

Abstract
In this study, we determined the regulation and potential function of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (HMGR) during skin repair in mice. Upon skin injury, healthy mice exhibited a biphasic increase in HMGR expression and activity with elevated levels at days 3 and 13 post-wounding. In situ hybridization revealed wound margin keratinocytes as a cellular source of HMGR expression. In vitro experiments using cultured HaCaT keratinocytes uncovered epidermal growth factor (EGF), transforming growth factor (TGF)-α, and insulin as potent co-inducers of HMGR activity and vascular endothelial growth factor (VEGF) in the cells. Insulin-, but not EGF-mediated VEGF protein expression was functionally connected to co-induced HMGR activity, as simvastatin restrictively interfered only with insulin-induced translation of VEGF mRNA by inhibition of eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) phosphorylation. Functional ablation of insulin-induced sterol regulatory element-binding protein (SREBP)-2 by siRNA abolished HMGR expression and insulin-triggered VEGF protein release from keratinocytes. Simvastatin also blocked proliferation of cultured keratinocytes. The observed inhibitory effects of simvastatin on keratinocyte VEGF expression and proliferation could be reversed by mevalonate, the product of HMGR enzymatic activity. In accordance, simvastatin-mediated inhibition of HMGR activity in acutely regenerating tissue of wounded mice was paralleled by a marked loss of VEGF protein expression and disturbances of normal proliferation processes in wound margin keratinocytes during skin repair.
The skin represents an essential requirement to enable survival in a terrestrial environment. Accordingly, one of the most important functions of skin architecture is prevention of an uncontrolled water or fluid loss from the body. This overall goal is achieved by formation of a competent permeability barrier in the skin, which is constituted in the stratum corneum by extracellular lipids embedding lipid-depleted corneocytes (1). These lipids consist mainly of cholesterol, fatty acids, and ceramides (2). Thus, it is not remarkable that skin tissue appears as an active site of cholesterol biosynthesis in rodents (3) and also in primates (4). More importantly, the epidermis in particular accounts for most of the cutaneous cholesterol synthetic activity and must be considered as a prime tissue of cholesterol biosynthesis (3). Thus, it came not as a surprise that perturbation of the cutaneous permeability barrier displayed an increased epidermal cholesterol biosynthesis (5) to counteract the subsequent transepidermal water loss, which actually serves as the signal to this regulation (5, 6).
The enzyme, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA)2 reductase (HMGR), is in the center of this process, as it catalyzes the conversion of HMG-CoA to l-mevalonic acid, which depicts the rate-limiting step of the cholesterol biosynthetic pathway in mammals (7). This notion holds true for the disturbed permeability conditions of the skin: epidermal HMGR protein expression, activity, and cholesterol content increased markedly upon experimental disruption of the permeability barrier and subsequently restored barrier functions (8), whereas topical application of the HMGR inhibitor lovastatin suppressed the recovery of barrier functions upon injury (9).
In addition to the above mentioned surficial area disturbances, a skin wound also presents a profound and severe disruption of the cutaneous barrier. Besides fibroplasia and angiogenesis, wound re-epithelialization in particular, originating from migrating and proliferating wound margin and hair follicle keratinocytes, is pivotal to a rapid and efficient closure of the injured tissue area (10, 11). However, the physiologic and disturbed role of HMGR in the regulation of highly dynamic epidermal tissue movements during normal and impaired skin repair remains largely unknown.
Statins interfere with HMGR enzymatic activity at nanomolar concentrations and efficiently displace the natural substrate HMG-CoA from the catalytic site (12). Besides their direct effects on cholesterol levels, however, it is now evident that statins by inhibiting l-mevalonic acid synthesis mediate a series of cholesterol-independent, pleiotropic effects by blocking subsequent formation of the central isoprenoid intermediates farnesyl- and geranylgeranyl pyrophosphate (13). In particular, statins interfere with isoprenylation of cell cycle-regulating Ras and Rho GTPases (14, 15) and thus exert antiproliferative effects in a variety of normal and transformed epithelial and mesenchymal cell types (16-19). For these reasons, HMGR functions might not only be restricted to the maintenance of the cutaneous permeability barrier in skin homeostasis, but might also contribute to the regulation of dynamic keratinocyte movements during acute wound healing.
In addition to migratory and proliferative responses, wound keratinocytes participate in angiogenic processes as they represent a central cellular source of vascular endothelial growth factor (VEGF) in response to a diverse variety of growth factors and cytokines at the wound site (20, 21). Particularly, diabetes-disturbed wound healing conditions in mice were characterized by an impaired keratinocyte proliferation (22), markedly reduced VEGF expression (21, 23), and the persistence of a severe insulin resistance at the wound site (24). Using the competitive HMGR inhibitor simvastatin and a mouse model of diabetes-disturbed skin repair, we here provide the basis that both keratinocyte proliferation and VEGF expression were dependent on the activity of an induced HMGR in vitro and during acute cutaneous regeneration in vivo.
EXPERIMENTAL PROCEDURES
Simvastatin—Simvastatin (Calbiochem, Darmstadt, Germany) was dissolved at a final concentration of 3.3 mg/ml. 4 mg of simvastatin was dissolved in 100 μl of ethanol and 150 μl of 0.1 n NaOH, incubated at 50 °C for 2 h, and adjusted to pH 7.0. The final volume was corrected by adding 1.2 ml of phosphate-buffered saline (PBS).
