCR from strain 2928-4 with oligos RE296 (5′-AATTAGGATCCCAGTACGGTATGGGTATTC-3′) and RE297 (5′-AATTAGGATCCGTCGTATGTTCTAAGTAGA-3′). Nucleotides corresponding to the initiation and termination codons are underlined. The amplified fragment was cloned downstream of the PGK1 promoter of vector pRE910, giving plasmid pRE914. Vector pRE910 is a derivative of pI2 (30) and has a 1.1-kb fragment containing the HIS3 marker inserted into the unique EcoRV site, interrupting the TRP1 gene of pI2.
RESULTS
SKI1 Contributes to the Efficient Generation of 20S RNA Virus in Vivo—We have recently established an in vivo launching system for 20S RNA virus from a yeast expression vector (19). The vector contains the entire 20S RNA cDNA sequence downstream of the PGK1 promoter. The hepatitis delta virus antigenomic ribozyme sequence is directly attached to the 3′-end of the viral genome in the vector to facilitate the creation of the precise viral 3′-end termini in the transcripts (Fig. 2A). Although the vector contained a non-viral sequence between the major transcription start site for the PGK1 promoter and the 5′-end of the viral genome, the virus generated had the authentic 5′-terminus without this non-viral vector sequence. Using this launching system, we examined the effects of host genes on virus generation. When a ski1Δ mutant strain was used as a host, we found that the standard launching plasmid failed to generate the virus (Fig. 2B, lane 4). Although this mutant strain showed severe defects in mating and meiosis, we found that the cells could support replication of 20S RNA when the virus was introduced by a cytoplasmic mixing (cytoduction) (not shown). Furthermore, two tetrads obtained from a cross with a 20S RNA-carrying strain produced a 4+:0 segregation of 20S RNA virus (Fig. 2C), indicating that the virus does not require SKI1 for its replication. Because the primary transcripts from the vector contain the 5′ cap structures as well as 47 nt of non-viral upstream sequences, we suspected that the failure in generating virus from the standard launching plasmid was caused by the inefficient removal of the non-viral 5′ sequence from the transcripts. When this upstream sequence was reduced from 47 to 19 nt or to 9 nt, the vectors successfully generated the virus in the ski1Δ strain (Fig. 2B, lanes 5 and 6). In particular, the shortest one generated the virus almost as efficiently as in the isogenic wild-type strain (Fig. 2B, lanes 3 and 6). Translation of p91 was not affected by these modifications in the upstream sequence (not shown). Once the virus was generated, ski1Δ cells could maintain it stably even after curing the launching plasmid. Next, we amplified a 5.5-kb DNA fragment encompassing the SKI1 gene from the wild-type strain 2928-4 and cloned it into a single copy centromeric vector. The authenticity of the gene was confirmed by DNA sequencing. When the ski1Δ strain was transformed with the vector expressing Ski1p, now the cells could generate 20S RNA virus from the standard launching plasmid (Fig. 2D, lane 2). Furthermore, cells transformed with the vector containing ski1-D206A, D208A failed to generate the virus under the same conditions (Fig. 2D, lane 3). These mutations are known to abolish the 5′-exonuclease activity of the gene product (10). These results indicate that the inability of the ski1Δ strain to generate the virus is indeed caused by the deletion of SKI1 and point out the importance of eliminating the non-viral 5′-upstream sequence for virus generation. Because decapping precedes 5′-exonuclease cleavage in mRNA turnover, we examined the effects on 20S RNA virus generation of the genes involved in mRNA decapping. Among them, we chose some components of the Lsm1p-7p/Pat1p complex (Lsm1p, Lsm6p, Lsm7p, and Pat1p), Dhh1p, an RNA helicase that is necessary for the decapping activity of Dcp1p-Dcp2p, and Edc1p, an enhancer of the decapping activity (31–33). We did not test deletions of DCP1 and DCP2 because these are essential genes. Strains deleted in LSM1, LSM6 (not shown), LSM7 (not shown), PAT1, or EDC1 could generate 20S RNA virus from the standard launching plasmid (Fig. 3A) or from a launching plasmid with a 9-nt upstream sequence (not shown). However, we were unable to launch 20S RNA in the dhh1Δ mutant from either plasmid (Fig. 3A, lane 5; and not shown). To examine whether the failure of the dhh1Δ strain to generate virus is caused by the inability to support 20S RNA replication or by its defect in RNA processing, we introduced 20S RNA virus into dhh1Δ cells by cytoduction. As shown in Fig. 3B, cytoductants received the virus from the donor strain and maintained it during the subsequent colony isolation procedure. We observed no effects of dhh1Δ on the level of 20S RNA introduced. This indicates that the dhh1Δ strain has no defects in supporting 20S RNA virus replication. These results demonstrate that host genes involved in RNA processing could affect 20S RNA virus generation in this launching system. Therefore, it is necessary to distinguish them from those genuinely involved in virus replication by genetic or other means.
FIGURE 2.

