

Abstract
A number of intracellular proteins that are protective after brain injury are classically thought to exert their effect within the expressing cell. The astrocytic metallothioneins (MT) are one example and are thought to act via intracellular free radical scavenging and heavy metal regulation, and in particular zinc. Indeed, we have previously established that astrocytic MTs are required for successful brain healing. Here we provide evidence for a fundamentally different mode of action relying upon intercellular transfer from astrocytes to neurons, which in turn leads to uptake-dependent axonal regeneration. First, we show that MT can be detected within the extracellular fluid of the injured brain, and that cultured astrocytes are capable of actively secreting MT in a regulatable manner. Second, we identify a receptor, megalin, that mediates MT transport into neurons. Third, we directly demonstrate for the first time the transfer of MT from astrocytes to neurons over a specific time course in vitro. Finally, we show that MT is rapidly internalized via the cell bodies of retinal ganglion cells in vivo and is a powerful promoter of axonal regeneration through the inhibitory environment of the completely severed mature optic nerve. Our work suggests that the protective functions of MT in the central nervous system should be widened from a purely astrocytic focus to include extracellular and intra-neuronal roles. This unsuspected action of MT represents a novel paradigm of astrocyte-neuronal interaction after injury and may have implications for the development of MT-based therapeutic agents.
The mechanisms by which certain protective proteins expressed by astrocytes affect neuronal regeneration are not well understood. As an example, we have demonstrated that mice lacking the ability to express a protein produced predominantly by astrocytes in the CNS,3 metallothionein isoforms I/II (MT-I/-II), exhibit significantly worse outcomes following a range of CNS injuries (1, 2). Likewise, MT-I/-II-deficient animals fare worse following stroke, experimental autoimmune encephalomyelitis (an experimental animal model of multiple sclerosis), and motor neuron disease (3-5, respectively). Hence, perturbation of an astrocytic protein has major consequences in the injured CNS, including an increase in apoptotic neurons and impaired neuronal regenerative growth. Indeed, genetically modified animals have been produced that exhibit the full range of possible astrocytic MT-I/-II expression, from null to overexpressing strains, and there is a robust correlation between MT-I/-II expression and the ability of the animal to recover from CNS insult or degenerative disease (1, 6, 7). These studies clearly demonstrate that MT-I/-II represented an important mechanism of protection and regeneration in the injured CNS.
There are a number of ways that MT-I/-II might conceivably enhance the ability of astrocytes to promote neuronal regeneration. Metallothioneins, as exemplified by family members MT-I/-II, are zinc-binding proteins that may have roles in metal homeostasis or free radical scavenging (for reviews see Refs. 8, 9), although their role in any tissue remains a matter of debate. Because they lack conventional secretion sequences (10) and demonstrably accumulate in the astrocytic cytoplasm after neuronal injury (11), the general consensus based upon more than 40 years of research is that MTs likely act within the expressing cell itself, and they may, for example, be part of the mechanism by which astrocytes handle toxic intermediates such as reactive oxygen molecules.
However, we have shown that the role of MT-I/-II is potentially more complex than simply acting within astrocytes per se. We recently reported that MT-I/-II strongly increases post-injury regenerative sprouting when added directly to injured neurons in culture (12). In these experiments, there are no glial or immune system cells present, indicating that extracellular MT-I/-II can act directly on injured neurons, i.e. outside the context of astrocytic cytoplasm. These experiments have since been replicated elsewhere showing that “exogenous” MT-I/-II strongly promotes regenerative neurite growth of cortical (12), dopaminergic, and hippocampal neurons (13) and retinal ganglion cells (14) suggesting that there is a robust and generic neuronal response to extracellular MT-I/-II.
Based upon these data and the existing literature, we have hypothesized a model to explain the role for extracellular MT-I/-II within the injured brain. We suggested that astrocytes respond to neural trauma by up-regulating MT-I/-II expression with the MT-I/-II being subsequently secreted, allowing direct interaction with neurons which promotes neuronal regeneration and survival following injury (15). Although there are numerous studies investigating MT-I/-II expression following injury, little is known about the mechanism(s) whereby MT-I/-II might interact with neurons to exert their neuroregenerative effect. To examine these mechanisms, we addressed four questions. (a) Is MT-I/-II released by astrocytes in culture and following brain injury? (b) Is the interaction of extracellular MT-I/-II with neurons receptor-mediated? (c) Can MT-I/-II transfer from astrocytes to neurons be observed directly? (d) Is uptake of exogenous MT-I/-II associated with axon regeneration in vivo? We find that astrocytes are capable of releasing MT-I/-II in culture and that these proteins can be detected within the extracellular environment of the injured brain. In tissue culture experiments we identify a receptor whereby extracellular MT-I/-II directly interacts with neurons and demonstrate the direct transfer of MT-I/-II from astrocytes to neurons. Finally, we demonstrate in vivo that exogenous MT-I/-II uptake is associated with robust axon regeneration following optic nerve transection. The results suggest that transfer of MT-I/-II from astrocytes to neurons is an important component of the response of the CNS to injury.
EXPERIMENTAL PROCEDURES
Metallothionein Protein—Because these studies have been performed in four different laboratories collaborating together, slightly different forms of MT protein have been used. The particular MT form that is used for each experiment has been specifically stated within the text and includes rabbit Zn-MT-IIA (Bestenbalt LLC), rabbit Zn-MT-II (Sigma catalog number M 9542), and a mixture of rabbit Zn-MT-I and Zn-MT-II (Sigma catalog number M 7641). There are small differences in the amino acid structure of these proteins, although all maintain the 20 conserved cysteine residues that characterize mammalian MTs. We have recently reported that MT proteins from a variety of sources, including mammalian and Drosophila MTs, appear to have a similar ability to promote wound healing following CNS injury (35), so we do not expect any issues in using the slightly different MT forms in this study.
Focal Injury to the Adult Rat Neocortex, Collection of Gelfoam, and Western Blotting—Injuries were made to the Par 1 region of the adult rat neocortex as reported previously (12). At the appropriate time, rats were re-anesthetized and transcardially perfused, and brains were then removed and post-fixed overnight in 4% paraformaldehyde at 4 °C. They were then sectioned at 50-μm thickness by vibratome (Leica VT1000E).
In some experiments, absorbent gelfoam, which had been used to seal the burr hole in the skull during the initial surgery, was recovered at several time points post-injury (three animals per group). Gelfoam pieces were pooled for each time point, and soluble proteins were removed from the gelfoam by the addition of 100 μl of PBS, followed by vortexing. Samples were briefly centrifuged at 15,000 rpm for 2 min, and the supernatant was collected and stored at -20° C for subsequent Western blotting.
Western blotting was performed as described previously (36), under reducing conditions to minimize any potential oxidation of MT proteins. In subsequent Western blots, similar procedures were performed with the same samples, but either mouse anti-GFAP (1:500, Chemicon) or rabbit anti-ferritin (1:1000, ICN) primary antibodies were used.