Animals—Female C57BL/6J and C57BL/6J-ob/ob mice were obtained from The Jackson Laboratories (Bar Harbor, ME) and maintained under a 12-h light/12-h dark cycle at 22 °C until they were at the age of 8 weeks. At this time, they were caged individually, monitored for body weight, and wounded as described below.
Treatment of Mice—Simvastatin (40 mg/kg body weight in 8.3% ethanol in PBS) (25, 26) was injected intraperitoneally (intraperitoneal) once a day for the indicated time periods. Control mice were treated with vehicle (8.3% ethanol in 0.5 ml of PBS).
Wounding of Mice—Wounding of mice was performed as described previously (27, 28). Mice were anesthetized with a single intraperitoneal injection of ketamine (80 mg/kg body weight)/xylazine (10 mg/kg body weight). The hair on the back of each mouse was cut, and the back was subsequently wiped with 70% ethanol. Six full-thickness wounds (5 mm in diameter, 3 to 4 mm apart) were made on the back of each mouse by excising the skin and the underlying panniculus carnosus. The wounds were allowed to form a scab. Skin biopsy specimens were obtained from the animals 1, 3, 5, 6, 7, and 13 days after injury. At each time point, an area that included the scab, the complete epithelial and dermal compartments of the wound margins, the granulation tissue, and parts of the adjacent muscle and subcutaneous fat tissue was excised from each individual wound. As a control, a similar amount of skin was taken from the backs of nonwounded mice. For each experimental time point, tissue from four wounds each from four animals (n = 16 wounds, RNA analysis) and from two wounds each from four animals (n = 8 wounds, protein analysis) were combined and used for RNA and protein preparation. Nonwounded back skin from four animals served as a control. All animal experiments were performed according to the guidelines and approval of the local Ethics Animal Review Board.
Separation of the Epidermis and Dermis Layer of Mouse Tail Skin—Mouse tail skin was incubated for 30 min at 37 °C in a solution of 2 m NaBr. The epidermis was separated from the underlying dermis, immediately frozen in liquid nitrogen, and used for subsequent analyses.
RNA Isolation and RNase Protection Analysis—RNA isolation and RNase protection assays were performed as described previously (28, 29). The cDNA probes were cloned using reverse transcriptase-polymerase chain reaction. The probes corresponded to nucleotides 2441-2722 (for human HMGR, M11058.1), nucleotides 2484-2779 (for murine HMGR, BC085083.1), nucleotides 3260-3566 (for human SREBP-2, NM_004599.2), nucleotides 139-585 (for murine VEGF, S38083), nucleotides 163-317 (for murine GAPDH, NM002046), nucleotides 339-498 (for human VEGF, Ref. 30), or nucleotides 961-1070 (for human GAPDH, M33197) of the published sequences.
Immunohistochemistry—Mice were wounded as described above. Animals were sacrificed at day 3 and day 6 after injury. Complete wounds of mice were isolated from the back, fixed in formalin and subsequently embedded in paraffin. 6-μm sections were subsequently incubated overnight at 4 °C with antisera raised against murine Ki67 (Dianova, Hamburg, Germany) and murine VEGF (Santa Cruz Biotechnology, Heidelberg, Germany). Finally, sections were counterstained with hematoxylin.
In Situ Hybridization—Mice were wounded as described above. Animals were sacrificed at day 5 after injury. Complete wounds of mice were isolated from the back and fixed in 4% paraformaldehyde/PBS. 6-μm serial sections were subsequently analyzed for HMGR mRNA expression using the HybriProbe Custom Design TriSeq-Kit according to the instructions of the manufacturer (Biognostik, Göttingen, Germany).
Cell Culture—Quiescent, confluent human HaCaT keratinocytes (31) were stimulated with EGF (10 ng/ml), TGF-α (10 ng/ml), or a combination of IL-1β (2 nm) and TNF-α (2 nm) in the presence or absence of wortmannin (200 nm), U0126 (10 μm), simvastatin (10 μm), or mevalonate (1 mm). Insulin and EGF were purchased from Roche Applied Science, IL-1β and TNF-α were from Peprotec (Hanau, Germany), respectively. Wortmannin and simvastatin were from Calbiochem (Darmstadt, Germany), U0126 was purchased from Alexis Biochemicals (Lörrach, Germany). Mevalonate was from Sigma (Taufkirchen, Germany).
Preparation of Protein Lysates and Western Blot Analysis—Cell culture samples were homogenized in lysis buffer (1% Triton X-100, 20 mm Tris/HCl pH 8.0, 137 mm NaCl, 10% glycerol, 1 mm DTT, 10 mm NaF, 2 mm Na3VO4, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 0.1 μm okadaic acid). Extracts were cleared by centrifugation (27, 32). Protein concentrations were determined using the BCA Protein Assay kit (Pierce). 50 μg of total protein from skin or cellular lysates was separated using SDS gel electrophoresis. After transfer to a nitrocellulose membrane, specific proteins were detected using antisera raised against non-phosphorylated and phosphorylated eukaryotic initiation factor 4 E-binding protein 1 (Thr-37/46) (Cell Signaling, Frankfurt, Germany), SREB-2 (BD Biosciences, Heidelberg, Germany), or β-actin (Sigma, Taufkirchen, Germany). A secondary antibody coupled to horseradish peroxidase and the enhanced chemiluminescence (ECL) detection system was used to visualize the proteins. phenylmethylsulfonyl fluoride, DTT, aprotinin, NaF, and Na3VO4 were from Sigma. Leupeptin and okadaic acid were from BioTrend (Köln, Germany). The ECL detection system was obtained from Amersham Biosciences (Freiburg, Germany).
