

Abstract
Glycolysis is essential to Trypanosoma brucei, the protozoan parasite that causes African sleeping sickness in humans and nagana in cattle. Hexokinase (HK), the first enzyme in glycolysis, catalyzes the phosphorylation of glucose to form glucose 6-phosphate. T. brucei harbors two HKs that are 98% identical at the amino acid level, T. brucei hexokinase 1 (TbHK1) and TbHK2. Recombinant TbHK1 (rTbHK1) has HK activity, whereas rTbHK2 does not. Unlike other eukaryotic HKs, TbHK1 is not subject to inhibition by ADP and glucose 6-phosphate. However, TbHK1 is inhibited by myristate, a critical fatty acid in T. brucei biology. We report here that rTbHKs, similar to authentic TbHK, form oligomers. Myristate dissociated these assemblies when incubated with either ATP or glucose. Furthermore, oligomer disruption was reversible by removal of myristate. Mixing of rTbHK1 and rTbHK2 monomers followed by reassembly yielded enzyme with an ∼3-fold increase in specific activity compared with similarly treated rTbHK1 alone. Surprisingly, reassembly of rTbHK2 with an inactive rTbHK1 variant yielded an active HK, revealing for the first time that rTbHK2 is competent for HK activity. Finally, pyrophosphate inhibits active reassembled rTbHK2 oligomers but not oligomeric rTbHK1, suggesting that the two enzymes have distinct regulatory mechanisms.
The African trypanosome Trypanosoma brucei is the causative agent of human African sleeping sickness and nagana in livestock. The parasite has a flexible host-dependent metabolism. Bloodstream form (BSF)3 parasites, found in the mammalian host, exclusively utilize glycolysis for ATP production, whereas procyclic form (PF) parasites found in the insect vector rely on the catabolism of amino acids for energy. Even though energy metabolism varies between life stages, glycolysis appears to be essential to both BSF and PF parasites, as the silencing, mislocalization, or inhibition of glycolytic enzymes is lethal in both stages (1–4).
Glycolysis in trypanosomes is unique in several ways. Although glycolysis in most eukaryotes is cytoplasmic, a majority of the enzymes required for glycolysis in T. brucei are compartmentalized in a specialized peroxisome called the glycosome. In higher eukaryotes, glycolysis is regulated through allosteric modulation of glycolytic enzymes, including hexokinase (HK), by enzyme products or other metabolic effectors. Interestingly, T. brucei HK does not appear to be regulated in this manner (5).
T. brucei expresses two hexokinases, TbHK1 and TbHK2, that are 98% identical at the amino acid level. Both have been detected in the glycosomes of PF and BSF parasites (6). It is unclear why T. brucei would need two nearly identical hexokinases, and historically, there has been little discrimination between the two genes and the enzymes they produce.
Recombinant TbHK1 (rTbHK1) exhibits HK activity in vitro (7, 8). Furthermore, RNA interference (RNAi) of TbHK1 leads to a reduction in HK activity in both PF and BSF parasites and is lethal to BSF parasites (4, 9). These observations have led to the presumption that TbHK1 is the main HK involved in glycolysis (9).
However, the function of TbHK2 has remained a mystery. To date, rTbHK2 has lacked detectable HK activity, suggesting that it might have an activity in vivo distinct from catalytically active TbHK1. PF parasites lacking TbHK2 (by knock-out) are morphologically distinct from the parental strain, and they display increased HK activity, an observation that has been attributed to an increase in TbHK1 protein expression found in the TbHK2-deficient cells (7). In the BSF, the role of TbHK2 is also unclear, although RNAi knockdown of TbHK2 results in the loss of cellular TbHK activity and is lethal, suggesting an important function (1).
Here, we show that TbHK1 and TbHK2 assemble into mixed high molecular mass complexes that exhibit enzymatic activities and inhibition profiles that are dramatically different from homogenous complexes composed of either TbHK1 or TbHK2 alone. Interestingly, TbHK1 activates TbHK2, and the enzymatic activity of the mixed complex, unlike that of TbHK1 alone, is regulated by pyrophosphate (PPi), suggesting a novel role for TbHK2 in the regulation of cell metabolism.
MATERIALS AND METHODS
TbHK Assays and Myristate Inhibition—rTbHK1, rTbHK2, and TbHK1(S160A) were expressed from pQE30 (Qiagen Inc., Valencia, CA) and protein-purified as described (7).
HK assays and inhibitor studies were performed as described with the modification that kinetic analyses were performed using KaleidaGraph 4.1 (Synergy Software, Reading, PA) with the Michaelis-Menten curve fit algorithm (7). For assays on cell lysates, 1 × 106 parasites were washed and lysed in 0.1% Triton X-100 and 0.1 m triethanolamine, pH 7.4, on ice and then assayed.
Native Gels and In-gel HK Assay—Native gel electrophoresis of rTbHK1 was performed on 4% polyacrylamide gels (4% bisacrylamide, 375 mm Tris-HCl, pH 8.8, 0.05% (v/v) TEMED, and 0.05% ammonium persulfate) resolved in Tris/glycine buffer (2.7 mm Tris-HCl and 192 mm glycine, pH 6.9). Samples (∼50 ng of protein) were diluted in native gel loading buffer (10% (v/v) glycerol, 2.7 mm Tris-HCl, pH 6.8, and 0.1% bromphenol blue), resolved, and silver-stained.
For in-gel HK assays, proteins were resolved on 4% native gels, and the gels were washed in ultrapure water (5 min, room temperature) and then incubated in the dark (15 min, room temperature) with 3 units/ml glyceraldehyde-6-phosphate dehydrogenase, 0.02 mg/ml phenazine methosulfate, 0.2 mg/ml nitro blue tetrazolium, and 0.3 mm NADP+ in HK assay buffer (45 mm Tris-HCl, pH 9.5, 5 mm glucose, 3 mm MgCl2, and 3 mm ATP) (10). Gels were then soaked in 10% acetic acid (15 min, room temperature) to quench the reaction. Glucose 6-phosphate, the product of HK, was visualized by the precipitated formazan on the gel.