Primary Astrocyte Cultures, Culture Media Collection, and MT-I/-II Radioimmunoassay—Primary astrocyte cultures were prepared as described previously (37). Astrocyte cultures were at least 98% pure (results not shown). Confluent astrocytes cultures were maintained in a total of 500 μl of culture medium and treated with either 10 units/ml IL-1α (Chemicon catalog number IL001), 10 μm ZnSO4, or 10 units/ml IL-1α + 10 μm ZnSO4 combined for the allotted time period, and media were collected, centrifuged at 13,000 × g (5 min), and stored at -20 °C. MT-I/-II was measured by radioimmunoassay as described previously (16).
Primary Neuron Cell Cultures—Cortical neuron cultures were prepared as reported previously (12) and maintained in a culture medium consisting of Neurobasal™ medium, supplemented with 10% fetal bovine serum, 0.1% (final concentration) B-27 supplement, 0.1 mm (final concentration) l-glutamine, and 200 units/ml gentamicin. At the appropriate time, cells were fixed with 4% paraformaldehyde for 20 min, and a permeabilizing immunocytochemistry method was used to visualize intracellular proteins, with most membrane-bound proteins lost using this protocol. This procedure was performed using an antibody diluent containing 0.03% Triton X detergent. For immunocytochemistry, mouse anti-MT (1:500; Dako), rabbit anti-megalin (1:500; Santa Cruz Biotechnology), and rabbit anti-Tau (1:5000; DAKO) antibodies were used. Secondary labeling was performed using appropriate AlexaFluor-488- or -594-conjugated secondary antibodies (Molecular Probes).
Cerebellar granule neurons were prepared from postnatal day 7 Wistar rats (Charles River, Sulzfeld, Germany) as described previously by Schousboe et al. (38). For the cerebellar granule neuron neurite outgrowth assay, MT-I/-II was applied to neurons immediately after plating and fixed 24 h after treatment with 4% (v/v) formaldehyde for 20 min and immunostained using primary rabbit antibodies against GAP-43. Alexa-Fluor secondary goat anti-rabbit Ig antibodies were used to visualize the cells. In some experiments, MT-I/-II was immobilized onto coverslips by incubation on Permanox plasticware overnight and washed three times with PBS, and the neurons were then plated onto these coverslips, and neurite outgrowth was assessed 24 h later. For the analysis of neurite outgrowth, we have used a computer-assisted fluorescence microscopy technique described previously (32). In brief, digital images of at least 200 neurons for each group in each individual experiment were obtained systematically; a frame was superimposed on each image, and the number of intersections between neurites and test lines in a given frame was counted and divided by the number of neuron bodies, whereby a relationship between the number of neurite intersections and neurons is obtained. The neurite growth value of control cells was then considered 100%, and all the experimental groups are compared with it. Statistical evaluation was performed by the Student's paired t test using Fig-P, 2.98 (Biosoft, Cambridge, UK).
Co-immunoprecipitation and Western Blotting Studies—To confirm the interaction between MT and megalin, co-immuno-precipitation experiments were performed whereby an MT antibody was used to pull down MT and MT complexes, which were then probed for the presence of megalin by Western blotting. Briefly, 14-day in vitro cortical neurons plated at a density of 1 × 106 cells/flask were treated with MT, and after 24 h the cells were lysed in 250 μl of ice-cold resuspension buffer (20 mm Tris, pH 7.4, 400 mm NaCl, 7.5 mm MgCl2, 0.2 mm EDTA, 1.0 mm dithiothreitol). The cell lysate was collected into an Eppendorf tube, to which 250 μl of dilution buffer (20 mm Tris, pH 7.4, 100 mm NaCl, 7.5 mm MgCl2, 0.2 mm EDTA, 0.75% Ipegal, 10% glycerol) was added. Lysates were pre-cleared by adding 50 μl of 50% protein-A-Sepharose slurry for 1 h at 4 °C with shaking on a rotational shaker to remove nonspecific proteins that potentially bind to the protein-A-Sepharose beads. The following steps were all performed in a 4 °C cold room. The cell lysate was centrifuged at 3000 rpm for 5 min to pellet the Sepharose beads, and the supernatant was transferred to a fresh Eppendorf tube. The MT antibody (10 μl; Dako) was added to the lysate and incubated at 4 °C with shaking for 1 h. To this, 50 μl of 50% protein-A-Sepharose was added, and the lysate was incubated overnight at 4 °C with shaking. The sample was centrifuged at 3000 rpm for 5 min, and the supernatant was transferred to a new Eppendorf tube. The beads were washed three times with 200 μl of dilution buffer, centrifuging each time at 3000 rpm for 5 min. The final pellet was resuspended in 15 μl of Nu-PAGE lithium dodecyl sulfate sample buffer (Invitrogen) and immediately used for Western blotting. Western blotting was performed as described previously (32), using the Nu-PAGE system (Invitrogen) and 10-20% gradient gels. For all Western blots, the total protein content of samples was determined by the Bradford assay, and equal protein amounts were loaded for all samples. Membranes were probed with a rabbit anti-megalin antibody (1:500; Santa Cruz Biotechnology) and goat anti-rabbit horseradish peroxidase secondary antibody (1:1000; Dako).
AlexaFluor-594 Labeling of Rabbit MT-IIA—Rabbit MT-IIA was chemically conjugated to AlexaFluor-594 using an AlexaFluor probe labeling kit (Molecular Probes). Following the conjugation reaction, AlexaFluor-594 bound MT-IIA (MT-594) was separated from excess AlexaFluor-594 dye by standard gel chromatography over a G-75 column. Western blotting analysis confirmed the separation of the fluorescently tagged MT-IIA from excess dye. The excess fluorescent dye, which becomes nonreactive during this process, was used in later studies as an experimental control.
Megalin siRNA Transfections into Neurons—In some experiments cortical neurons at a density of 1 × 106 cells were transfected with two pre-designed siRNA molecules targeting different regions of the rat megalin gene (Ambion catalog numbers 199330 and 199331). Transfections were performed by electroporation using a rat hippocampal neuron protocol and the Nucleofector™ (Amaxa) upon neurons at the end of the culturing process and immediately prior to plating out. 6 nm siRNA was used for each transfection. As a control, neurons underwent the entire transfection protocol but in the absence of siRNA. Western blotting 24 h later confirmed that transfection with either siRNA molecule greatly reduced expression of megalin (results not shown).