Preparation of Microsomes—Mice were wounded as described above. Animals were sacrificed at days 3, 5, 6, 7, and 13 after injury. Total wound tissue was isolated from the back and homogenized in liquid nitrogen. Homogenized frozen wound tissue was resuspended in 0.5 ml of homogenizing buffer (50 mm Tris pH 7.4, 1.15% KCl, 1 mm EDTA, 5 mm glucose, 0.1 mm DTT, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, 0.1 μm okadaic acid). 0.5 ml of homogenizing buffer was used to lyse HaCaT keratinocytes from two 100-mm dishes. Resuspended wound tissue and keratinocyte lysates were subsequently sonicated for 5 min. The homogenates were centrifuged (1000 × g, 15 min, 4 °C), the supernatants were removed and re-centrifuged at 100,000 × g for 1 h at 4 °C. Pellets were resuspended in 100 μl of assay buffer (50 mm potassium phosphate buffer pH 7.4, 5 mm DTT, 5 mm EDTA, 0.2 m KCl). Protein concentrations were determined using the BCA Protein Assay kit.
HMGR Activity Assay—HMGR activity was determined by incubating 150 μg of microsomal protein (see above) in 70 μl of assay buffer (50 mm potassium phosphate buffer, pH 7.4, 5 mm DTT, 5 mm EDTA, and 0.2 m KCl), and 20 μl of buffer 1 (32.5 mg/ml glucose 6-phosphate, 9 mg/ml NADPH, 500 mm potassium phosphate buffer, pH 7.4, 30 μm DTT). HMGR enzymatic activity was initiated by addition of 10 μl of buffer 2 (0.025 μCi/reaction 3-hydroxy-3-methyl [3-14C]glutaryl coenzyme A, 0.5 mm 3-hydroxy-3-methyl-glutaryl coenzyme A, 4.2 mm potassium phosphate at pH 5.5, 0.7 units/reaction of glucose-6-phosphate dehydrogenase) and incubated at 37 °C for 90 min. The reaction was terminated by the addition of 10 μl of HCl (32% w/v). We added (±) mevalonolactone (10 mm) and 0.025 μCi/reaction (R/S)-[5-3H(N)]mevalonolactone as internal standard. The acidified reaction mixture was incubated at 37 °C for 30 min to assure lactonization of the mevalonate. Samples were centrifuged at 1000 × g for 15 min. The supernatants were applied on dry silica gel impregnated glass fiber sheets (Pall Corporation, VWR, Darmstadt, Germany) and resolved by thin layer chromatography with acetone/benzol (1:1, v/v). Regions corresponding to [5-3H (N)]mevalonolactone and [5-14C (N)]mevalonolactone were analyzed. 3-Hydroxy-3-methyl [3-14C]glutaryl coenzyme A was from Amersham Biosciences, (R/S)-[5-3H(N)] mevalonolactone was from PerkinElmer (Wiesbaden, Germany), glucose 6-phosphate, NADPH, glucose-6-phosphate dehydrogenase, mevalonolactone, and HMG-CoA were obtained from Sigma.
[methyl-3H]Thymidine Incorporation Assay—HaCaT keratinocytes (104 cells per well, 24-well plates) were grown in Dulbecco's modified Eagle's medium for 24 h and subsequently incubated with simvastatin (10 μm), mevalonate (1 mm), and 1 μCi/ml [methyl-3H]thymidine (GE Healthcare, Freiburg, Germany). The medium was removed after 24 h, and cells were washed twice with PBS. Cells were treated with ice-cold trichloroacetic acid (5%) at 4 °C for 30 min, lysed with 1 n NaOH, and [methyl-3H]thymidine incorporation was assessed using a scintillation counter (Beckman, Krefeld, Germany).
Enzyme-linked Immunosorbent Assay (ELISA)—Quantification of human and mouse VEGF protein was performed using the human (BIOSOURCE, Nivelles, Belgium) or the mouse (R&D systems, Wiesbaden, Germany) VEGF ELISA kits according to the instructions of the manufacturer.
Silencing of SREBP-2 Expression by siRNA—2 × 105 HaCaT keratinocytes were grown in 6-well plates to reach 40-60% confluency. Cells were subsequently transfected twice with small interfering RNA (siRNA, 50 nm final concentration) using Oligofectamine® (Invitrogen) and OptiMEM (Invitrogen) as described by the manufacturer.
Determination of Simvastatin (SV) and Its Active Hydroxy Acid Form (SVA) in Mouse Plasma Samples—Aliquots of mouse plasma samples were extracted by methanol precipitation. HPLC analysis was done under gradient conditions using a Gemini 5μ C18 column (Phenomenex, Aschaffenburg, Germany). MS and MS/MS analyses were performed on a 4000 Q TRAP triple quadrupole mass spectrometer with a Turbo V source (Applied Biosystems, Darmstadt, Germany) in the positive and negative ion mode for SV and SVA, respectively. Precursor-to-product ion transitions of m/z 436 → 285 for SV and m/z 435 → 319 for SVA were used for the MRM with a dwell time of 200 ms. Concentrations of the calibration standards, quality controls, and unknowns were evaluated by Analyst software (version 1.4; Applied Biosystems, Darmstadt, Germany). Variations in accuracy and intra-day and inter-day precision (n = 6 for each concentration, respectively) were <15% over the range of calibration.