Two-dimensional Gel Electrophoresis and Western Blotting of rTbHK1 Native Gels—The molecular masses of the components released from rTbHK1 oligomers following myristate treatment were determined using two-dimensional gel electrophoresis, where the first dimension was a 4% native gel, and the second was a 10% SDS-polyacrylamide gel. The 4% gels were run as described above. Lanes of the 4% native gel were excised, placed on the stacking gel of a 10% SDS-polyacrylamide gel, and overlaid with 5× SDS-PAGE loading buffer prior to application of current to resolve proteins in the second dimension.
For Western blotting, proteins were resolved by native gel and transferred in Tris/glycine buffer to nitrocellulose preequilibrated in the same buffer. Membranes were blocked with 1% nonfat milk in 1× TNT (5 mm Tris-HCl, pH 8.0, 75 mm NaCl, and 0.025% Tween) prior to incubation with mouse anti-RGS-His6 antibody (1:2000, 1 h, room temperature; Qiagen), which was detected with a horseradish peroxidase-conjugated secondary antibody (1:10,000, 1 h, room temperature) and chemiluminescent substrate (Pierce).
Disassembly and Reassembly of rTbHK Oligomers—Recombinant hexokinases (rTbHK1, rTbHK2, and TbHK1(S160A)) were incubated (1 h, room temperature) with fatty acids solubilized in DMSO, including stearate (octadecanoic acid, C18), myristate (C14), and decanoate (C10) in 0.1 m triethanolamine, pH 7.4, supplemented with 5 mm ATP/Mg2+ or 5 mm glucose. Myristate was removed by washing the sample in an Amicon Ultra centrifugal filter device (3000 molecular weight cutoff, Millipore Corp., Billerica, MA) after dilution in wash buffer (20 mm NaH2PO4, 0.5 m (NH4)2SO4, 0.1% Triton X-100, and 250 mm imidazole). Seven volumes of wash buffer were passed through the filter. Oligomer disassembly was confirmed by native gel electrophoresis, and the concentration of recovered protein was determined using a Pierce BCA kit. Recovery was confirmed by a Coomassie Blue-stained SDS-polyacrylamide gel. To reassemble oligomers, myristate-treated monomeric rTbHK1 and rTbHK2 were mixed prior to washing on the Amicon filtration unit.
RESULTS
Myristate and Lonidamine (LND) Inhibit TbHK from Parasite Lysate, but Only Myristate Disrupts Oligomerization—Little is known concerning the regulation of T. brucei HK in vivo. Unlike many HKs, TbHK is not inhibited by glucose 6-phosphate or ADP (5). To investigate potential regulators, we explored rTbHK1 inhibition with a spectrum of metabolites, including amino acids (alanine, proline, glutamine, leucine, and threonine), carbohydrates (galactose, N-acetylglucosamine, and inositol), phospholipids (phosphatidylinositol, phosphatidylethanolamine, and phosphatidic acid), small molecules (lactate, malate, succinate, butyrate, glycerol, glycerol 3-phosphate, and acetyl-CoA), and glycolytic intermediates (fructose 6-phosphate, fructose 1,6-bisphosphate, pyruvate, and phosphoenolpyruvate (PEP)) as potential TbHK1 inhibitors. None of these impacted rTbHK1 activity at a 10 mm final concentration under our standard assay conditions.
Previously, we found that medium to long chain (C10–18) free fatty acids inhibited rTbHK1, with myristate being the most potent (7). Authentic TbHK from BSF 90-13 lysate is also sensitive to myristate, being completely inhibited by 1 mm free fatty acid in an in vitro assay (Fig. 1A). Likewise, LND, another potent inhibitor of rTbHK1, inhibited authentic HK activity from BSF lysates (4).
FIGURE 1.
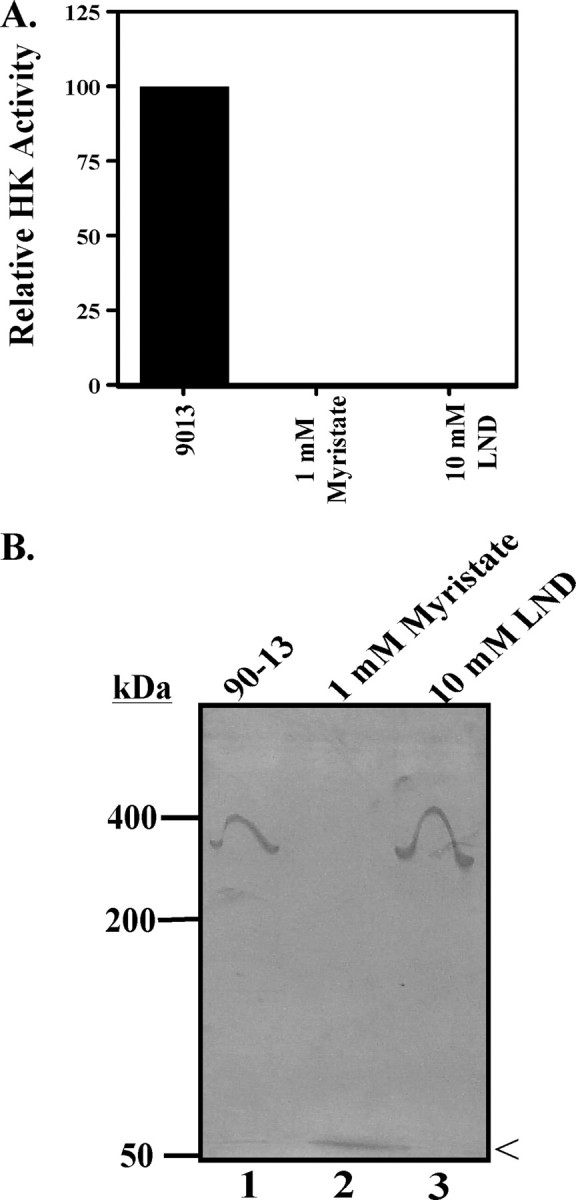
Impact of myristate on TbHK in BSF lysates. A, myristate inhibits HK activity from BSF cell lysate. Myristate (1 mm) or LND (10 mm) was incubated with 5 × 105 BSF cell equivalents for 30 min at room temperature, and HK assays were performed as described under “Materials and Methods.” B, native gel analysis of BSF 90-13 trypanosome lysate without (lane 1) or with incubation with 1 mm myristate (lane 2) or 10 mm LND (lane 3) is shown. < indicates the migration of HK activity in the myristate-treated lane.