Astrocyte Transfection Studies—The coding sequence for human MT-IIA was removed from a pre-existing bacterial expression vector (12) and inserted into the pEGFP vector (BD Biosciences) such that the plasmid expresses an MT-IIA-GFP fusion protein. The pEGFP vector served as the control for all transfection studies. For transfection, a minimum of 1 × 106 astrocytes was transfected using the Amaxa Nucleofector™ and the Nucleofector™ rat astrocyte transfection kit (VPG-1006). In preliminary studies, transfection efficiencies were ∼30%, and immunocytochemistry confirmed co-localization of MT immunolabeling with MT-GFP (results not shown). Following transfection, the astrocytes were seeded into 14-day in vitro cortical neuron cultures (5 × 104 cells) at a seeding density of 1 × 104 cells and maintained for up to 7 days. At the appropriate time, cells were fixed with 4% paraformaldehyde, and immunocytochemistry was performed as discussed previously. For neuronal immunolabeling, a rabbit anti-Tau antibody (DAKO) was used at a concentration of 1:5000. For detection of MT-GFP and GFP in culture medium, samples were collected and concentrated ∼10-fold using Centricon-3 ultrafiltration columns (Millipore). Briefly, 500 μl of media was placed into each spin column and centrifuged at 10,000 × g for 90 min. Concentrated samples were used for Western blotting studies, using the protocol described earlier. A monoclonal anti-GFP antibody (BD Biosciences) was used at a concentration of 1:4000, detected with a secondary goat anti-mouse horseradish peroxidase-conjugated antibody (DAKO) used at a concentration of 1:1000.
Complete Optic Nerve Transection Surgeries—Adult male Hooded Wistar rats were deeply anesthetized; the right optic nerve was exposed intraorbitally, and the nerve sheath was cut transversely except for the ventral aspect that was left intact to avoid damage to the ophthalmic blood vessels. The nerve parenchyma was lifted using hooked forceps and completely transected with iridectomy scissors ∼1 mm from the back of the eye. The cut ends of the nerve sheath were then sutured together (10-0 thread). The completeness of transection was confirmed by anterograde labeling. Animals with ischemic retinae (determined ophthalmoscopically) were euthanized. Rabbit MT-I/-II (Sigma) or control solutions (PBS or ZnSO4) were administered via a stereotactically positioned 30-gauge needle attached to a 10-μl Hamilton syringe. A final volume of 2.5 μl was administered via two injection sites (nasal and temporal). Eight animals received MT-I/-II injections, and eight received vehicle (saline).
Statistical Analysis—All data are expressed as mean ± S.E. For cell culture experiments, statistical evaluation was performed by the Student's paired t test using either SigmaStat (Systat Software) or Fig-P version 2.98 (Biosoft, Cambridge, UK). RGC counts and axon measurements were analyzed using analysis of variance (StatView, USA) and p values calculated using the Scheffe post hoc test.
RESULTS
Detection of Extracellular MT-I/-II Released from Cultured Cortical Astrocytes—To test the hypothesis that astrocytes are capable of releasing MT-I/-II, primary cortical astrocytes were maintained in vitro, and the presence of MT-I/-II in the culture media was assessed using an MT-I/-II-specific radioimmunoassay (RIA) that is capable of detecting picogram amounts (16, 17). In confluent astrocyte cultures, MT-I/-II was detected in the medium at a range of 15.6 ± 0.49 pg of MT/μg of total protein (average over 12 different experiments). In these cultures, intracellular MT-I/-II expression as observed by immunocytochemistry was low in most astrocytes. Astrocytes were then exposed to either zinc (ZnSO4, 10 μm) or interleukin-1 (recombinant human IL-1, 10 units/ml) or both. Co-treatment with zinc and interleukin-1 has been shown previously to lead to accumulation of MT-I/-II in a granular form near the cell membrane (18), feasibly suggesting MT-I/-II localization within secretory vesicles. When either zinc or IL-1 was added, there was no observable increase in extracellular MT-I/-II within the culture media over a 72-h period (Fig. 1A). However, the zinc and IL-1 co-treatment resulted in a statistically significant (p < 0.01; analysis of variance) increase in extracellular MT-I/-II within culture media, with levels increasing 2.5-fold by 72 h (Fig. 1A). The amount of extracellular MT-I/-II was calculated as a percentage of the total MT-I/-II protein in the cultures, and the levels increased from 6% in the control cultures to 13% 72 h after zinc and IL-1 co-treatment. No cell lysis was observed at any time point using lactate dehydrogenase assays (data not shown). To confirm these observations, astrocytes were cultured in flasks (1 × 106 cells/flask) and treated with either zinc or IL-1 alone, or zinc and IL-1 together (at the concentrations used above). After 72 h the culture medium was collected, concentrated using Millipore Ultrafiltration columns, and the presence of MT in culture medium detected by Western blotting (Fig. 1B). Although there was a low level of MT detectable within the culture medium of untreated cells, this was mildly increased following treatment with zinc or IL-1. However, the dual zinc and IL-1 treatment induced much greater levels of MT secretion by astrocytes (Fig. 1B), in accordance with the RIA results above. Note that equal amounts of protein were loaded for each sample. In parallel experiments, from the same media samples we performed Western blots for albumin and noted no change in albumin levels within the culture medium (Fig. 1B).
FIGURE 1.

A, culture media were collected from primary astrocyte cultures and MT-I/-II levels measured by radioimmunoassay. In uninduced cultures, MT levels were 15.6 ± 0.49 pg per 1 μg of total protein in media. Treatment with either 10 μm ZnSO4 (▪) or 10 units of IL-1 (♦) did not increase levels of MT-I/-II in the media. However, treatment with both ZnSO4 and IL-1 (⋄) resulted in significantly increased levels of MT-I/-II being detected in the culture media. B, Western blotting of culture medium from astrocytes confirmed that treatment with zinc and IL-1 together induces secretion of MT-I/-II by astrocytes but no change in secretion of albumin. All Western blot media samples were collected 72 h after treatment. All RIA measurements are represented as mean ± S.E. values from at least three different experiments.
Detection of Extracellular MT-I/-II Following Focal Cortical Injury—To address the issue of whether MT-I/-II is found extracellularly following brain injury, we used a model of focal cortical injury to the adult rat brain as described previously (11, 12). Absorbent gelfoam was placed over the pial surface above the lesion (Fig. 2A) at the time of injury and left in place for 1-14 days. Proteins were extracted from the gelfoam and analyzed by SDS-PAGE and Western blotting for MT-I/-II, GFAP, and ferritin (Fig. 2B). The latter two proteins were examined as a control for the presence of cells or cell debris (astrocytes and microglia, respectively) that may have invaded the gel foam. An adult rat brain homogenate was used as a positive control for all antisera. As shown in Fig. 2B, MT-I/-II was initially undetectable, but it could be clearly detected in the gelfoam extract at both 7 and 14 days post-injury. The absence of signature intracellular proteins from the two major classes of MT-expressing cells in the CNS, astrocytes and microglia, suggests that the MT-I/-II, which had accumulated in the gelfoam, was derived from an extracellular pool. The adult rat brain homogenate was positive for all three antigens, confirming that Western blotting was successful.
FIGURE 2.

A, schematic diagram describing the focal injury model and the placement of gelfoam above the lesion site. B, Western blotting analysis of protein extracted from gelfoam recovered from cortical brain injury sites, using antibodies against MT-I/-II, GFAP (a marker of astrocytes), and ferritin (a marker of activated microglia). Lane 1, 1 day; lane 2, 4 day; lane 3, 7 day; lane 4, 14 day; lane 5, brain homogenate.