The SKI1 5 ′-exonuclease has a major role in the 5 ′ processing of the primary transcripts to generate 20S RNA virus in vivo. A, diagrams of 20S RNA transcripts produced in vivo from the standard launching plasmid pRE763 or derivatives thereof. The 20S RNA genome sequence is depicted with the bold line and numbered 1–2514. Thin lines indicate the non-viral sequences flanking the viral genome. The hepatitis delta virus antigenomic ribozyme sequences (R) attached to the 3′-end of the viral genome are boxed, and the curved arrow indicates the cleavage site. The standard launching plasmid has 47 nt as the non-viral upstream sequence in the transcripts, while the derivatives have shorter (19 and 9 nt) upstream sequences. B, ski1Δ strain failed to generate 20S RNA virus from the standard launching plasmid. A SKI1 (lanes 1–3) or ski1Δ (lanes 4–6) strain was transformed with the standard launching plasmid (lanes 1 and 4) or with a plasmid having a shorter upstream sequence (19 or 9 nt) (lanes 2, 3, 5, and 6). Phenol-extracted lysates were separated on 1.3% agarose gels and blotted onto nylon membranes. RNA on the membranes was detected by Northern hybridization with a 20S RNA specific probe. The position of 20S RNA is indicated. C, 20S RNA virus does not require SKI1 for its replication. RNA from one tetrad (A–D) and the parental strains was extracted, and the viral RNA in the extracts was analyzed as described in B. D, SKI1 on a centromeric vector complements the ski1Δ strain to generate 20S RNA virus from the standard launching plasmid. The ski1Δ strain was first transformed with a centromeric vector alone (lane 1), or the vector with SKI1 (lane 2), or ski1-D206A, D208A (lane 3). Then the transformants were again transformed with the standard launching plasmid. 20S RNA virus generated was detected as described in B.
FIGURE 3.

Effects of genes involved in mRNA decapping on 20S RNA launching. A, control strain BY4741 (lane 1) or a strain deleted in LSM1 (lane 2), PAT1 (lane 3), EDC1 (lane 4), or DHH1 (lane 5) was transformed with the standard launching plasmid. 20S RNA virus generated was detected as described in the legend to Fig. 2B. B, 20S RNA virus was introduced from strain 1101 (Donor) into a dhh1Δ strain (Recipient) by cytoduction. 20S RNA in two cytoductants and in the donor and recipient strains was analyzed as described in Fig. 2B. Ethidium bromide staining of the gel is also shown (EtBr). The positions of 20S RNA and 25S and 18S rRNAs are indicated.
Differential Effects of the SKI1 Gene on Yeast RNA Viruses—The SKI1 gene product suppresses the copy number of L-A dsRNA virus. We observed that the amount of L-A virus is 5–10 times higher in the ski1Δ strain than in the isogenic SKI1 wild-type strain (Fig. 4A, lanes 1 and 4). SKI1 on a centromeric vector reduced the amount of L-A to the wild-type level, while ski1-D206A, D208A showed no such suppressive activity (Fig. 4A, lanes 2 and 3). We subcloned the SKI1 open reading frame into a2 μm based multicopy vector, downstream of the constitutive PGK1 promoter. The expression of the SKI1 transcripts is at least 50 times higher than that from the centromeric vector or from the single gene in the yeast chromosome (Fig. 4B). When the ski1Δ strain was transformed with the SKI1 multicopy vector, the transformants lost the L-A virus at a high frequency (7 of 10) (Fig. 4C, lanes 3–12). We confirmed the loss of L-A virus by curing the plasmid. By contrast, transformants with the vector alone (not shown) or with ski1-D206, D208A retained a high amount of L-A virus as the original non-transformed cells (Fig. 4C, lanes 1 and 2). These results demonstrate a strong anti-L-A virus activity of the SKI1 5′-exonuclease. We also analyzed the effects of SKI1 on 20S RNA virus. Because the copy number of L-A virus is very high in the ski1Δ strain, its presence affects the copy number of 20S RNA in the same cell, perhaps by competing for resources such as nucleotide precursors, energy, etc. In the following experiments, therefore, we used an L-A-o strain. SKI1 on the centromeric vector had almost no effect on 20S RNA virus, and SKI1 even on the multicopy vector only slightly reduced the amount of 20S RNA virus (Fig. 5A). In the latter case, we did not observe any segregation of 20S RNA-free colonies. In contrast to L-A virus, therefore, 20S RNA virus is quite resistant to SKI1.
FIGURE 4.

The SKI1 5 ′-exonuclease has a strong anti-L-A virus activity. A, suppression of L-A virus by a single-copy of the SKI1 gene. L-A viral RNA was extracted and analyzed as described in the legend to Fig. 2B except that an L-A positive strand specific probe was used. Lane 1, ski1Δ strain; lane 2, ski1Δ strain with ski1-D206A, D208A in a centromeric vector; lane 3, ski1Δ strain with SKI1 in a centromeric vector; lane 4, SKI1 strain BY4741. B, expression of SKI1 transcripts from a centromeric or a multicopy vector. RNA was extracted from late logarithmic phase cells and separated in a denaturing agarose gel. After blotting, SKI1 transcripts were detected with a SKI1 specific probe. The lower panel is overexposed to reveal SKI1 transcripts from a single copy gene. Lane 1, SKI1 strain BY4741; lane 2, ski1Δ strain; lane 3, ski1Δ strain transformed with a SKI1 centromeric vector; lane 4, ski1Δ strain transformed with a SKI1 multicopy vector. C, curing of L-A virus by SKI1 overexpression. A ski1Δ strain was transformed with a SKI1-overexpressing multicopy vector (lanes 3–12) or the vector with ski1-D206A, D208D (lane 2). L-A virus was analyzed as described in A. Lane 1, the non-transformed ski1Δ strain.