TbHK activity has been previously isolated as an ∼300-kDa hexameric protein complex containing an unknown mixture of TbHK1 and TbHK2 from BSF parasites (11). Because HK activity can be recovered after electrophoresis (due to loss of inhibitor during processing), we were able to examine the effect of inhibitor on the oligomeric state using an in-gel HK assay. Lysates were resolved on 4% native gels, and HK in-gel assays were performed. Untreated parasites yielded HK activity that migrated on the native gel as an oligomeric complex consistent with a hexamer with an apparent molecular mass of ∼300 kDa (Fig. 1B, lane 1). Interestingly, lysates treated with myristate lacked detectable activity at the ∼300-kDa position but instead had HK activity at ∼50 kDa, consistent with migration of the monomer of the enzyme. LND-treated cell lysate yielded HK activity corresponding to an ∼300-kDa species, suggesting that LND does not disrupt the complex. Of note, in assays performed on lysates in solution, both myristate and LND were potent TbHK1 inhibitors.
rTbHK Forms Oligomers—Oligomerization is not limited to authentic TbHK, as native gel analysis of purified rTbHK1 and rTbHK2 revealed that the proteins behave as oligomeric complexes, consistent with an apparent mass of ∼300 kDa (Fig. 2A, lanes 1 and 11). The apparent mass of the complex was confirmed by gel filtration chromatography, with TbHK2 eluting between β-amylase (200 kDa) and apoferritin (440 kDa) (data not shown). These high molecular mass complexes were not the result of covalent linkage of subunits, as oligomers were disrupted by treatment with 0.1% SDS (data not shown).
FIGURE 2.

rTbHK1 and rTbHK2 formoligomers. A, native gel(4% acrylamide) analysis of rTbHK1 (50 ng; lane1) after treatment (30 min, room temperature) with DMSO (lane 2), myristate (500 μm; lanes 3–5), or LND (10 mm; lanes 8–10) in the presence of 5 mm ATP/Mg2+ (lanes 3 and 8), 5 mm glucose (lanes 4 and 9), or no substrate (lanes 5 and 10) is shown. Additionally, untreated rTbHK1 (lane 6), untreated rTbHK2 (lane 11), DMSO treated rTbHK1 (lane 7), and rTbHK2 treated with ATP and myristate (lane 12) are included. Samples were then resolved and silver-stained. B, treatment of rTbHK1 oligomers with increasing salt can disrupt the complex. rTbHK1 (50 ng) was incubated with increasing NaCl(upper panels), KCl(middle panels), or MgCl2 (lower panels), and the impact was assessed by native gel (upper gel) or 10% SDS-polyacrylamide gel (lower gel). Salt concentrations were 250 mm, 500 mm, 1 m, and 2.5 m. Aberrant migration (upon SDS-PAGE) was likely due to the high salt concentrations.
Interestingly, incubation of purified rTbHK1 with myristate (500 μm) in the presence of either ATP or glucose led to a disruption of the complex (Fig. 2A, lanes 3 and 4), whereas incubation with a longer fatty acid, stearate, or a shorter fatty acid, decanoate (neither of which appreciably inhibited TbHK1 activity) (7), under similar conditions had little impact on the oligomers (data not shown). Incubation with fatty acid alone (in the absence of either glucose or ATP) or with vehicle (DMSO) did not impact oligomerization (Fig. 2A, lanes 2 and 5). rTbHK2 oligomers were also disrupted by myristate in the presence of ATP (Fig. 2A, lane 12).
Similar studies were carried out with LND, another potent rTbHK1 inhibitor. Incubation of rTbHK1 with LND in the presence or absence of ATP/Mg2+ or glucose had no impact on the ∼300-kDa complex compared with untreated rTbHK1 (Fig. 2A, lanes 6–10). This result is similar to that observed when LND was used to inhibit endogenous TbHK followed by in-gel HK assays (Fig. 1B, lane 3).
Incubation of recombinant oligomers with different salts had distinct impacts on the complexes. Incubation with NaCl and KCl (from 250 mm to 2.5 m) had minimal impact on oligomers (Fig. 2B). Similar treatment with the divalent salt MgCl2, which is also required for activity, disrupted oligomers at higher concentrations. To control for possible loss of protein in sample handling, proteins were also analyzed by SDS-PAGE, which revealed minimal loss of protein during either treatment (Fig. 2B).