Immobilized Extracellular MT-I/-II Is Not Neuritogenic—We have recently demonstrated that extracellular MT-I/-II can promote neurite outgrowth and axon regeneration, both in culture (12-14) and in vivo following an injury to the brain (12) or to a peripheral (sciatic) nerve (19). To investigate whether it is necessary for extracellular MT to be internalized to mediate neurite outgrowth of cultured neurons as we have reported previously (12-14), rabbit MT-I/-II was bound to Permanox-coated slides. This technique allows the tethered protein to interact with the cell membrane of adhered cells but blocks intracellular import. Compared with the neuritogenic activity of untethered MT-I/II, immobilized MT-I/II was relatively inactive (Fig. 3). Addition of mobile MT-I/-II restored neurite outgrowth in the presence of immobilized MT-I/-II (Fig. 3). As an experimental control, a previously described peptide (P2d) was used (20). The P2d peptide has significant neuritogenic effects that are mediated even if the P2d is immobilized to the Permanox substrate (Fig. 3). The P2d peptide binds and signals through a cell surface glycoprotein (neural cell adhesion molecule, NCAM), and thus is expected to function from outside the cell by means of surface membrane receptors (20).
FIGURE 3.

Rabbit MT-II was exposed to Permanox cover slides, resulting in an immobile MT-II substrate upon which cerebellar granule neurons were plated. Immobilized MT-II had no effect on neurite outgrowth, even at 81 μg/ml, whereas immobilized P2d peptide, which interacts directly with the neural cell adhesion molecule, did promote neurite outgrowth. Addition of labile MT-II to media of cultures that had immobilized MT-II (9 μg/ml) promoted neurite outgrowth. Data are expressed as mean values ± S.E. **, p < 0.01; ***, <0.005, when compared with the control cultures. ctrl, control. Scale bar = 10 μm.
Identification of Megalin as a Receptor Responsible for MT Uptake in Cultured Neurons—One explanation for the failure of immobilized MT-I/-II to promote neuritogenic growth is that this process requires neuronal uptake. To investigate this, the interaction of exogenous MT-I/-II with cultured neurons was examined using detergent-permeabilizing immunohistochemical protocols to visualize intracellularly localized proteins (Fig. 4). Using standard permeabilizing immunocytochemical techniques (fixation with 4% paraformaldehyde followed by incubation with antibodies in the presence of Triton X-100), faint and diffuse immunostaining was observed throughout the cell body within untreated neurons (Fig. 4A), which was probably nonspecific because identical outcomes were observed 24 h after treatment with saline (Fig. 4C) or when the primary antibody was excluded from the immunolabeling process (Fig. 4D). However following 24 h of incubation with extracellular MT-I/-II, intense MT-I/-II immunoreactivity was specifically distributed, forming a crescent shape within the cell, which was excluded from the nucleus and also in neuritic processes (Fig. 4B). Based upon the immunostaining methodology, these observations suggest that the intense MT-I/-II immunoreactivity within neurons most likely represents internalized MT-I/-II. This was confirmed using MT-I/-II bound to AlexaFluor-594 (MT-594). After addition to neuronal cultures, MT-594 associated with neurons also in a punctate crescent-shaped distribution after 24 h (Fig. 4, E and G), whereas AlexaFluor-594 dye alone was not internalized by neurons (Fig. 4F). Immunostaining with an anti-MT-I/-II antibody shows co-localization of MT-594 fluorescence and MT-I/-II protein (results not shown). We did observe occasional uptake of MT-594 by contaminating astrocytes within these primarily neuronal cultures (<98%), although the distribution within the cell was different (results not shown).
FIGURE 4.
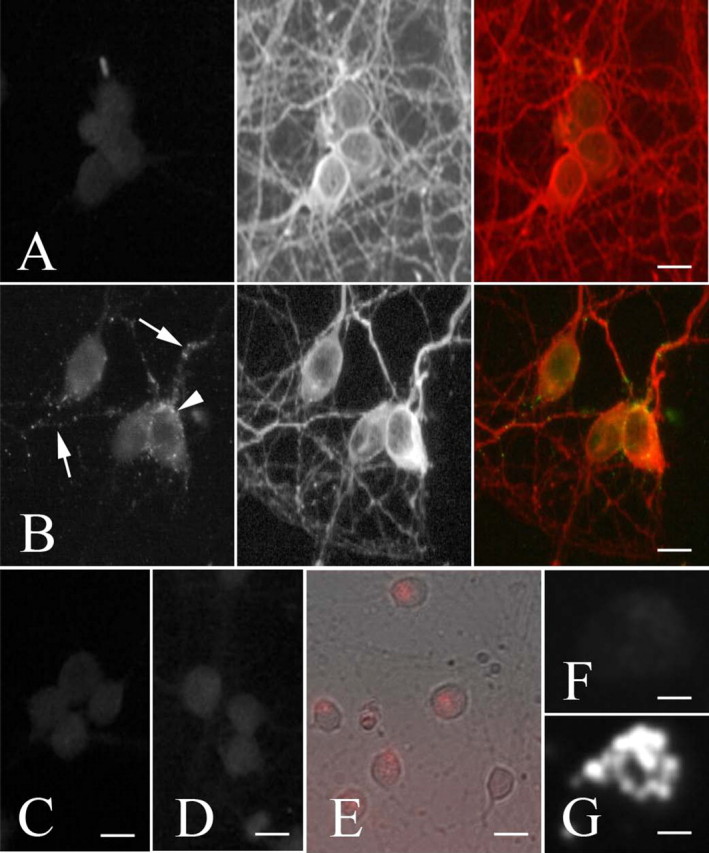
Using standard permeabilizing immunocytochemical techniques, faint and diffuse MT (green) immunostaining was observed within untreated neurons (A) co-labeled for the microtubule-associated protein Tau (red). However after 24 h, intense MT-IIA immunoreactivity was present in a very defined distribution within neurons, forming a crescent shape within the cell (arrow-head), and excluded from the nucleus, and was also found within neuronal processes (arrows) (B). A similar faint and diffuse MT staining pattern was observed following treatment with saline (C) or when the primary antibody was excluded from the immunolabeling protocol (D). When MT-IIA bound to AlexaFluor-594 (MT-594) was applied to neurons, MT-594 (red) associated with neurons in a punctate crescent-shaped distribution after 24 h (E, higher magnification G), whereas AlexaFluor-594 dye alone was not associated with neurons (F). Scale bars = 10 μm (A-E), 5 μm (F and G).