FIGURE 5.
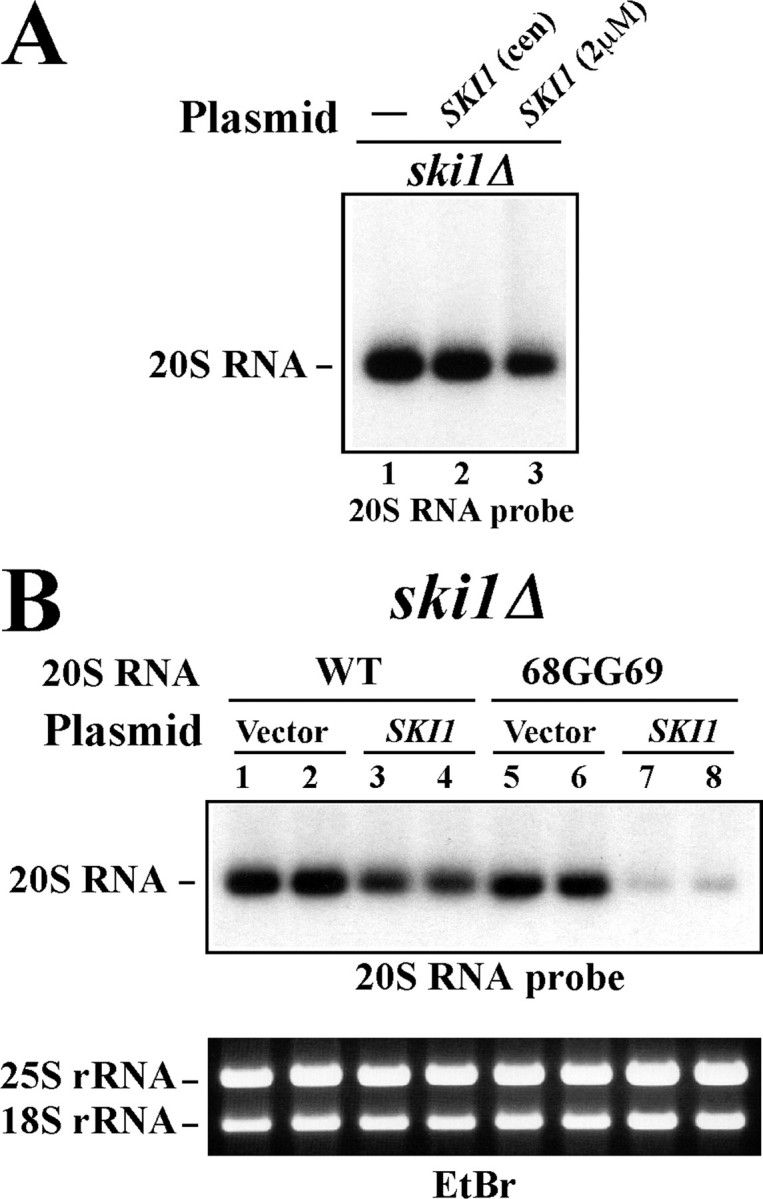
Effects of SKI1 on 20S RNA virus. A, wild-type 20S RNA virus is quite insensitive to SKI1 overexpression. ski1Δ strain carrying wild-type 20S RNA virus was transformed with a SKI1 centromeric vector (lane 2) or with a SKI1 multicopy vector (lane 3). Lane 1, 20S RNA virus-carrying ski1Δ strain without plasmid as control. B, 20S RNA with the 69GG69 mutation becomes sensitive to SKI1 suppression. ski1Δ strain carrying wild-type 20S RNA virus (lanes 1–4) or virus with the 68GG69 mutation (lanes 5–8) was transformed with a SKI1-overexpressing multicopy vector (lanes 3, 4, 7, and 8) or the vector alone (lanes 1, 2, 5, and 6). Two transformants from each experiment were analyzed. 20S RNA was detected as described in the legend to Fig. 2B. The lower panel shows 25S and 18S rRNAs stained with ethidium bromide as loading control. The photograph was taken before blotting. The strains used in A and B have no L-A virus.
The 5′-Terminal Structure Contributes to Resistance of 20S RNA Virus to SKI1—We wondered why 20S RNA and L-A viruses differentially respond to the SKI1 5′-exonuclease. The dsRNA genome of L-A virus is encapsidated into intracellular viral particles. Isolated particles protect the RNA genome from RNase A treatment (27). EM studies indicated that the capsid has holes with a size large enough for newly synthesized transcripts to pass through into the cytoplasm, but too small for the entry of globular proteins (34, 35). Thus, it is the non-encapsidated cytoplasmic stage of L-A positive strand transcripts that appears to be vulnerable to the 5′-exonuclease.