Disassembly Yields HK Monomers—To further resolve the impact of myristate destabilization on rTbHK oligomers, samples were analyzed by two-dimensional gel electrophoresis with native gel electrophoresis in the first dimension and SDS-PAGE in the second. This revealed that untreated rTbHK1 migrates as an ∼300-kDa complex that resolves into an ∼50-kDa species in the second denaturing dimension (indicated by * in Fig. 3A). However, incubation of rTbHK1 with myristate (1 mm) led to a decrease in the ∼50-kDa species arising from the ∼300-kDa complex (80% by densitometer) with a corresponding increase in a species migrating in the first dimension at the dye front that is ∼50 kDa in the SDS-PAGE dimension (indicated by [caret] in Fig. 3A). Western blotting of the 4% native gel with an antibody to the N-terminal epitope tag revealed an increase in the abundance of the monomeric (∼50 kDa) protein in response to increasing myristate (Fig. 3B). A corresponding decrease in the ∼300-kDa species in the native gel was noted by silver stain, but the oligomer was not transferred efficiently enough for Western blotting purposes.
FIGURE 3.

Oligomers disassemble into monomers. A, two-dimensional electrophoresis of untreated (left panel) or myristate (1 mm)-treated (right panel) rTbHK1 oligomers. Proteins were resolved on 4% native gels that were then inserted into the stacking gel of a 10% SDS-polyacrylamide gel. Proteins from the oligomer (*) or the monomer ([caret]) are indicated. The gels shown are representative of three experiments from two purifications of rTbHK1. B, Western blotting of rTbHK1 oligomers treated with increasing myristate (0–750 μm) resolved by 4% native gel. Following transfer, the blot was incubated with a primary mouse monoclonal antibody (1:2000) against the RGS-His6 epitope tag used in the purification. Following incubation with horseradish peroxidase-conjugated secondary antibody (1:10,000), the membrane was developed using SuperSignal West Pico chemiluminescent substrate (Pierce).
Disassembly Is Reversible—To determine whether the disassembly of rTbHK oligomers is reversible, we incubated rTbHK1 or rTbHK2 with myristate in the presence of ATP/Mg2+ and then washed the myristate out of the samples by centrifugal filtration (see the scheme in Fig. 4A). Oligomers were treated with myristate (or DMSO carrier) and analyzed by native gel electrophoresis (Fig. 4B) or by SDS-PAGE to confirm equal loading (lower panels in all sections of Fig. 4B). rTbHKs treated with myristate were disassembled (Fig. 4B, upper center panel) compared with samples treated with DMSO (Fig. 4B, lower center panel). As anticipated, myristate treatment also inhibited rTbHK1 by >95% (data not shown).
FIGURE 4.

TbHK oligomer reassembly reveals that TbHK2 is an active hexokinase. A, schematic illustrating the disassembly and reassembly of oligomeric rTbHKs. B, native gel (4%) and SDS-PAGE analysis of TbHKs prior to incubation (left panels) with or without DMSO or myristate. Samples were then analyzed to assess dissociation (middle panels) and again after centrifugal filtration (right panels).
Samples were then subjected to centrifugal filtration, washed exhaustively to remove myristate, and then subjected to electrophoresis on native gels to examine reassembly. Both rTbHK1 and rTbHK2 yielded reassembled ∼300-kDa complexes after myristate disruption and centrifugal filtration. These oligomers were similar to complexes found in untreated samples (Fig. 4B, right panels).
Oligomer Reassembly Alters Enzymatic Activity of the Complex—Following centrifugal filtration to remove myristate, rTbHK1 reassembled into oligomers with specific activity similar to untreated or DMSO-treated rTbHK1 samples (Fig. 5B, left panel). Furthermore, the reassembled rTbHK1 had Km values for glucose and ATP of 0.05 ± 0.003 and 0.25 ± 0.005 mm, respectively, similar to untreated rTbHK1 (0.06 mm 0.28 mm) (8). On the other hand, reassembled rTbHK2 remained inactive, as was found in the untreated sample (Fig. 5B, left panel).
FIGURE 5.

Reassembly of mixed oligomers reveals that TbHK2 has HK activity. A, schematic representation for assembly of mixed complexes. B, hexokinase activity of untreated, DMSO-treated, or myristate-dissociated and reassembled rTbHK1 and rTbHK2 (left panel) and rTbHK1(S160A) (right panel). Equal amounts (250 ng) of rTbHK1 and rTbHK2 were used throughout. Please note that the differences in HK activities of DMSO-treated rTbHK1 in the left and right panels were due to differences in the specific activity of proteins used.
Mixing rTbHK1 and rTbHK2 homohexamers without first disrupting the complexes had no impact on HK activity (Fig. 5B, left panel) (7). However, mixing equal amounts of myristate-treated (disassembled) rTbHK1 and rTbHK2 (Fig. 5A) prior to centrifugal filtration yielded an ∼3-fold increase in enzymatic activity compared with the controls (Fig. 5B, left panel). The heteromeric oligomer had similar affinity for glucose compared with rTbHK1, with Km = 0.035 ± 0.003 mm, whereas affinity for ATP was increased slightly, with Km = 0.12 ± 0.01 mm. Disassembly and reassembly were confirmed as before using native gel electrophoresis (data not shown).
TbHK2 Assembled with a Catalytically Inactive Mutant, TbHK1(S160A), Has HK Activity—To determine whether the increase in mixed oligomer activity was due to a change in the specific activity of rTbHK1 or the result of some “activation” of rTbHK2 (which heretofore was inactive), rTbHK2 was reassembled with an inactive rTbHK1 variant, rTbHK1(S160A), which lacks phosphotransferase activity (7). First, the impact of centrifugal filtration and reassembly was assessed for rTbHK1(S160A). This variant lacked detectable HK activity, similar to rTbHK2 reassembled as a homohexamer (Fig. 5B, right panel). Interestingly, mixing rTbHK2 and rTbHK1(S160A) (to yield the rTbHK2-rTbHK1(S160A) complex) resulted in detectable HK activity (Fig. 5B, right panel) that had similar kinetic properties compared with rTbHK1, with Km values for glucose and ATP of 0.45 ± 0.006 and 0.19 ± 0.018 mm, respectively, similar to those found for rTbHK1 (7). Mixing myristate-treated rTbHK1(S160A) with disassembled rTbHK1 had little impact on activity.