Statistical Analysis—Data are shown as means ± S.D. Data analysis was carried out using the unpaired Student's t test with raw data. The statistical comparison between more than two groups was carried out by analysis of variance (ANOVA, Dunnett's method).
RESULTS
Induction and Localization of HMGR Expression and Activity in Normal and Wounded Skin—It has long been reported that epidermal HMGR expression, activity and subsequent cholesterol synthesis increase upon perturbation of the cutaneous permeability barrier and that enzyme activity pivotally contributes to restoration of a disturbed barrier function (8, 9). As shown in Fig. 1, we now investigated regulation and function of HMGR upon skin repair in a mouse model of excisional wounding. Following injury, HMGR displayed a biphasic increase in mRNA expression (Fig. 1a) and enzyme activity (Fig. 1b) with developing maxima at acute (days 3 and 5 post-wounding) and late repair (day 13 post-wounding). Moreover, we observed a marked and constitutive presence of HMGR mRNA expression in non-wounded skin, which appeared to be predominantly localized to the dermis layer (Fig. 2, a-c). This finding is consistent with the published data (3), which connect dermal HMGR to pilosebaceous epithelium that remains embedded in the dermis during epidermal-dermal separation. However, moderately elevated levels of HMGR activity in the dermis appeared to be not significant compared with activity levels assessed for the isolated epidermal compartment (Fig. 2c). As a next step, we performed an in situ hybridization of 5-day wound tissue to again confirm the established localization of HMGR in keratinocytes, as functional antibodies against HMGR protein are still not available. As shown in Fig. 2d, wound margin keratinocytes exhibit particular signals for HMGR-specific mRNA and confirm keratinocytes as a source of HMGR, also during cutaneous wound healing.
FIGURE 1.
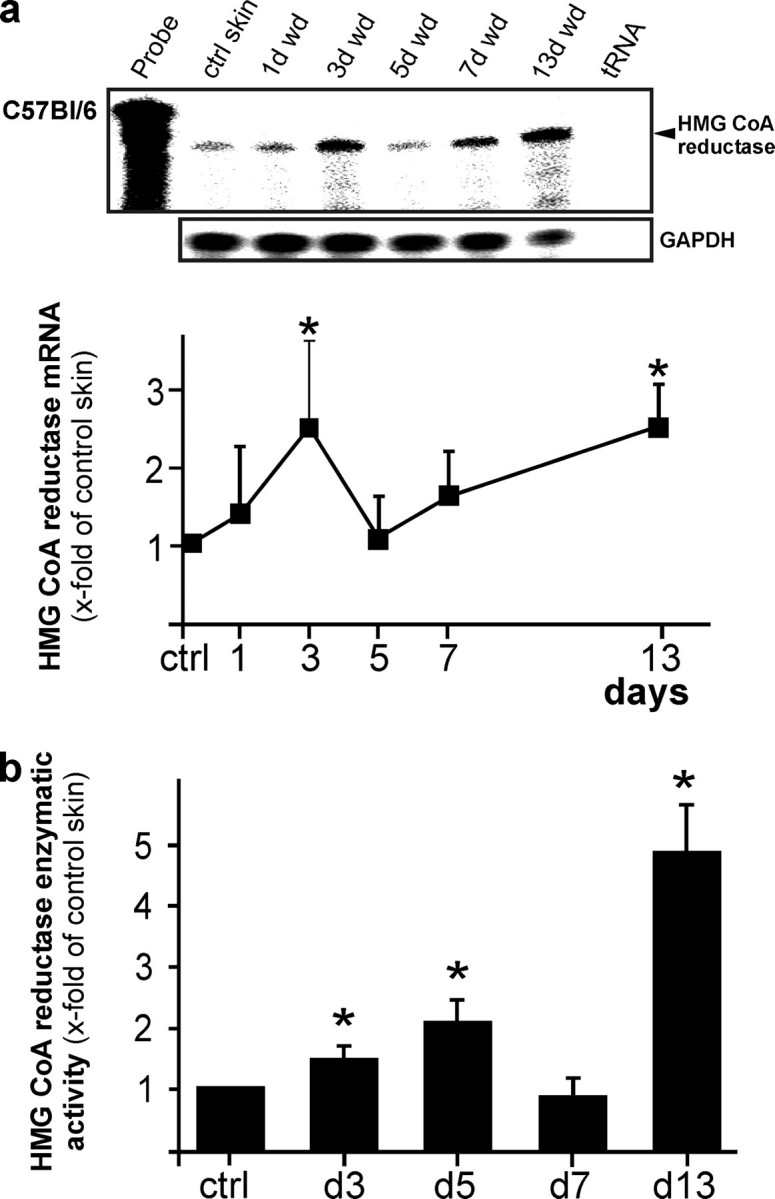
Biphasic regulation of HMGR in skin repair. a, regulation of HMGR mRNA expression in skin wounds as assessed by RNase protection assay. The time after injury is indicated at the top of each lane. Ctrl skin refers to back skin biopsies of non-wounded mice. 1000 cpm of the hybridization probe were used as site marker. Hybridization against GAPDH was used as a loading control. A quantification of HMGR mRNA (PhosphorImager PSL counts per 15 μg of total wound RNA) is shown in the lower panel.*, p < 0.05 (ANOVA, Dunnett's method) compared with control skin. Bars indicate the mean ± S.D. obtained from wounds (n = 48) isolated from animals (n = 12) from three independent animal experiments. b, HMGR activity assays of wound tissue assessed using 3-methyl [3-14C]glutaryl coenzyme A as substrate. *, p < 0.05 (ANOVA, Dunnett's method) compared with control skin. Bars indicate the mean ± S.D. obtained from wounds (n = 24) isolated from animals (n = 12) from three independent animal experiments.