Intercellular Transfer of MT-IIA from Astrocytes to Neurons in Culture—Given our observations of astrocytic secretion of MT-I/-II and neuronal uptake of extracellular MT-I/-II, we aimed to observe the intercellular transfer of MT from astrocytes to neurons in culture. Cortical astrocytes were transfected with a MT-IIA-GFP fusion protein expressing plasmid and seeded into 14-day in vitro cortical neuron cultures. After 4 days, MT-IIA-GFP could be visualized not only in transfected astrocytes but also in surrounding neurons (Fig. 5A). At higher magnification, it appeared that MT-GFP had accumulated within neurons in a crescent-shaped distribution similar to that observed in the MT-594 studies and also to a lesser degree in neuronal processes (Fig. 5B), although the latter was not always clearly visible. We did not readily observe MT-IIA-GFP in neurons 1 or 2 days (results not shown) after seeding astrocytes into neuronal cultures of similar density, suggesting the gradual accumulation of MT-IIA-GFP in neurons. In parallel experiments, astrocytes transfected with a GFP-expressing plasmid were seeded into neuronal cultures of similar cell density. However, no GFP was observed within neurons, up to 7 days later (Fig. 5C). Cell homogenates were collected from the cultures, and Western blotting confirmed that in cultures with MT-IIA-GFP expressing astrocytes, the MT-IIA-GFP was present as the fusion protein, with no nonfusion GFP detectable (Fig. 5D). In separate experiments, media were collected from MT-IIA-GFP or GFP-transfected astrocytes over 24 h, and Western blotting was used to determine the levels of secretion of both proteins from astrocytes. Although there was some GFP detectable in culture media from GFP-transfected astrocytes, there was appreciably greater levels of MT-IIA-GFP detected in media from transfected astrocytes (Fig. 5E), indicating the secretion of MT-IIA-GFP by astrocytes.
FIGURE 5.

Astrocytes expressing MT-IIA-GFP fusion protein were seeded into 14-day in vitro cortical neuron cultures (immunolabeled against the cytoskeletal protein Tau). After 4 days, MT-GFP was visualized not only in transfected astrocytes (arrow) but also in surrounding neurons (arrowheads, A). At higher magnification, MT-GFP appeared in a crescent-shaped distribution within neurons (inset, B). In parallel experiments, GFP-expressing astrocytes transfected were seeded into neuronal cultures of similar density (demonstrated by nuclear staining) (A and B); however, no GFP was observed within neurons 7 days later (C). Cell lysates were collected from the cultures at 1 and 4 days after seeding of astrocytes, and Western blotting confirmed that MT-GFP was present as the fusion protein (D). The size difference between GFP and MT-GFP reflects the additional 7-kDa mass in the fusion protein (D). In separate experiments, media were collected from MT-IIA-GFP or GFP expressing astrocytes over 24 h, and Western blotting was used to determine that MT-GFP is secreted from astrocytes, with low levels of GFP secretion also detected (E). Scale bar = 15 μm (A) and 22.5 μm (B and C).
Neuronal Uptake of MT Is Mediated by Interaction with the Megalin Receptor—It has recently been reported that the uptake of extracellular MT-I/-II by renal cells is mediated by the endocytic receptor megalin (21, 22), raising the possibility of a similar import mechanism within CNS neurons. Using immunohistochemistry, we observed megalin expression by cortical neurons throughout the cell body and processes (Fig. 6, A and B). To investigate the possibility that megalin is involved in neuronal uptake of extracellular MT-I/-II, cortical neurons were pretreated with the competitive megalin ligand RAP (1 or 2 μm for 15 min) followed by addition of MT-594 into the culture medium. After 24 h, cells were fixed and counterstained with the nuclear stain Hoechst S769121 (Molecular Probes), and the percentage of cells containing MT-594 was determined. In this series of experiments, ∼78% of all cortical neurons contained MT-594 (n = 4 experiments). In the presence of either 1 or 2 μm RAP, this dropped significantly to 54% (p < 0.01) or 37% (p < 0.01), respectively.
FIGURE 6.

Immunocytochemistry was used to visualize megalin expression in cultured cortical neurons. Megalin expression was observed throughout the cell bodies and processes of neurons 2 (A) and 24 h (B) after plating. To confirm the involvement of megalin in mediating neuronal uptake of extracellular MT, 10 μg/ml MT was applied into the culture medium, and cell lysates were collected 24 h later. Using co-immunoprecipitation techniques, MT-megalin complexes were isolated from the cellular lysates following MT treatment and visualized by Western blotting. No such MT-megalin complexes were observed by Western blotting in vehicle (saline)-treated neurons, or in a sample where no cell lysate was used (C). In a final series of experiments, cortical neurons were transfected with siRNA targeting megalin (Ambion pre-designed siRNA), and MT-mediated neurite outgrowth was assessed after 24 h. Both siRNA molecules blocked the ability of MT to promote neurite outgrowth (D). Experiments were performed three times, and the data are presented as mean ± S.E. from the replicate experiments. Scale bars = 15 μm (A) and 7.5 μm (B).
To confirm the interaction between MT and megalin, co-immunoprecipitation experiments were performed. After pull-down with an anti-MT antibody, Western blotting revealed a single band corresponding to megalin for cell samples collected from neurons treated with 10 μg/ml MT-IIA for 24 h (Fig. 6C), and given that there is little MT-IIA immunostaining on the cell surface at this time (Fig. 4A), this suggests that MT-IIA and megalin are present as a complex within cortical neurons. Unexpectedly, neurons not treated with exogenous MT-IIA exhibited a faint megalin band, indicating that endogenous MT (expressed by cells present in the culture) was also present in a complex with megalin inside cortical neurons.
As a final confirmation that megalin mediates the action of exogenous MT upon neurons, we transfected cortical neurons with Ambion pre-designed siRNA targeting megalin (catalog numbers 199330 and 199331) and after 24 h applied MT into the culture medium. In the absence of megalin siRNA, exogenous MT promoted neurite outgrowth by more than 30% after 24 h (Fig. 6D). However, transfection with either siRNA molecule blocked the action of MT (Fig. 6D).
MT-I/-II Is a Potent Promoter of Axonal Regeneration Following Complete Optic Nerve Transection—Given our data regarding the requirement for internalization of MT-I/-II by neurons for neuritogenic activity (Fig. 3), a suitable model of in vivo axon regeneration was required to investigate MT-I/-II-mediated regeneration. The transected optic nerve model was considered appropriate because of the anatomical separation between RGC cell bodies and the site of lesion and the absence of axonal regeneration following transection. In preliminary experiments, we performed immunohistochemistry to determine the levels of endogenous MT-I/-II within the uninjured and injured eye. No MT-I/-II staining was observed within RGCs, either in uninjured animals or 4 weeks after injury (Fig. 7A), and there was only a minor increase in noncellular MT-I/-II immunoreactivity (no co-localization with RGCs, RGC axons, glial end feet, or displaced amacrine cells; results not shown) in the eye at 4 weeks post-injury suggesting only limited, at best, production of endogenous MT-I/-II following transection. We assessed RGC uptake of MT-I/-II by injecting MT-594 into the vitreous of the eye of adult rats. At 2 h post-injection, retinal whole mounts were made, and intracellular MT-594 was found within the retinal ganglion cell layer of the retina (Fig. 7B). Hoechst staining demonstrated that MT-594 was not found in other cellular layers (Fig. 7C). Immunostaining with the neurofilament marker Tuj1 was co-localized with the presence of MT-594, confirming that RGCs rapidly take up MT-594 (Fig. 7, D and E). The presence of MT-594 persisted 4 h after injection; however, by 24 h there was little MT-594 within the retinal ganglionic cell layer (results not shown). These experiments were performed in three rats per time point, with similar results observed in all animals. Immunohistochemistry demonstrated that most RGCs also express megalin (Fig. 7F), a finding we have recently demonstrated in vitro (14). To confirm the involvement of megalin in mediating MT uptake by RGCs, MT or saline was injected into one eye of adult rats, and after 2 h both injected and noninjected retinae were removed and processed for co-immunoprecipitation (as per the cortical neuron samples, see “Experimental Procedures”). Western blotting demonstrated the presence of MT-megalin complexes in the MT-injected retina but not the saline-injected or noninjected eyes (Fig. 7G).