The 5′-terminal region of the L-A positive strand contains an AU-rich sequence and does not show strong secondary structures (Fig. 1C). By contrast, 20S RNA has a strong secondary structure at the 5′-end, and the 5′-terminal nucleotide is buried at the bottom of the stem (Fig. 1B). Furthermore, the first four nucleotides at the 5′-terminus are G. It is known that oligo G tracts and strong secondary structures inhibit the progression of the 5′ SKI1 exonuclease (13, 36). We therefore, hypothesized that this strong secondary structure contributes to the insensitivity of 20S RNA virus to SKI1 suppression. To examine this hypothesis, we introduced mutations in the launching plasmid to destabilize the 5′-end structure of the 20S RNA genome. Because the ski1Δ strain failed to generate 20S RNA virus from the standard launching plasmid (Fig. 2B), we used the plasmid having a shorter (9 nt) non-viral upstream sequence. The first mutant plasmid (G69) contains a single substitution at the position 69 (C to G) and creates a single mismatch at the lower part of the stem (Fig. 6A). This mutation does not alter the amino acid sequence of p91. The second plasmid (68GG69) harbors two substitutions (68UC69 to GG), destabilizing the stem more profoundly and encodes p91 modified at codon 19, from wild-type Val to Gly (Fig. 6A). We transformed the SKI1 strain with these plasmids as well as with the control plasmid having the wild-type 20S RNA sequence. Transformants with the first plasmid (G69) generated 20S RNA virus, but its amount was less compared with the control (Fig. 6B, lanes 1 and 2). Their ratio remains similar after curing the plasmid (not shown), indicating that the virus with this mutation is less stable. More significantly, the second plasmid (68GG69) failed to generate virus (Fig. 6B, lane 3). Because this second plasmid encodes a modified p91, its failure to generate virus could be attributed to the amino acid change (V19G) in the protein. To eliminate this possibility, the following two experiments were carried out. We previously showed that transcripts containing the 20S RNA negative strand genome did not generate virus because p91, essential for replication, could not be decoded from them. When an active p91 was provided from a second vector, the negative strand transcripts generated 20S RNA virus (19). We introduced the V19G mutation into the p91 coding sequence on the second vector and found that the modified p91 could generate virus from the negative strand transcripts (not shown), indicating that the protein is capable of, at least, a first round of positive strand synthesis. More direct evidence comes from the experiments shown in Fig. 6B, lanes 4–6. Here, a ski1Δ strain was transformed with the same three plasmids. Not only G69 but also 68GG69 mutant plasmids generated virus, and the amounts of 20S RNA in these transformants are indistinguishable from the one having the wild-type viral sequence. We confirmed that viruses generated from G69 and 68GG69 mutant plasmids in the ski1Δ strain were not revertants. Viral RNA was isolated from plasmid-cured cells, and the 5′-end region was amplified by reverse transcription-PCR. Sequencing five independent cDNA clones indicated that the viruses retained the respective mutations originally introduced in the launching plasmids. Thus p91 with the V19G mutation is active and has no defects in replication. Furthermore, the ski1Δ strain harboring a SKI1-expressing centromeric plasmid failed to generate virus from the 68GG69 mutant plasmid and generated virus from the G69 mutant plasmid but in a lesser amount, as the wild-type SKI1 strain did (compare lanes 1–3 to lanes 7–9 in Fig. 6B). Therefore, SKI1 is responsible for the instability of the mutant viruses and in the absence of the gene product, even 20S RNA virus with the 68GG69 mutation can propagate normally as the wild-type virus does.
FIGURE 6.

Mutant viruses with destabilized 5 ′-terminal stem structures become susceptible to SKI1 suppression. A, 5′-terminal stem structures of WT and G69 and 68GG69 mutant 20S RNA viruses. Only the lower half of the 5′-terminal stem-loop structure is shown. Nucleotides are numbered from the 5′-end. Small bars indicate the coding frame of p91 and the initiation codon (start) is boxed. Note that the 68GG69 mutation causes an amino acid change at codon 19 from Val to Gly (indicated with circles). The free energy of each structure (nt 1–71) is shown. B, wild type SKI1 (lanes 1–3) or ski1Δ strain (lanes 4–6), or the ski1Δ strain complemented with a SKI1 centromeric vector (lanes 7–9) was transformed with 20S RNA launching plasmids containing the wild-type sequence (lanes 1, 4, and 7) or with the G69 (lanes 2, 5, and 8) or 68GG69 (lanes 3, 6, and 9) mutation. 20S RNA virus generated was analyzed as described in the legend to Fig. 2B.
20S RNA Virus with the 68GG69 Mutation Becomes Vulnerable to SKI1 Suppression—20S RNA virus with the 68GG69 mutation was generated from a launching plasmid in the ski1Δ strain but not in the SKI1 strain. Here we asked whether this mutant virus after its generation in the ski1Δ strain is still susceptible to SKI1. We cured the launching plasmid from the ski1Δ strain that had generated the mutant virus. Then the cells were transformed with a SKI1-expressing multicopy vector or with the vector alone (Fig. 5B, lanes 5–8). As control, the ski1Δ strain with wild-type 20S RNA virus was processed similarly (Fig. 5B, lanes 1–4). As observed earlier (Fig. 5A), wild-type virus was only slightly affected by the overexpression of SKI1 (Fig. 5B, compare lanes 3 and 4 with lanes 1 and 2). By contrast, the mutant virus was reduced by more than 90% by SKI1 (Fig. 5B, compare lanes 5 and 6 with lanes 7 and 8). Thus the mutant virus becomes more susceptible to SKI1 compared with the wild-type virus, and this vulnerability is not related with the launching process from a vector. These results confirm that the integrity of the 5′-end structure is critical for the virus to resist SKI1 suppression. Unlike in the case of L-A virus, however, the cells harboring the mutant 20S RNA viruses did not produce virus-free colonies by the overexpression of SKI1.