FIGURE 2.
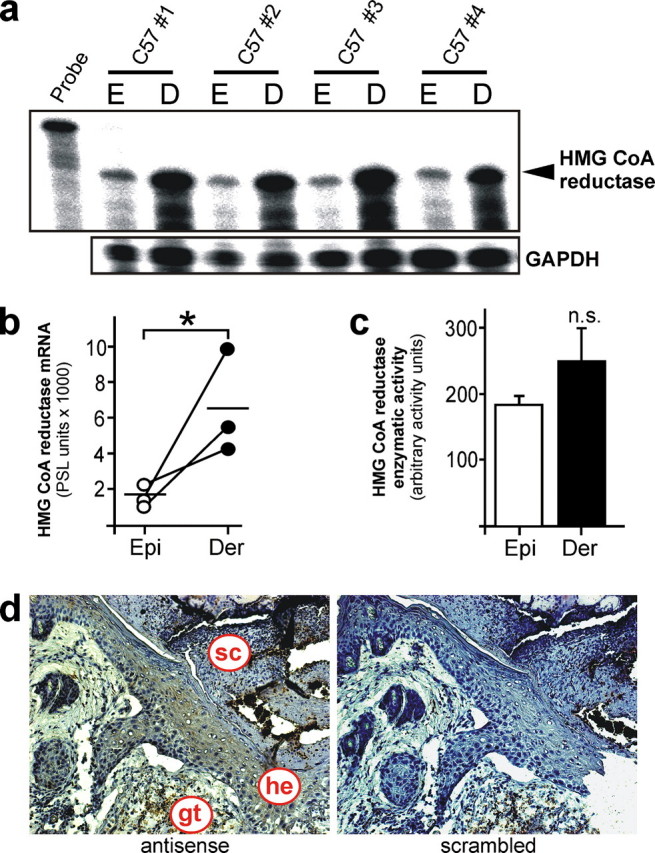
Localization of HMGR expression in non-wounded and wounded skin. a, expression of HMGR mRNA in epidermis (E) and dermis (D) layer of non-wounded skin tissue as assessed by RNase protection assay. 1000 cpm of the hybridization probe were added to the lane labeled Probe. Hybridization against GAPDH was used as a loading control. A quantification of HMGR mRNA (PhosphorImager PSL counts per 15 μg of total RNA) is shown in b.*, p < 0.05 (unpaired Student's t test) as indicated. Bars indicate the mean ± S.D. obtained from epidermal (Epi) - dermal (Der) separations (n = 5) isolated from individual animals (n = 5). c, HMGR activity assays of epidermis (Epi) and dermis (Der) using 3-methyl [3-14C]glutaryl coenzyme A as substrate. n.s., not significant (unpaired Student's t test) as indicated. Bars indicate the mean ± S.D. obtained from epidermal (Epi) - dermal (Der) separations (n = 5) isolated from individual animals (n = 5). d, in situ hybridization of 5-day wound tissue using HMGR-specific antisense or nonspecific scrambled oligonucleotides. gt, granulation tissue; he, hyperproliferative epithelium; sc, scab.
Regulation of HMGR Expression and Activity by Wound-derived Mediators in Keratinocytes—As keratinocytes appeared to contribute to cutaneous HMGR expression and activity in skin repair (Fig. 2d), we now assessed potential mediators that might serve as inducers of HMGR expression and enzymatic activity in acute skin wounds. To do so, we stimulated cultured HaCaT keratinocytes with EGF, TGF-α, and a combination of cytokines and insulin. As given in Fig. 3, all stimuli emerged as potent inducers of HMGR mRNA expression in keratinocytes. Notably, induction of HMGR mRNA was not rapid, with elevated mRNA levels starting to appear upon 16-24 h of stimulation. In accordance, we observed a subsequent, marked and simvastatin-inhibitable induction of HMGR activity in the cells upon EGF-, TGF-α-, and insulin treatment (Fig. 4). By contrast, a cytokine-stimulated increase in HMGR mRNA expression was not followed by an increase in enzymatic activity (Figs. 3c and 4). Leptin, TGF-β1, and keratinocyte growth factor had no effect on keratinocyte HMGR expression and activity (data not shown).
FIGURE 3.

Growth factors and insulin stimulate HMGR expression in keratinocytes. HaCaT keratinocytes were starved for 24 h and subsequently treated with (a) EGF (10 ng/ml), (b) TGF-α (10 ng/ml), (c) a combination of cytokines (2 nm IL-1β and TNF-α), or (d) insulin (2 μg/ml) for the indicated time periods. Induction of HMGR mRNA expression was assessed by RNase protection assay. 1000 counts of the hybridization probe were used as a size marker. Simultaneous hybridization against GAPDH was used as a loading control (upper panels). A quantification of HMGR mRNA (x-fold induction) is shown in the lower panels.*, p < 0.05; #, p < 0.05; n.s., not significant (unpaired Student's t test) as compared with control. Bars indicate the mean ± S.D. obtained from three (n = 3) independent cell culture experiments.