FIGURE 7.

A, immunohistochemistry demonstrated that MT-I/-II is not present within RGCs (present as empty spaces in the tissue; arrows) either pre- or 4 weeks post-injury. Fluorescence was observed in the RGC and inner plexiform layers in a noncellular distribution, in a similar pattern and intensity to the no primary antibody negative control, suggesting that this is nonspecific staining. There appeared to be a mild increase in MT-I/-II immunoreactivity 4 weeks after injury, in a noncellular distribution. To investigate RGC uptake of MT-I/II, fluorescently tagged rabbit MT-IIA (MT-594) was intravitreally injected into the eye of adult rats. At 2 h post-injection, MT-594 was observed within cells at the upper layer of the retina, most likely RGCs (B). The cellular organization of the retina was clearly distinguished by Hoechst dye (C). Immunohistochemistry using Tuj1, a specific marker of RGCs, confirmed that the MT-594 containing cells are RGCs (D). At higher magnification (E), it is clear that although MT-594 is localized within the cell body of RGCs (arrows), some RGCs contained no MT (arrowhead). Immunohistochemistry confirmed that RGCs express megalin (F). In a separate series of experiments, MT or saline was injected into the retina, and whole retinae were collected 2 h later, homogenized, and processed for co-immunoprecipitation to detect MT-megalin complexes. Western blotting revealed the presence of MT-megalin complexes only in the MT-injected retina, not saline-injected or noninjected eyes of the same animals (G). GCL, ganglionic cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; AB, antibody.
At 4 weeks after complete optic nerve transection, GAP-43 immunohistochemistry showed that optic axons within the nerve sheath of vehicle-treated animals had retracted back toward the eye from the lesion site (Fig. 8A). Remaining axons exhibited punctate staining, suggesting degeneration. Strikingly, however, in animals that received a single intravitreal injection of MT-I/-II, GAP-43+ve optic axons filled the proximal nerve, extending up to the transection site (Fig. 8B). Although this may represent either axon regeneration or lack of “die back” from the lesion site, the swollen shape of the nerve was engorged with axons, many of which had turned upon themselves at the transection site, suggesting a robust regenerative response. In five of eight animals, axons had regenerated well beyond the transection site and into the distal nerve (Fig. 8, B and D). Axons were present across the width of the nerve and projected an average of 346 ± 288 μm (a total of 1346 ± 288 μm from the base of the retina) beyond the transection site (Fig. 8E), with the longest extending 720 μm. These observations were confirmed with DiI tracing; in vehicle-treated animals, there was an absence of axons at the transection site or within the distal nerve (Fig. 8C), but following MT-I/II, axons were observed up to 1000 μm beyond the transection site (Fig. 8D). Interestingly, injection of MT-I/-II at the injury site (presumably limiting uptake of MT-I/-II by RGC cell bodies) immediately after optic nerve transection provided a mild benefit but did not promote axon regeneration up to or across the lesion site (Fig. 8E).
FIGURE 8.

GAP-43-labeled axons in the proximal optic nerve of PBS-injected (A) and rabbit MT-I/-II-injected (B) animals at 4 weeks after optic nerve transection surgery. Arrows indicate site of transection. Confocal microscopy at 4 weeks after optic nerve transection found abrupt termination of DiI-labeled axons within the proximal nerve (C), well before the transection site (large arrow). Intravitreal injection of MT-I/-II resulted in a number of regenerating axons crossing the lesion site (large arrow) and extending into the distal nerve (D). Leading axons extend ∼1000 μm into the distal nerve (small arrows). DiI back-labeled axons were also measured from their origin site at the base of the retina (E). Although intravitreal injection of PBS marginally improved average axonal length (E), a single intravitreal injection of rabbit MT-I/-II resulted in significantly improved axonal regeneration, with the average axonal length ∼3 times greater than vehicle-treated animals. Injection of MT-I/-II at the injury site did not promote axon regeneration (E). The distance from the lesion site to the base of the retina (1000 μm) is indicated by the segmented line.
DISCUSSION
Extracellular MT-I/-II within the Injured Brain and Astrocytic Release of MT-I/-II—We demonstrate that cultured astrocytes are capable of releasing MT-I/-II in response to combined zinc and IL-1α stimulation by up to 2.5-fold at 72 h. Such stimulation encourages astrocytic accumulation of MT-I/-II within vesicles (18). Interestingly, zinc and, to a lesser extent, IL-1α are powerful inducers of MT-I/-II synthesis in astrocytes, but either one alone does not promote MT-I/II release. Both zinc and IL-1α would plausibly be present following a CNS lesion. The levels of MT-I/-II attained in astrocyte media in our studies are below the 0.1-5.0 μg/ml range at which extracellular MT-I/-II promotes neurite outgrowth in culture (12-14). However, these levels are diluted out over the total medium volume, and it is likely that the local concentrations of MT-I/-II at the junctions between astrocytes and neurons in vivo are much higher. Combinations of agents that are more potent in their ability to trigger MT-I/-II release may also exist.
Although we and others have demonstrated secretion of MT from cultured cells (23-25), we show here for the first time that extracellular MT-I/-II unambiguously accumulates in the extracellular environment of the injured brain. Direct quantitation was not possible in vivo, but levels had increased sufficiently to be visualized by Western blotting. Our in vitro data suggest regulated astrocytic MT-I/-II release to the extracellular environment. Although we suggest that regulated release may also occur in our brain injury model, lysed astrocytes are another possibility. However, release via lysis is unlikely for two reasons; MT-I/-II was only detected after 4 days by which time the majority of astrocytic death had occurred, and GFAP was not detected in the extracellular space, as would be expected if the MT-I/-II had been derived from lysed cells.
Observation of Intercellular Transfer of MT from Astrocytes to Neurons—Although we showed in separate experiments that MT-I/-II is released by astrocytes, and taken up by neurons, it was important to demonstrate directly that transfer occurred between astrocytes and neurons. Using an MT-GFP fusion protein that can be readily distinguished from endogenous MT-I/-II, we demonstrated unambiguously that transfer occurs from astrocytes to neurons in co-cultures. MT-GFP first appears in the culture medium and accumulates in neurons over several days, correlating with the time course of MT-I/-II accumulation as measured by RIA. We are confident that the green fluorescence observed in neurons represents MT-GFP and not GFP cleaved from the fusion protein for three reasons. First, we found that MT-GFP is released into the culture medium by transfected astrocytes; second, Western blots did not reveal any cleaved GFP within the co-cultures; and third, GFP alone did not undergo intercellular transfer under the same culture conditions. The results further support our model that astrocytes release MT, which is subsequently internalized by neurons.