The G69 and 68GG69 Mutations Do Not Directly Affect the Formation of Ribonucleoprotein Complexes between 20S RNA and the RNA Polymerase p91—The genomic RNA of 20S RNA virus forms a ribonucleoprotein complex in vivo with its RNA polymerase p91. We have recently shown that p91 interacts with the RNA at the 5′- and 3′-end regions, and to a lesser extent, at the central region. The 5′ cis site is present at the second stem-loop structure (nt 72–104) from the 5′-end (Fig. 1A). Because the two mutations G69 and 68GG69 destabilize the 5′-terminal stem-loop structure, and because they are located close to the 5′ cis site for formation of complexes, we examined whether these mutations affect the formation of ribonucleoprotein complexes. Transcripts containing the 20S RNA genome with the wild-type sequence or with mutations were expressed in vivo from a vector. The HDV ribozyme in the vector had been inactivated by replacing GGG 3′ to the cleavage site with AAA; thus the vector could not generate the virus (19, 37). The transcripts, however, served as mRNAs to translate p91 as well as substrates to form complexes with the decoded p91. We analyzed 20S RNA transcripts in these complexes by a pull-down assay using anti-p91 antiserum followed by Northern blot. In the ski1Δ strain, the antiserum immunoprecipitated transcripts with the wild-type sequence and also with the G69 or 68GG69 mutation, and their amounts were almost indistinguishable (Fig. 7A, right panel). This indicates that neither mutation affects the formation of complexes with p91. These results are consistent with previous data (Fig. 6B) showing that both mutations did not affect virus generation from a launching vector in a ski1Δ strain. In a SKI1 strain, however, the G69 mutation reduced the formation of complexes by 70% and the 68GG69 mutation by 90%. This may suggest that the SKI1 product directly impedes p91 to form complexes with the mutant RNAs. However, considering that the gene product has 5′-exonuclease activity and that these mutant RNAs have no defects in forming complexes with p91 in the ski1Δ strain, however, a more feasible explanation is that the effects of SKI1 on complexes are secondary. It is likely that the amount of mutant transcripts available for formation of complexes is lower compared with that of the wild-type sequence because of a greater susceptibility of the mutant RNAs to the Ski1p digestion. Alternatively, transcripts with the wild-type 20S RNA sequence in the complexes are more resistant to the Ski1p digestion by stopping the enzyme at the viral 5′-end, by virtue of the intact 5′-terminal secondary structure, whereas the destabilized 5′-terminal structures in the mutant RNAs fail to stop Ski1p and allow the enzyme to further digest the RNA, eventually releasing p91 from the 5′ cis site. At any rate, the results suggest that the interaction of p91 with the 5′ cis site for complex formation does not protect the viral 5′-end from the SKI1 5′-exonuclease. These explanations are consistent with the following observations. When the transcripts in the lysates (total RNA) from the SKI1 strain were detected with a full-size probe for 20S RNA, we found no significant difference in the amounts of mutant and wild-type RNAs (Fig. 7A, left panel). However, a 5′ oligo probe complementary to the first 22 nt of the viral genome detected 68GG69 mutant transcripts 30% less compared with the wild-type transcripts (Fig. 7A, left panel). As expected, the same oligo probe showed no noticeable differences between the wild type and 68GG69 mutant transcripts in the ski1Δ lysates (Fig. 7A, right panel). These results indicate that a small fraction of 68GG69 mutant transcripts lack the 5′-terminal region of the viral sequence in the SKI1 background. Finally, we confirmed that the G69 and 68GG69 mutations did not significantly affect the expression of p91 from vectors in the SKI1 or ski1Δ strain (Fig. 7B). Therefore, the decrease in the formation of complexes with these mutant transcripts in the SKI1 strain should be attributed to the vulnerability of the mutant RNA transcripts to the SKI1 product.
FIGURE 7.

5 ′-terminal mutations G69 and 68GG69 do not directly affect the formation of ribonucleoprotein complexes and p91 translation. A, effects of the G69 and 68GG69 mutation on the formation of ribonucleoprotein complexes. The SKI1 (left panels) or ski1Δ (right panels) strain was transformed with a ribozyme-inactivated plasmid expressing the wild-type 20S RNA sequence (WT) or 20S RNA with the G69 (G69) or 68GG69 (68GG69) mutation. Because these plasmids have an inactive ribozyme, they do not generate 20S RNA virus. Lysates prepared from logarithmically growing cells were incubated with anti-p91 antiserum (Anti-p91) or preimmune serum (Preimmune). RNA was extracted from the immunoprecipitates, blotted, and hybridized with a full-size 20S RNA probe. RNA directly extracted from the lysates (Total RNA) was processed similarly and detected with three different probes: a full-size 20S RNA probe (Full-size 20S RNA), a 22-nt 20S RNA 5′-end oligo probe (5′-end), and a 25S rRNA-specific oligo probe (25S rRNA). 25S rRNA serves as loading control. The diagrams below illustrate the two 20S RNA specific probes that were used. B, Western blot of p91 present in the cell lysates used in A.