TbHK2 Is Inhibited by Pyrophosphate—Risedronate, a bisphosphonate analog of pyrophosphate, inhibits Trypanosoma cruzi HK (12, 13) but lacks activity against rTbHK1 (4). Interestingly, the HK activity resulting from the rTbHK2-rTbHK1(S160A) complex was slightly sensitive to 5 mm risedronate (Fig. 6A), a concentration that had no impact on rTbHK1 in complex with rTbHK1(S160A) (Fig. 6A). PPi itself was an even more potent inhibitor of the rTbHK2-rTbHK1 (S160A) complex (70% inhibition at 5 mm). Like risedronate, PPi had a negligible effect on TbHK1 complexed with rTbHK1(S160A) (Fig. 6A).
FIGURE 6.
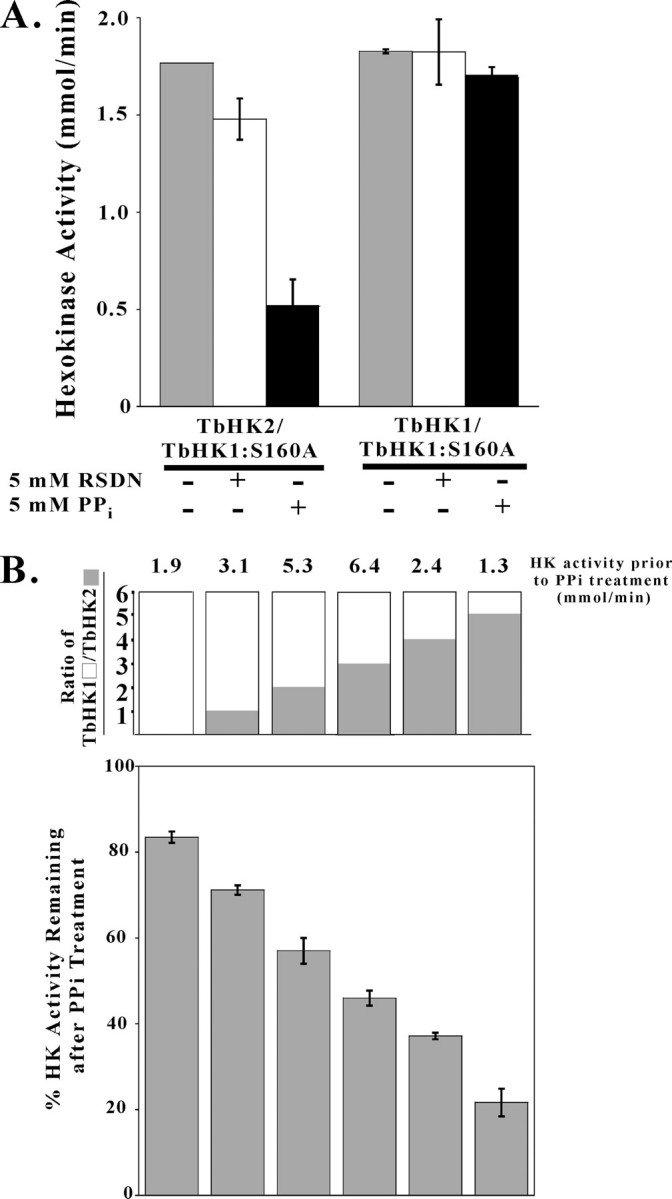
Pyrophosphate inhibits rTbHK2. A, reassembled rTbHK2-rTbHK1(S160A) complex or rTbHK1-rTbHK1(S160A) complex was incubated with risedronate (RSDN; 5 mm) or PPi (5 mm) prior to assaying for HK activity. B, complexes generated with a higher percentage of rTbHK2 have increased sensitivity to PPi. Proteins were reassembled with increasing molar amounts of rTbHK2 (while total protein was kept constant at 500 ng/assay) and then assayed for HK activity in the presence or absence of PPi (5 mm). Reassembly yielded TbHK1/TbHK2 ratios of 6:0, 5:1, 4:2, 3:3, 2:4, and 1:5, with untreated samples having ratios of 1.9, 3.1, 5.3, 6.4, 2.4, and 1.3 mmol/min HK activity, respectively. Results are plotted as the percent of this untreated activity remaining after PPi inhibition.
Altering the ratio of active rTbHK1 monomer to inactive rTbHK2 monomer (both generated by myristate disruption of homohexamers) prior to reassembly altered the sensitivity of the mixed complexes to PPi (Fig. 6B). As more rTbHK2 was included in the complexes, the HK activity became more sensitive to PPi inhibition relative to untreated control complexes, suggesting that rTbHK2 is an important determinant in PPi sensitivity.