FIGURE 4.

Growth factors and insulin stimulate HMGR activity in keratinocytes. HaCaT keratinocytes were starved for 24 h and subsequently treated with EGF (10 ng/ml), TGF-α (10 ng/ml), insulin (2μg/ml), or a combination of cytokines (2 nm IL-1β and TNF-α) in the absence or presence of simvastatin (10 μm) for the indicated time periods. HMGR activity was assessed using 3-methyl [3-14C]glutaryl coenzyme A as substrate. **, p < 0.01; *, p < 0.05 (unpaired Student's t test) compared with control.Barsindicatethemean±S.D.obtainedfromthree(n=3) independent cell culture experiments.
Signaling Pathways in EGF- and Insulin-stimulated HMGR Expression and Activity—We next researched the impact of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways on EGF- and insulin-induced HMGR regulation. We did so, as PI3K/Akt and MAPK have been shown to be central signaling molecules in the transmission of signals from activated insulin (33) or EGF (34) receptors. Here we show that the EGF-triggered HMGR-specific responses were completely blocked by U0126, an inhibitor of the mitogen extracellular kinase (MEK)-1/2 and subsequent activation of p42/44, but not by interference with the PI3K/Akt pathway (Fig. 5a). In addition to MEK-1/2 activation, insulin-mediated HMGR activation was also decisively dependent on PI3K/Akt signaling, as demonstrated using wortmannin as an inhibitor of this signaling pathway (Fig. 5b).
FIGURE 5.

Dependence of EGF- and insulin-mediated HMGR expression on MAPK and PI3K activation. Quiescent human HaCaT keratinocytes were stimulated with EGF (a) or insulin (b) for 24 h in the absence or presence of U0126 (10 μm) or wortmannin (WTM, 200 nm) as indicated and subsequently analyzed for HMGR mRNA expression by RNase protection assay (left panels) and activity using 3-methyl [3-14C]glutaryl coenzyme A as substrate (right panels). **, p < 0.01; *, p < 0.05 (unpaired Student's t test) as compared with the respective control. §§, p < 0.01; §, p < 0.05 (unpaired Student's t test) as compared with stimulated cells w/o inhibitor. Bars indicate the mean ± S.D. obtained from three (n = 3) independent cell culture experiments.
Insulin-stimulated VEGF Expression from Keratinocytes Is Dependent on HMGR Activity—Keratinocytes provide a prime source of VEGF production during acute wound healing (20) (see also Fig. 9f, left panel) and markedly express VEGF in response to EGF, cytokines (21) or insulin.3 In accordance with VEGF, wound keratinocytes did express HMGR in vivo (Fig. 2d), and enzyme activity was observed upon EGF and insulin stimulation (Figs. 3 and 4). Therefore, we hypothesized a functional connection between induced HMGR activity and VEGF biosynthesis in the cells. To again support this notion, we showed a marked increase in wound VEGF protein content exactly at those time points of acute skin repair in wild-type mice (29 ± 21 pg/50 μg of total skin protein (non-wounded) versus 121 ± 17 pg/50 μg total wound protein (day 3 wound tissue; p < 0.05 compared with non-wounded skin) and 141 ± 9 pg/50 μg total wound protein (day 5 wound tissue; p < 0.01 compared with non-wounded skin), when HMGR exhibited its first peak of activity at days 3 and 5 post-wounding. To determine a possible functional connection between HMGR activity and VEGF in keratinocytes, we investigated EGF- and insulin-inducible HMGR (Figs. 3 and 4) in the regulation of the co-induced pro-angiogenic factor VEGF in the cells. As shown in Fig. 6a, insulin had no major effect on VEGF mRNA expression in cultured HaCaT keratinocytes and, in addition, simvastatin failed to reduce VEGF mRNA levels in the presence or absence of insulin. Also, simvastatin failed to inhibit EGF-induced VEGF mRNA expression in the cells (data not shown). However, when examined at the protein level, EGF and insulin both mediated a marked expression of VEGF protein accumulation, although induction of VEGF protein was inhibited by simvastatin only in the case of insulin stimulation (Fig. 6b). Moreover, it is important to note that insulin treatment of keratinocytes appeared to initiate a potent translation of VEGF protein from the observed constitutive pool of VEGF-specific mRNA species (Fig. 6a), which could be nearly completely blocked by interfering with HMGR enzymatic activity using simvastatin (Fig. 6, b and c). Addition of mevalonate, which represents the direct product of HMGR enyzmatic activity (7), completely abrogated the inhibitory effect and excluded a potential HMGR-independent action of simvastatin on VEGF translation (Fig. 6c). To research an underlying mechanism driving the observed functional connection between insulin-induced HMGR activity and VEGF translation, we focused on the eukaryotic initiation factor 4E-binding protein (4E-BP1), which represents a prominent target of the PI3K/Akt/mTOR pathway and has been shown to control VEGF production from keratinocytes (35, 36). For this purpose, we stimulated quiescent HaCaT keratinocytes with insulin (Fig. 6d) or EGF (Fig. 6e) in the presence or absence of wortmannin or simvastatin. Wortmannin, which has been used to demonstrate PI3K/Akt-driven 4E-BP1 function in the control of translation, effectively suppressed insulin-(Fig. 6d), but not EGF-(Fig. 6e) mediated 4E-BP1 phosphorylation and subsequent VEGF protein release (EGF: 6004 ± 480 pg/ml versus EGF/wortmannin: 5802 ± 252 pg/ml; insulin: 1082 ± 391 pg/ml versus insulin/wortmannin: 125 ± 122 pg/ml) from keratinocytes. More importantly, again insulin-, but not EGF-mediated phosphorylation of 4E-BP1 appeared to be markedly reduced upon simvastatin treatment (Fig. 6, d and e), suggesting that HMGR activity essentially participates in insulin-triggered activation of 4E-BP1 and subsequent VEGF protein production (Fig. 6, b and c).