DISCUSSION
In this work we have studied the effects of SKI1 on 20S RNA virus on two different aspects. One is on the launching process of 20S RNA virus from a vector and the other on the virus per se. These effects should be clearly distinguished.
The primary transcripts from the launching vector contain the 20S RNA viral genome flanked by non-viral sequences at both the 5′- and 3′-ends. The transcripts have 5′ cap structures and poly(A) tails at their 3′-ends as well. Virus generated from the vector has no such extra sequences. Thus these sequences must be eliminated during the launching process.
The importance of the 5′-end processing for in vivo launching was demonstrated by the failure of a ski1Δ strain to generate 20S RNA virus from our standard launching plasmid. 20S RNA virus, however, does not require the SKI1 gene product for its replication because the ski1Δ strain can support virus replication when 20S RNA is introduced by the cytoplasmic mixing that occurs during mating. Furthermore, the ski1Δ strain successfully generated the virus when the 5′-upstream sequence in the primary transcripts was reduced from 47 nt in the standard launching plasmid, to 19 nt or to 9 nt. Thus, a shorter 5′-upstream sequence is critical to generate the virus in a ski1Δ genetic background. Finally, a centromeric vector containing SKI1 but not ski1-D206A, D208A complemented the defect of the ski1Δ strain to generate virus from the standard launching plasmid. These results indicate that the SKI1 5′-exonuclease plays a major role in the 5′-processing of the primary transcripts. In the absence of SKI1 there is still a minor activity that is capable of eliminating a shorter 5′-upstream sequence. This suggests the involvement of a 5′-exonuclease rather than an endonuclease in the latter activity. S. cerevisiae has the essential RAT1 gene that encodes a nuclear 5′-exonuclease with a high degree of similarity to the SKI1 enzyme (38). Recently it has been suggested that yeast mitochondria have a 5′-exonuclease involved in apocytochrome b mRNA processing whose activity is governed by PET127 (39). These exonucleases could be candidates for the minor 5′-upstream processing activity found in our launching system. The SKI1 5′-exonuclease utilizes RNA with a 5′-monophosphate as substrate but cannot act on 5′-capped RNA (13). Thus the cap structure must be eliminated from the primary transcripts prior to the SKI1 action. Numerous genes are known to promote decapping. We chose strains deleted in genes belonging to the Lsm1p-7p/Pat1p complex, EDC1, or DHH1. None of them except for the dhh1Δ strain significantly affected virus generation from the standard launching plasmid and also from a plasmid with a shorter upstream sequence (9 nt). Although the dhh1Δ strain failed to generate virus from either plasmid, this strain could propagate and maintain the virus when it was introduced by cytoduction. Thus, the 20S RNA virus does not require DHH1 for replication. Similar to the case of SKI1, the DHH1 product appears to affect the processing of primary transcripts from the plasmid. Because Dhh1p is known to stimulate the decapping activity of a Flag-purified Dcp1p preparation in vitro (33), our results suggest that dhh1Δ affects the decapping activity of the Dcp1p-Dcp2p complex more severely than do the other mutations examined. Because the launching plasmid with the shorter upstream sequence failed to generate the virus in a dhh1Δ strain, the minor 5′-exonuclease activity observed in the ski1Δ strain appears to require decapping of the primary transcripts prior to its action.
Concerning the 3′-end, we previously observed that the HDV ribozyme directly attached to the 3′-end of the viral genome in the vector was critical for virus generation (19). The inactivation of the ribozyme or elimination of the ribozyme sequence from the vector resulted in the failure of virus launching. However, insertion of a few nucleotides between the 3′-end of the viral genome and the ribozyme cleavage site did not affect virus generation although the insertion of a G-rich sequence noticeably reduced its efficiency. In either case, the virus generated had none of the extra sequence at the 3′-end. The transcription termination site for the FLP gene of the 2 μm DNA is located 0.7-kb downstream of the 3′-terminus of the viral genome in the vector. Therefore, there is an activity capable of eliminating a short extra sequence but not a longer one from the viral 3′-end. This suggests the involvement of a 3′-exonuclease such as the exosome rather than an endonuclease in this process. Thus, our 20S RNA virus launching system may provide a convenient and sensitive method to analyze the processing of mRNAs at the 5′- and 3′-ends in vivo.
Unlike its generation from a launching vector, 20S RNA virus does not require SKI1 for replication. The virus even shows insensitivity to the antiviral activity of SKI1; the overexpression of SKI1 from a multicopy vector or its deletion did not affect 20S RNA virus significantly. It is in stark contrast to the L-A virus. The deletion of SKI1 resulted in the derepression of L-A copy number 5–10-fold (in the presence of the satellite RNA M1, this effect is less evident4 because of suppression of L-A copy number by M1 (40)). Furthermore, overexpression of SKI1 suppressed L-A virus and generated virus-free mitotic segregants at a high frequency. The 5′-terminal region of L-A positive strand is AU-rich and has no obvious secondary structure, whereas the 20S RNA genome has a strong secondary structure at the 5′-terminus, with the terminal nucleotide buried at the bottom of a long stem. This prompted us to hypothesize that the secondary structure at the 5′-terminus contributes to the resistance of 20S RNA virus to the SKI1 anti-viral activity. We introduced two mutations into the viral genome to disturb the 5′-terminal structure and tested this hypothesis. As expected these mutations made the virus susceptible to SKI1. Especially, the mutant that had the more disturbed 5′ structure (68GG69) could not be generated from a launching vector in the SKI1 wild-type background. In a ski1Δ strain, however, these mutant viruses were generated and replicated the same as the wild-type virus, indicating that the SKI1 5′-exonuclease is responsible for the instability of the mutant viruses in the wild-type strain. Furthermore, the 68GG69 mutant virus launched in a ski1Δ strain was greatly suppressed by overexpression of SKI1, whereas the wild-type virus was not. This indicates that the suppression of the mutant virus by SKI1 is not related to the launching process of the virus. We also demonstrated that these mutations had no direct effects on translation to decode p91 or on the formation of complexes with p91. Thus these data confirm our hypothesis that the secondary structure at the 5′-terminus is important to protect the 20S RNA genome from the SKI1 5′-exonuclease.