Cellular HK Pyrophosphate Sensitivity Is Influenced by Parasite Carbon Source—The PPi sensitivity of the rTbHK2-rTbHK1(S160A) complex (Fig. 6A) and the observation that complexes containing a higher rTbHK2 percentage (Fig. 6B) were more sensitive to PPi suggested that this inhibitor could be used as a probe to determine the relative composition of TbHK complexes from T. brucei. To date, the lack of sensitive TbHK1 and TbHK2 antibodies has made it extremely challenging to investigate relative amounts of the nearly identical polypeptides. BSF 90-13 and TbHK2–/– PF 29-13 cells (which lack TbHK2) (7) had HKs with similar sensitivity to PPi, suggesting that the BSF may rely more on TbHK1 for cellular HK activity (Fig. 7A). However, parental PF 29-13 cells had HK activity that was composed of a higher percentage of PPi-sensitive HK, suggesting that TbHK2 plays a more important role in the metabolism of the PF parasite. The PF 29-13 HK activity was dynamic, with growth under different nutrient conditions leading to HK activity with differing PPi sensitivity (Fig. 7B). Parasites grown in medium with very little glucose (SDM-80) (14) had TbHK activity that was sensitive to PPi inhibition (with TbHK activity inhibited by >80%) (Fig. 7B). Addition of glycerol (1 mm) to the SDM-80 medium did not alter this sensitivity, suggesting that under conditions using a carbon source distinct from glucose, TbHK2 is predominantly responsible for cellular TbHK activity. However, inclusion of glucose (1 mm) or transfer of the parasites to a glucose-rich medium (SDM-79, which has ∼9 mm glucose) led to an increase in resistance to PPi, suggesting that under glucose-rich conditions, the cellular TbHK activity results from a greater TbHK1 contribution. In contrast, we have found that TbHK2–/– PF 29-13 cells grown under these varied conditions did not have an appreciable change in cellular HK sensitivity to PPi (data not shown), supporting the idea that alteration of the relative amounts of TbHK1 and TbHK2 in the cell leads to changes in PPi sensitivity.
FIGURE 7.
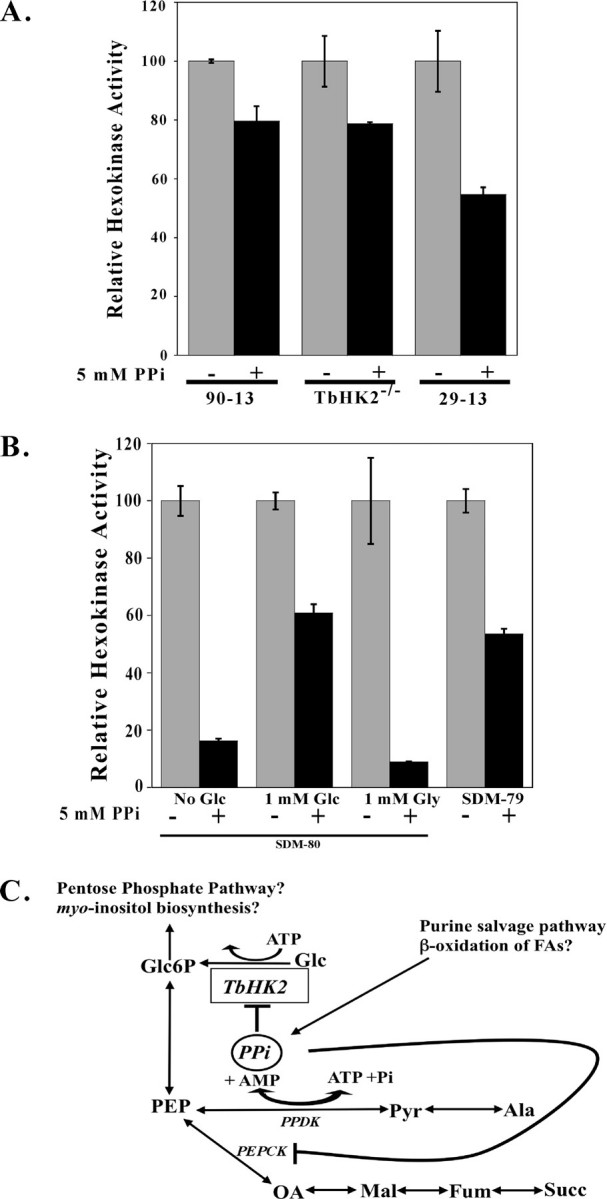
HK activity from cell lysates is inhibited by PPi. A, BSF 90-13, TbHK2–/– PF 29-13, or PF 29-13 cell lysates (from 5 × 105, 1 × 106, and 4 × 106 cell equivalents to keep the HK amount in the assays similar) (7) were incubated with PPi and then assayed for HK activity. These cell lysates have specific activities of 0.097, 0.124, and 0.053 units/mg, respectively. B, under different environmental conditions, T. brucei HK activity sensitivity to PPi changes. PF 29-13 trypanosomes were adapted to growth in SDM-80 without glucose supplemented with 9% heat-inactivated dialyzed serum (Sigma) and 1% heat-inactivated serum (14). The medium was then supplemented with glucose (Glc; 1 mm) or glycerol (Gly; 1 mm), or parasites were shifted into SDM-79 (a glucose-rich medium). After 24 h, cell lysates were prepared, incubated with PPi (5 mm, 10 min on ice), and assayed for HK activity. Lysates (untreated) had 0.045, 0.057, 0.027, and 0.053 units/mg HK activity, respectively. C, a schematic of interconnection of metabolic pathways in the glycosome is shown. Pathways labeled with a question mark are suggested by glycosome proteomics and genomic analysis. Glc6P, glucose 6-phosphate; PEPCK, PEP carboxykinase; PPDK, pyruvate-phosphate dikinase; Pyr, pyruvate; Ala, alanine; Fum, fumarate; Mal, malate, OA, oxalacetate; Succ, succinate; FAs, fatty acids.
DISCUSSION
T. brucei expresses two 98% identical HKs in both PF and BSF life stages (6). Our initial investigations into the functions of the two proteins revealed that TbHK2 was a glycosomal protein that lacked detectable HK activity in vitro (7). However, PF parasites lacking TbHK2 had altered morphology, retarded growth, and increased cellular HK activity (presumably from TbHK1), suggesting that the protein serves some function for T. brucei. Recently, Albert et al. (1) found through RNAi silencing of TbHK2 in BSF parasites that the gene is essential, with RNAi triggering a reduction in cellular glycolysis, leading to BSF death, again emphasizing the important role of TbHK2 in the parasite.