The Mechanism of MT-Neuronal Interactions—We have presented several different lines of evidence to suggest that MT-I/-II is internalized by cultured neurons, and that internalization is required for MT-I/-II-dependent neurite growth. First, MT-I/-II bound to Permanox substrate fails to promote neurite outgrowth, suggesting that although MT-I/-II can access surface receptors on the neurons, such access is not sufficient for bioactivity. Second, MT-I/-II accumulates at the neuronal membrane for 24 h before being internalized. Third, MT-IIA-GFP undergoes intercellular transfer from astrocytes to neurons in vitro. Finally, the competitive megalin receptor ligand (RAP) blocks a significant proportion of MT-I/-II binding to neurons, and inhibiting megalin expression using siRNA blocked the ability of MT to promote neurite outgrowth.
Previous reports provide additional evidence that the megalin receptor is involved in MT-I/-II uptake (30-60%) by other cell types, including kidney tubule cells (21, 22). Megalin and exogenously applied MT co-localize within kidney cells, and MT uptake is reduced in the presence of the megalin ligand RAP or anti-megalin antibodies. Surface plasma resonance studies also suggest that there is one binding site within the α-domain (21). Taken together, the results support our observations of neuronal MTI-/-II uptake, at least some of which is mediated by the megalin receptor, which can then in turn promote neurite outgrowth. We note that at the time of writing a recent report has been published indicating that interaction with megalin is required for MT to promote neurite outgrowth of cerebellar granule neurons (26), further supporting our hypothesis.
MT-I/-II Promotes Axonal Regeneration Following Complete Optic Nerve Transection—The optic nerve provides an elegant injury model for directly investigating the effect of MT-I/-II on injured CNS neurons in vivo. Another major advantage is that endogenous MT-I/-II was not detected in retinal ganglion cells or other retinal cells, even after optic nerve transection, therefore allowing direct assessment of exogenous MT-I/-II. A single intravitreal injection of MT-I/-II following complete optic nerve transection results in axonal regeneration up to 1000 μm past the transection site, an extent significantly greater than that seen after injection of vehicle alone. Our observations are supported by previous data showing that axonal regeneration is impaired in peripheral nerves of MT-I/-II-deficient mice (19). We also demonstrate RGC uptake of MT-594 at the soma as well as megalin expression on RGCs and the presence of MT-megalin complexes in the retina following MT injection by co-immunoprecipitation and Western blotting; conversely, MT-II injection at the injury site does not promote axon regeneration. We also have recently reported that MT-I/-II promotes neurite outgrowth of cultured RGCs and that this is mediated via megalin, which is located primarily on RGC somata (14). Taken together, our in vitro and in vivo studies suggest that uptake of extracellular MT-II (mediated in part by megalin) by RGC somata is required for improved axon regeneration.
A Novel Interaction between Injured Neurons and Astrocytes Promotes Axonal Regeneration That Is Mediated by MT-I/-II—It is well accepted that astrocytes contribute to the inhibitory environment following CNS lesions, via the proliferation and migration of astrocytes into a lesion site to form a glial scar as well as via the expression of inhibitory molecules (see review by Yiu and He (27)). However, astrocytes also have a protective role because targeted ablation to remove dividing reactive astrocytes in the immediate vicinity of the injury causes widespread cellular degeneration and tissue disruption (28). Protection is attributable to neurotrophin production and free radical scavenging (29-31).
Here we add to the known repertoire of astrocytes and present a novel neuroregenerative mechanism employed by these glial cells following injury. We have provided a series of experiments demonstrating for the first time that astrocytes respond to injury by releasing a protective protein, in this instance MT, to the extracellular milieu. The protective protein is internalized by neurons over a protracted period, which are then able to elicit a regenerative response. Our results correlate well with published observations of MT-I/-II physiology within the injured brain, where we have shown previously that MT expression is up-regulated 4 days after cortical injury (11) correlating with the onset of regenerative sprouting (33, 34). It will be interesting to investigate whether other proteins with a known protective role inside astrocytes are similarly released following injury and interact with neurons. Our results also suggest that MT-I/-II may represent an attractive target for therapeutic intervention to promote axonal regeneration.
New Insight into the Function of Metallothionein within the Injured Brain—Understanding the intracellular, and more recently reported extracellular, physiological functions of MTs in the injured brain has posed a significant challenge. Here we have provided a series of experiments that support the hypothesis that extracellular MT-I/-II represents an important event following CNS injury. Extracellular MT-I/-II thus promotes axon regeneration in vitro and in vivo, a response mediated by neuronal uptake of MT-I/-II via the megalin receptor. Although the individual pieces of evidence suggest that this mechanism occurs physiologically, conclusive demonstration is understandably difficult. Nevertheless, we show the intercellular transfer of MT-I/-II from cultured astrocytes to neighboring neurons, providing strong evidence to support our hypothesis. In summary, our findings represent a major advance in the understanding of the physiological function of extracellular MT-I/-II within the injured brain. Extracellular function does not preclude the intracellular roles of MT-I/-II, such as free radical scavenging and metal binding, and it is likely that there is a complex combination of intra- and extracellular roles of MT-I/-II following brain injury.
This work was supported in part by Australian Research Council Discovery Project Grant DP0556630 (to R. S. C.), a grant from the Australian Alzheimers Research Foundation (to A. K. W.), a Research Grant from the Clive and Vera Ramaciotti Foundation, and by Ministerio de Ciencia y Tecnología and Feder Grant SAF2005-00671 (to J. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: CNS, central nervous system; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; GAP-43, growth-associated protein-43; GFAP, glial fibrillary acidic protein; GFP, green fluorescent protein; IL-1α, interleukin-1α; MT, metallothionein; PBS, phosphate-buffered saline; RAP, receptor-associated protein; RGC, retinal ganglion cell; RIA, radioimmunoassay; siRNA, short interfering RNA.