In a SKI1 strain the 69G and 68GG69 mutations reduced the formation of complexes to 30 and 10% (basal level), respectively, compared with the wild-type RNA, whereas the same mutations did not affect the formation of complexes in a ski1Δ strain. This indicates that the interaction of p91 with the 5′ cis site (nt 72–104) does not protect the viral genome from the SKI1 5′-exonuclease. Therefore, the 5′-terminal secondary structure itself is accountable for the resistance of 20S RNA virus to the Ski1p digestion. One of the major roles for the positive strand 20S RNA genome during its replication cycle is to translate p91. The initiation codon of p91 is located at the 5′-side of the 5′-terminal stem structure (Fig. 1B). It may be speculated that the intricate secondary structure (nt 1–71) at the 5′-terminus has dual roles: one to protect the viral genome from 5′-exonucleases and the other to promote translation of p91 from an RNA template that appears to have no 5′ cap structure. 23S RNA virus, a virus closely related to 20S RNA virus found in the same host, also possesses a very similar secondary structure at the 5′-terminus of the 23S RNA genome (3). The initiation codon for its RNA polymerase p104 is located in the middle of the 5′-terminal long stem. This virus is also resistant to SKI1 overexpression.4 Therefore, the 5′-terminal structure of the 23S RNA genome may have similar dual roles. In the 20S RNA/p91 complex, if p91 bound to the 5′-terminal structure, then such binding might interfere with p91 translation. Sitting on the adjacent structure (nt 72–104) instead, p91 may, by taking advantage of its proximity, sense or influence translational events from the initiation codon. Previously, we observed a weak protection from hydroxyl radical cleavages in the ribonucleoprotein complexes at positions 61 and 62, just opposite to the initiation codon in the 5′-terminal stem (18). Progression of the translating ribosome may displace p91 from the 5′ cis site. Although the interactions of p91 at the 5′ and 3′ cis sites are coordinated, the remaining interaction with the central cis site would prevent the complete dissociation of p91 from the viral genome. 20S RNA virus has a single viral protein p91 and exists as a ribonucleoprotein complex composed of the viral genome and p91 in 1:1 stoichiometry. Through interactions at the 5′, central, and 3′ cis sites, p91 may sense the current status of the complex on translation or replication (negative strand synthesis). This suggests that the ribonucleoprotein complex is not static but dynamic in structure and function.
RNA viruses, as obligatory intracellular parasites, have to propagate in the cell without being destroyed or neutralized by the host defense. Here, we have shown that the SKI1 gene has a strong anti L-A virus activity. The overexpression of the gene product could even eliminate the virus from the cells. Although the wild-type 20S RNA virus is quite resistant to SKI1, mutant viruses modified at the 5′-end became susceptible to SKI1 suppression. Our results suggest that SKI1 is part of the host defense against RNA viruses by virtue of its exonuclease activity. Perhaps L-A virus alleviates its deadly effects by sequestering the genome into intracellular particles, whereas 20S RNA virus has evolved such a way that the RNA genome itself becomes resistant to the enzyme by having a strong secondary structure at the 5′-end. It was reported that in vitro transcripts of L-A virus had diphosphates at their 5′-ends (41). Our preliminary data suggest that 20S RNA genome has neither a capped structure nor a monophosphate at the 5′-end.4 It remains to be clarified whether these viral genomes are directly subjected to SKI1 degradation or need to be processed prior to its action.
Supplementary Material
Acknowledgments
We thank Pilar Gómez Jiménez for invaluable technical support.
This work was supported in part by Grants BFU2004-00373 and BFU2007-60057 from the Spanish Ministry of Education and Science. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: nt, nucleotides; dsRNA, double-stranded RNA; WT, wild type; EM, electron microscopy.