FIGURE 9.
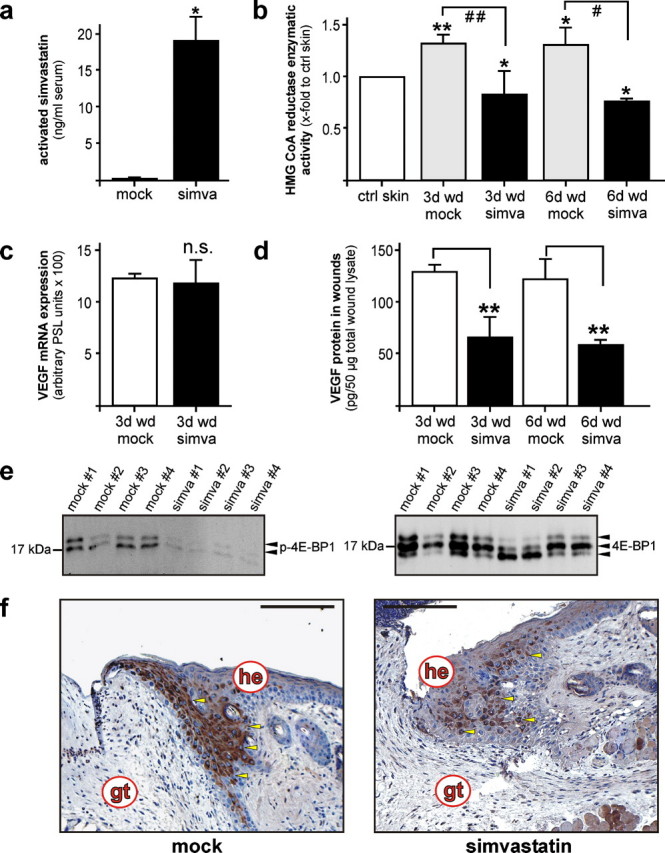
Wound VEGF levels are dependent on HMGR activity in acute skin wounds. a, bioavailability of simvastatin 3 h after systemic application. *, p < 0.05 (unpaired Student's t test) as compared with non-treated animals (mock). Bars indicate the mean ± S.D. obtained from three (n = 3) individual mice. b, HMGR activity assays of 3-day and 6-day wound tissue assessed using 3-methyl [3-14C]glutaryl coenzyme A as substrate in control (mock) and simvastatin-treated (simva) mice. **, p < 0.01; *, p < 0.05 (unpaired Student's t test) as compared with control skin. ##, p < 0.01; #, p < 0.05 as indicated by the brackets. Bars indicate the mean ± S.D. obtained from wounds (n = 6) isolated from three individual animals (n = 3). c, VEGF mRNA expression in 3-day skin wounds from non-(mock) or simvastatin-(simva) treated mice as indicated. A quantification of RNase protection assays (PhosphorImager PSL counts per 15 μg of total wound RNA) is shown. n.s., not significant (unpaired Student's t test) as compared with mock-treated animals. Bars indicate the mean ± S.D. obtained from wounds (n = 12) isolated from four (n = 4) individual animals (n = 4). d, ELISA analysis of wound VEGF protein from untreated (mock) and simvastatin-treated (simva) mice. **, p < 0.01 (unpaired Student's t test) as indicated by brackets. Bars indicate the mean ± S.D. obtained from wounds (n = 6) isolated from three individual animals (n = 3). e, immunoblot showing the presence of phosphorylated 4E-BP1 in wound lysates from individual untreated (mock) and simvastatin-treated (simva) mice as indicated (left panel). Total 4E-BP1 is shown as a control (right panel). f, localization of VEGF protein expression in 6-day wound tissue of untreated (mock) and simvastatin-treated (simva) mice. All sections were stained with the avidin-biotin-peroxidase complex system using 3,3-diaminobenzidine tetrahydrochloride as a chromogenic substrate. Nuclei were counterstained with hematoxylin. gt, granulation tissue; he, hyperproliferative epithelium. Bars, 50 μm.
FIGURE 6.