References
- 1.Penkowa, M., Carrasco, J., Giralt, M., Moos, T., and Hidalgo, J. (1999) J. Neurosci. 1 2535-2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giralt, M., Penkowa, M., Lago, N., Molinero, A., and Hidalgo, J. (2002) Exp. Neurol. 173 114-128 [DOI] [PubMed] [Google Scholar]
- 3.Campagne, M. L., Thibodeaux, H., van Bruggen, N., Cairns, B., Gerald, R., Palmer, J. T., Williams, S. P., and Lowe, D. G. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 12870-12875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Penkowa, M., and Hidalgo, J. (2001) Exp. Neurol. 170 1-14 [DOI] [PubMed] [Google Scholar]
- 5.Puttaparthi, K., Gitomer, W. L., Krishnan, U., Son, M., Rajendran, B., and Elliott, J. L. (2002) J. Neurosci. 22 8790-8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Penkowa, M., Espejo, C., Martinez-Caceres, E. M., Poulsen, C. B., Montalban, X., and Hidalgo, J. (2001) J. Neuroimmunol. 119 248-260 [DOI] [PubMed] [Google Scholar]
- 7.Xie, T., Tong, L., McCann, U. D., Yuan, J., Becker, K. G., Mechan, A. O., Cheadle, C., Donovan, D. M., and Ricaurte, G. A. (2004) J. Neurosci. 24 7043-7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hidalgo, J., Aschner, M., Zatta, P., and Vasak, M. (2001) Brain Res. Bull. 55 133-145 [DOI] [PubMed] [Google Scholar]
- 9.West, A. K., Chuah, M. I., Vickers, J. C., and Chung, R. S. (2004) Rev. Neurosci. 15 157-166 [DOI] [PubMed] [Google Scholar]
- 10.Palmiter, R. D. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 8428-8430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung, R. S., Adlard, P. A., Dittmann, J., Vickers, J. C., Chuah, M. I., and West, A. K. (2004) J. Neurochem. 88 454-461 [DOI] [PubMed] [Google Scholar]
- 12.Chung, R. S., Vickers, J. C., Chuah, M. I., and West, A. K. (2003) J. Neurosci. 23 3336-3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohler, L. B., Berezin, V., Bock, E., and Penkowa, M. (2003) Brain Res. 992 128-136 [DOI] [PubMed] [Google Scholar]
- 14.Fitzgerald, M., Nairn, P., Bartlett, C. A., Chung, R. S., West, A. K., and Beazley, L. D. (2007) Exp. Brain Res. 183 171-180 [DOI] [PubMed] [Google Scholar]
- 15.Chung, R. S., and West, A. K. (2004) Neuroscience 123 595-599 [DOI] [PubMed] [Google Scholar]
- 16.Gasull, T., Rebollo, D. V., Romero, B., and Hidalgo, J. (1993) J. Immunoassay 14 209-225 [DOI] [PubMed] [Google Scholar]
- 17.Hidalgo, J., Garcia, A., Olivia, A. M., Giralt, M., Gasull, T., Gonzalez, B., Milnerowicz, H., Wood, A., and Bremner, I. (1994) Chem. Biol. Interact. 93 197-219 [DOI] [PubMed] [Google Scholar]
- 18.Kikuchi, Y., Irie, M., Kasahara, T., Sawada, J., and Terao, T. (1993) FEBS Lett. 317 22-26 [DOI] [PubMed] [Google Scholar]
- 19.Ceballos, D., Lago, N., Verdu, E., Penkowa, M., Carrasco, J., Navarro, X., Palmiter, R. D., and Hidalgo, J. (2003) Cell. Mol. Life Sci. 60 1209-1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedersen, M. V., Kohler, L. B., Ditlevsen, D. K., Li, S., Berezin, V., and Bock, E. (2004) J. Neurosci. Res. 75 55-65 [DOI] [PubMed] [Google Scholar]
- 21.Klassen, R. B., Crenshaw, K., Kozyraki, R., Verroust, P. J., Tio, L., Atrian, S., Allen, P. L., and Hammond, T. G. (2004) Am. J. Physiol. 287 F393-F403 [DOI] [PubMed] [Google Scholar]
- 22.Wolff, N. A., Abouhamed, M., Verroust, P. J., and Thevenod, F. (2006) J. Pharmacol. Exp. Ther. 318 782-791 [DOI] [PubMed] [Google Scholar]
- 23.Trayhurn, P., Duncan, J. S., Wood, A. M., and Beattie, J. H. (2000) Horm. Metab. Res. 32 542-547 [DOI] [PubMed] [Google Scholar]
- 24.Trayhurn, P., Duncan, J. S., Wood, A. M., and Beattie, J. H. (2000) Am. J. Physiol. 279 R2329-R2335 [DOI] [PubMed] [Google Scholar]
- 25.Uchida, Y., Gomi, F., Masumizu, T., and Miura, Y. (2002) J. Biol. Chem. 277 32353-32359 [DOI] [PubMed] [Google Scholar]
- 26.Ambjørn, M., Asmussen, J. W., Lindstam, M., Gotfryd, K., Jacobsen, C., Kiselyov, V. V., Moestrup, S. K., Penkowa, M., Bock, E., and Berezin, V. (2008) J. Neurochem. 104 21-37 [DOI] [PubMed] [Google Scholar]
- 27.Yiu, G., and He, Z. (2006) Nat. Rev. Neurosci. 7 617-627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faulkner, J. R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B., and Sofroniew, M. V. (2004) J. Neurosci. 24 2143-2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sofroniew, M. V., Howe, C. L., and Mobley, W. C. (2001) Annu. Rev. Neurosci. 24 1217-1281 [DOI] [PubMed] [Google Scholar]
- 30.Chen, Y., and Swanson, R. A. (2003) J. Cereb. Blood Flow Metab. 23 137-149 [DOI] [PubMed] [Google Scholar]
- 31.Liberto, C. M., Albrecht, P. J., Herx, L. M., Yong, V. W., and Levison, S. W. (2004) J. Neurochem. 89 1092-1100 [DOI] [PubMed] [Google Scholar]
- 32.Rønn, L. C., Ralets, I., Hartz, B. P., Bech, M., Berezin, A., Berezin, V., Moller, A., and Bock, E. (2000) J. Neurosci. Methods 100 25-32 [DOI] [PubMed] [Google Scholar]
- 33.King, C. E., Jacobs, I., Dickson, T. C., and Vickers, J. C. (1997) Neuroreport 8 1663-1665 [PubMed] [Google Scholar]
- 34.King, C. E., Canty, A. J., and Vickers, J. C. (2001) Neuropathol. Appl. Neurobiol. 27 115-126 [DOI] [PubMed] [Google Scholar]
- 35.Penkowa, M., Tio, L., Giralt, M., Quintana, A., Molinero, A., Atrian, S., Vasak, M., and Hidalgo, J. (2006) J. Neurosci. Res. 83 974-984 [DOI] [PubMed] [Google Scholar]
- 36.Chung, R. S., Holloway, A. F., Eckhardt, B. L., Harris, J. A., Vickers, J. C., Chuah, M. I., and West, A. K. (2002) Biochem. J. 365 323-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vincent, A. J., Taylor, J. M., Choi-Lundberg, D. L., West, A. K., and Chuah, M. I. (2005) Glia 51 132-147 [DOI] [PubMed] [Google Scholar]
- 38.Schousboe, A., Meier, E., Drejer, J., and Hertz, L. (1989) in A Dissection and Tissue Culture Manual of the Nervous System (Shahar, A., de Vellis, J., Vernadakis, A., and Haber, B., eds) pp. 203-206, Alan R. Liss, Inc., New York