R. Esteban and T. Fujimura, unpublished results.
References
- 1.Rodríguez-Cousiño, N., Esteban, L. M., and Esteban, R. (1991) J. Biol. Chem. 266 12772–12778 [PubMed] [Google Scholar]
- 2.Matsumoto, Y., and Wickner, R. B. (1991) J. Biol. Chem. 266 12779–12783 [PubMed] [Google Scholar]
- 3.Rodríguez-Cousiño, N., Solórzano, A., Fujimura, T., and Esteban, R. (1998) J. Biol. Chem. 273 20363–20371 [DOI] [PubMed] [Google Scholar]
- 4.Widner, W. R., Matsumoto, Y., and Wickner, R. B. (1991) Mol. Cell Biol. 11 2905–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-Cuéllar, M. P., Esteban, L. M., Fujimura, T., Rodríguez-Cousiño, N., and Esteban, R. (1995) J. Biol. Chem. 270 20084–20089 [DOI] [PubMed] [Google Scholar]
- 6.Parker, R., and Song, H. (2004) Nat. Struct. Mol. Biol. 11 121–127 [DOI] [PubMed] [Google Scholar]
- 7.Wilusz, C. J., Wormington, M., and Peltz, S. W. (2001) Nat. Rev. Mol. Cell Biol. 2 237–246 [DOI] [PubMed] [Google Scholar]
- 8.Mitchell, P., Petfalski, E., Shevchenko, A., Mann, M., and Tollervey, D. (1997) Cell 91 457–466 [DOI] [PubMed] [Google Scholar]
- 9.Jacobs Anderson, J. S., and Parker, R. (1998) EMBO J. 17 1497–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solinger, J. A., Pascolini, D., and Heyer, W. D. (1999) Mol. Cell Biol. 19 5930–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens, A. (1980) J. Biol. Chem. 255 3080–3085 [PubMed] [Google Scholar]
- 12.Tucker, M., and Parker, R. (2000) Annu. Rev. Biochem. 69 571–595 [DOI] [PubMed] [Google Scholar]
- 13.Stevens, A. (2001) Methods Enzymol. 342 251–259 [DOI] [PubMed] [Google Scholar]
- 14.Toh-e, A., Guerry, P., and Wickner, R. B. (1978) J. Bacteriol. 136 1002–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wickner, R. B. (1986) Annu. Rev. Biochem. 55 373–395 [DOI] [PubMed] [Google Scholar]
- 16.Johnson, A. W., and Kolodner, R. D. (1995) Mol. Cell Biol. 15 2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solórzano, A., Rodríguez-Cousiño, N., Esteban, R., and Fujimura, T. (2000) J. Biol. Chem. 275 26428–26435 [DOI] [PubMed] [Google Scholar]
- 18.Fujimura, T., and Esteban, R. (2007) J. Biol. Chem. 282 19011–19019 [DOI] [PubMed] [Google Scholar]
- 19.Esteban, R., Vega, L., and Fujimura, T. (2005) J. Biol. Chem. 280 33725–33734 [DOI] [PubMed] [Google Scholar]
- 20.Ridley, S. P., Sommer, S. S., and Wickner, R. B. (1984) Mol. Cell Biol. 4 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickner, R. B. (1980) Cell 21 217–226 [DOI] [PubMed] [Google Scholar]
- 22.Wejksnora, P. J., and Haber, J. E. (1978) J. Bacteriol. 134 246–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortimer, R. K., and Hawthorne, D. C. (1975) Methods Cell Biol. 11 221–233 [DOI] [PubMed] [Google Scholar]
- 24.Conde, J., and Fink, G. R. (1976) Proc. Natl. Acad. Sci. U. S. A. 73 3651–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varshney, U., Lee, C.-P., and RajBhandary, U. L. (1991) J. Biol. Chem. 266 24712–24718 [PubMed] [Google Scholar]
- 26.Esteban, R., Fujimura, T., and Wickner, R. B. (1989) EMBO J. 8 947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimura, T., Esteban, R., Esteban, L. M., and Wickner, R. B. (1990) Cell 62 819–828 [DOI] [PubMed] [Google Scholar]
- 28.Maniatis, T., Fritsch, E. F., and Sambrook, J. (1993) Molecular Cloning: A Laboratory Manual, pp. 202–203, Cold Spring Harbor, NY
- 29.Sikorski, R. S., and Hieter, P. (1989) Genetics 122 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wickner, R. B., Icho, T., Fujimura, T., and Widner, W. R. (1991) J. Virol. 65 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnerot, C., Boeck, R., and Lapeyre, B. (2000) Mol. Cell Biol. 20 5939–5946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coller, J. M., Tucker, M., Sheth, U., Valencia-Sánchez, M. A., and Parker, R. (2001) RNA 7 1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher, N., and Weis, K. (2002) EMBO J. 21 2788–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng, R. H., Caston, J. R., Wang, G.-J., Gu, F., Smith, T. J., Baker, T. S., Bozarth, R. F., Trus, B. L., Cheng, N., Wickner, R. B., and Steven, A. C. (1994) J. Mol. Biol. 244 255–258 [DOI] [PubMed] [Google Scholar]
- 35.Caston, J. R., Trus, B. L., Booy, F. P., Wickner, R. B., Wall, J. S., and Steven, A. C. (1997) J. Cell Biol. 138 975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Decker, C. J., and Parker, R. (1993) Genes Dev. 7 1632–1643 [DOI] [PubMed] [Google Scholar]
- 37.Esteban, R., and Fujimura, T. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 2568–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson, A. W. (1997) Mol. Cell Biol. 17 6122–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fekete, Z. F., Ellis, T. P., Schonauer, M. S., and Dieckmann, C. L. (2008) J. Biol. Chem. 283 3767–3772 [DOI] [PubMed] [Google Scholar]
- 40.Ball, S. G., Tirtiaux, C., and Wickner, R. B. (1984) Genetics 107 199–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nemeroff, M. E., and Bruenn, J. A. (1987) J. Biol. Chem. 262 6785–6787 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.