The altered rate of glycolysis in the RNAi BSF parasites suggests that TbHK2 may be playing a role in the regulation of HK activity. Mammals have multiple HKs, with three ∼100-kDa isoforms (Types I, II, and III) consisting of two monomer-like HKs fused together (15). Although the C-terminal domains are catalytically active, the N-terminal monomer-like domains of the Type I and III isoforms are enzymatically inactive, serving to regulate the function of the C-terminal domain by binding glucose 6-phosphate. Authentic TbHK activity likely consists of mixed multimers of TbHK1 and TbHK2, as the original activity purified with a molecular mass consistent with a hexameric complex (11). Like the inactive N-terminal domains of mammalian Type I and III HKs, TbHK2 could regulate TbHK1 with which it is complexed, perhaps by altering the enzymatic activity of TbHK1 after binding some regulatory molecule.
We found previously that free fatty acids were inhibitors of TbHK1 (7), with myristate being the most potent identified to date. Here, we have found that in addition to being an inhibitor of TbHK1, the fatty acid also disrupts oligomers of TbHKs. Because fatty acids are likely found in the glycosome (the site of ether lipid biosynthesis and potentially fatty acid oxidation), similar dynamic reassembly could occur in vivo. Regulatory changes in activity as a result of modulation of complex composition are found throughout biology. Bax, for example, actively facilitates apoptosis via mitochondrial permeabilization as a homodimer while being inactive when dimerized with other cellular components (16). Authentic TbHK activity could be similarly influenced by changes in oligomer composition; as the ratio of complex components is changed during reassembly, the HK activity of the oligomer could change. Indeed, the precedent for allosteric regulation in T. brucei by a catalytically inactive enzyme homolog has recently been established, with S-adenosylmethionine decarboxylase being activated by dimerization with an inactive homolog (17).
However, the story here is different from that of T. brucei S-adenosylmethionine decarboxylase. Disruption of rTbHK oligomers isolated from Escherichia coli followed by reassembly does indeed alter TbHK activity, but in a most surprising way. First, reassembly of catalytically inactive rTbHK2 with a catalytically incompetent rTbHK1 variant (rTbHK1(S160A), which has an altered conserved residue essential for transfer of the phosphoryl group) yields an active rTbHK2 complex. Interestingly, reassembly of rTbHK2 with itself does not activate the assembly, suggesting that a specific interaction between rTbHK2 and rTbHK1 is required for activation to occur. Second, mixed oligomers of rTbHK1 and rTbHK2 have different kinetic properties compared with the parent homohexamers, including increased activity and increased affinity for ATP (Km = 0.12 mm), which more closely resembles values reported for authentic T. brucei HK activity (Km = 0.11 mm) (18) than the rTbHK1 homohexamer (Km = 0.28 mm) (8).
Why myristate? Myristate is very important to the biology of the BSF parasite, serving as the sole fatty acid in the mature glycosylphosphatidylinositol anchor of the variant surface glycoprotein (19). It is tempting to speculate that the large amount of myristate that is either taken up or synthesized by the BSF parasites for glycosylphosphatidylinositol biosynthesis might influence TbHK activity. Glycosylphosphatidylinositol biosynthesis and maturation occur on the endoplasmic reticulum (20), a compartment distinct from glycosomes. However, there is communication between the two organelles, as some proteins transit from one compartment to the other. In yeast, peroxisome biogenesis requires the anterograde movement of proteins from the endoplasmic reticulum to the organelle. In trypanosomes, a glycosylphosphatidylinositol-specific phospholipase C has been found to translocate from glycosomes (which are specialized peroxisomes) to the endoplasmic reticulum in response to cellular stress (21). These observations suggest that glycosomes may receive other cargo, including myristate, from the endoplasmic reticulum.
Alternatively, free fatty acids within the glycosome that originate from the action of glycosomal resident enzymes could be regulating TbHK. In addition to housing ether lipid biosynthesis proteins and enzymes involved in β-oxidation of lipids, lysophospholipase A has been recently identified to be a glycosomal resident protein in both BSF and PF parasites (6). The fatty acids, including myristate, liberated by the action of this protein could be important to TbHK regulation.
In agreement with work on other trypanosomatid HKs, TbHK2 is inhibited (albeit weakly) by bisphosphonates, which are polyphosphate analogs. However, PPi is a more potent inhibitor of TbHK2 while having little effect on TbHK1 activity. Of note, HK activity from BSF cell lysates is less sensitive to PPi inhibition than is HK activity from PF lysates, suggesting that the PF stage parasites may rely on TbHK2 more heavily for metabolic needs.
A number of glycosomal enzymes produce PPi, including hypoxanthine-guanine phosphoribosyltransferase (in the purine salvage pathway). Interestingly, glycosomes lack pyrophosphatase activity (22) but do harbor a pyruvate-phosphate dikinase that may consume PPi in the conversion of PEP to pyruvate (23). PPi inhibits other glycosomal enzymes, including PEP carboxykinase (from T. cruzi) (24) (Fig. 7C). PPi may play a central role in the regulation of the metabolism of PEP by limiting its consumption by either PEP carboxykinase (to succinate) or TbHK2 (to glucose and ATP, although we have yet to demonstrate that the enzyme is capable of catalyzing the reaction in this direction). This would result in directing PEP into amino acid biosynthesis via pyruvate-phosphate dikinase or into other biosynthetic pathways through glucose 6-phosphate.
PPi could alternatively link the coordination of glycolysis and amino acid metabolism, as conditions that favor PPi production from pyruvate-phosphate dikinase (amino acid metabolism) would inhibit TbHK2. PPi has been recognized as a potential regulatory molecule in other systems, with alteration in PPi levels in rat liver triggering changes in blood glucose levels (25). Our observation that TbHK activity in lysates of PF parasites grown under low glucose conditions (when the cells are metabolizing amino acids) is more sensitive to PPi inhibition suggests that TbHK2, and not TbHK1, is the primary HK in these cells, in part because it is a regulable HK.
Acknowledgments
We thank Drs. Roberto Docampo, Kimberly Paul, and Tina Saxowsky for helpful comments on the manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: BSF, bloodstream form; PF, procyclic form; HK, hexokinase; TbHK, T. brucei HK; rTbHK, recombinant TbHK; RNAi, RNA interference; LND, lonidamine; PEP, phosphoenolpyruvate; PPi, pyrophosphate; DMSO, dimethyl sulfoxide; rTbHK2-rTbHK1(S160A) complex, rTbHK2 reassembled with catalytically inactive rTbHK1 variant S160A; TEMED, N,N,N′,N′-tetramethylethylenediamine.
References
- 1.Albert, M. A., Haanstra, J. R., Hannaert, V., Van Roy, J., Opperdoes, F. R., Bakker, B. M., and Michels, P. A. (2005) J. Biol. Chem. 280 28306–28315 [DOI] [PubMed] [Google Scholar]
- 2.Furuya, T., Kessler, P., Jardim, A., Schnaufer, A., Crudder, C., and Parsons, M. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 14177–14182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drew, M. E., Morris, J. C., Wang, Z., Wells, L., Sanchez, M., Landfear, S. M., and Englund, P. T. (2003) J. Biol. Chem. 278 46596–46600 [DOI] [PubMed] [Google Scholar]
- 4.Chambers, J. W., Fowler, M. L., Morris, M. T., and Morris, J. C. (2008) Mol. Biochem. Parasitol. 158 202–207 [DOI] [PubMed] [Google Scholar]
- 5.Nwagwu, M., and Opperdoes, F. R. (1982) Acta Trop. 39 61–72 [PubMed] [Google Scholar]
- 6.Colasante, C., Ellis, M., Ruppert, T., and Voncken, F. (2006) Proteomics 6 3275–3293 [DOI] [PubMed] [Google Scholar]
- 7.Morris, M. T., DeBruin, C., Yang, Z., Chambers, J. W., Smith, K. S., and Morris, J. C. (2006) Eukaryot. Cell 5 2014–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambers, J. W., Morris, M. T., Smith, K. S., and Morris, J. C. (2008) Biochem. Biophys. Res. Commun. 365 420–425 [DOI] [PubMed] [Google Scholar]
- 9.Morris, J. C., Wang, Z., Drew, M. E., and Englund, P. T. (2002) EMBO J. 21 4429–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai, H. J., and Wilson, J. E. (1995) Arch. Biochem. Biophys. 316 206–214 [DOI] [PubMed] [Google Scholar]
- 11.Misset, O., Bos, O. J., and Opperdoes, F. R. (1986) Eur. J. Biochem. 157 441–453 [DOI] [PubMed] [Google Scholar]
- 12.Hudock, M. P., Sanz-Rodriguez, C. E., Song, Y., Chan, J. M., Zhang, Y., Odeh, S., Kosztowski, T., Leon-Rossell, A., Concepcion, J. L., Yardley, V., Croft, S. L., Urbina, J. A., and Oldfield, E. (2006) J. Med. Chem. 49 215–223 [DOI] [PubMed] [Google Scholar]
- 13.Sanz-Rodriguez, C. E., Concepcion, J. L., Pekerar, S., Oldfield, E., and Urbina, J. A. (2007) J. Biol. Chem. 282 12377–12387 [DOI] [PubMed] [Google Scholar]
- 14.Lamour, N., Riviere, L., Coustou, V., Coombs, G. H., Barrett, M. P., and Bringaud, F. (2005) J. Biol. Chem. 280 11902–11910 [DOI] [PubMed] [Google Scholar]
- 15.Cardenas, M. L., Cornish-Bowden, A., and Ureta, T. (1998) Biochim. Biophys. Acta 1401 242–264 [DOI] [PubMed] [Google Scholar]
- 16.Er, E., Lalier, L., Cartron, P. F., Oliver, L., and Vallette, F. M. (2007) J. Biol. Chem. 282 24938–24947 [DOI] [PubMed] [Google Scholar]
- 17.Willert, E. K., Fitzpatrick, R., and Phillips, M. A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 8275–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willson, M., Sanejouand, Y. H., Perie, J., Hannaert, V., and Opperdoes, F. (2002) Chem. Biol. 9 839–847 [DOI] [PubMed] [Google Scholar]
- 19.Ferguson, M. A., and Cross, G. A. M. (1984) J. Biol. Chem. 259 3011–3015 [PubMed] [Google Scholar]
- 20.Vidugiriene, J., and Menon, A. K. (1994) J. Cell Biol. 127 333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanya, S., and Mensa-Wilmot, K. (2006) FEBS J. 273 2110–2126 [DOI] [PubMed] [Google Scholar]
- 22.Michels, P. A., Chevalier, N., Opperdoes, F. R., Rider, M. H., and Rigden, D. J. (1997) Eur. J. Biochem. 250 698–704 [DOI] [PubMed] [Google Scholar]
- 23.Bringaud, F., Baltz, D., and Baltz, T. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 7963–7968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acosta, H., Dubourdieu, M., Quinones, W., Caceres, A., Bringaud, F., and Concepcion, J. L. (2004) Comp. Biochem. Physiol. B Biochem. Mol. Biol. 138 347–356 [DOI] [PubMed] [Google Scholar]
- 25.Veech, R. L., Cook, G. A., and King, M. T. (1980) FEBS Lett. 117 (suppl.) K65–K72 [DOI] [PubMed] [Google Scholar]