Insulin-mediated VEGF induction is dependent on HMGR activity. Quiescent human HaCaT keratinocytes were stimulated with insulin (a) for 24 h in the absence or presence of simvastatin (10 μm) and subsequently analyzed for VEGF mRNA expression by RNase protection assay. b, VEGF protein production from EGF- and insulin-treated HaCaT cells was analyzed by ELISA. **, p < 0.01 (unpaired Student's t test) as compared with control. ##, p < 0.01 (unpaired Student's t test) as indicated by the brackets. Bars indicate the mean ± S.D. obtained from three (n = 3) independent cell culture experiments. c, quiescent HaCaT keratinocytes were treated with insulin (2 μg/ml) in the absence or presence of simvastatin (10 μm), mevalonate (1 mm), or a combination of both as indicated and analyzed for VEGF protein production by ELISA. **, p < 0.01; *, p < 0.05 (unpaired Student's t test) as compared with control. ##, p < 0.01 (unpaired Student's t test) as compared with insulin stimulation alone. Bars indicate the mean ± S.D. obtained from three (n = 3) independent cell culture experiments. Immunoblots of total cellular protein from insulin-(d) or EGF-(e) treated HaCaT cells were analyzed for phospho-4E-BP1 (Thr-37/46) in the absence or presence of wortmannin (WTM, 200 nm) or simvastatin (sim, 10 μm) at the indicated time points. β-Actin was used to control equal loading of blots.
Insulin-induced SREBP-2 Controls HMGR Expression and Subsequent VEGF Synthesis—SREBPs control the transcription of enzymes required for cholesterol and lipid metabolism and play critical roles in insulin-dependent gene expression (37). Here we focused on the role of SREBP-2 in insulin-induced HMGR expression, activity, and VEGF production, as SREBP-2 has been reported to be the predominant isoform in keratinocytes in the absence of SREBP-1 (38). Consistent with insulin- and EGF-induced HMGR expression (Fig. 3), the anti-serum detected an increase of the 125-kDa unprocessed form of SREBP-2 in insulin- and EGF-treated keratinocytes after 16 and 24 h of stimulation (Fig. 7a). To assess a functional consequence of SREBP-2 induction, we abolished basal and insulin-induced expression of SREBP-2 using siRNA. SREBP-2-specific siRNA nearly completely abrogated SREBP-2 mRNA and protein expression (Fig. 7b). More importantly, abrogation of SREBP-2 potently interrupted insulin-induced HMGR expression and subsequent VEGF synthesis (Fig. 7c).
FIGURE 7.

SREBP-2 is essential for insulin-mediated coinduction of HMGR and VEGF expression. a, quiescent HaCaT keratinocytes were treated with insulin (2 μg/ml) (upper panel) and EGF (10 ng/ml) (lower panel) for the indicated time points. Total cellular protein was subsequently analyzed by immunoblot for SREBP-2 expression. RNase protection assays and immunoblots showing siRNA-mediated abrogation of basal and insulin-stimulated SREBP-2 mRNA (b, upper panel) and protein (b, lower panel) and its effects on basal and insulin-stimulated HMGR mRNA (c, upper panel) and VEGF expression (ELISA) (c, lower panel) in HaCaT cells after 48 h of siRNA pretreatment and 24 h of subsequent insulin stimulation as indicated. *, p < 0.05 (unpaired Student's t test) compared with control. A hybridization for GAPDH and detection of β-actin were used as respective loading controls.
HMGR Activity Participates in the Control of Keratinocyte Proliferation—Blockade of the cholesterol biosynthetic pathway by statins is now established to interfere with the mitogenic potential of cells (14). As keratinocyte proliferation is important to skin repair (10, 11), we examined the role of HMGR activity on cell proliferation in cultured HaCaT keratinocytes in vitro. Although exponentially growing keratinocytes exhibited a lower HMGR activity compared with confluent cells (Fig. 8a), we could nevertheless demonstrate clearly that HMGR activity in dividing cells was substantially necessary to serve a normal mitogenic potential of keratinocytes, as simvastatin treatment markedly interfered with proliferation of exponentially growing keratinocytes (Fig. 8b). Moreover, simvastatin-induced inhibition of proliferation could, at least partially, be rescued by mevalonate, the product of HMGR enzymatic activity (7), demonstrating a direct effect of HMGR in this process.
FIGURE 8.

HMGR activity in the control of keratinocyte proliferation. a, HaCaT keratinocytes were grown to 60% or 100% confluency, respectively. Cells were subsequently lysed and HMGR activity was assessed using 3-methyl [3-14C]glutaryl coenzyme A as substrate. *, p < 0.05 (unpaired Student's t test) as compared with control. Bars indicate the mean ± S.D. obtained from three (n = 3) independent cell culture experiments. b, HaCaT keratinocytes were grown exponentially in Dulbecco's modified Eagle's medium (DMEM) and treated with simvastatin (10 μm), mevalonate (1 mm), or a combination of both as indicated for 24 h. Non-treated cells served as control (DMEM). Proliferation rates were assessed by analysis of [3H]thymidine incorporation. **, p < 0.01 (unpaired Student's t test) as compared with control (DMEM). ##, p < 0.01 as indicated by the bracket. Bars indicate the mean ± S.D. obtained from three (n = 3) independent cell culture experiments.
Inhibition of HMGR Activity during Acute Wound Healing Interferes with Keratinocyte VEGF Production and Proliferation at the Wound Site—To scrutinize the observed roles of HMGR activity on keratinocyte VEGF synthesis and mitogenic activity, we treated wounded wild-type mice with simvastatin at concentrations (40 mg/kg) that have been established in different murine models of inflammation (25, 26). Systemic application of simvastatin resulted in a robust bioavailability of the drug as assessed by MS/MS analyses of plasma samples of mock- and simvastatin-administered mice (Fig. 9a). More importantly, systemic levels of simvastatin caused a marked and significant reduction of HMGR enzymatic activity in 3-day and 6-day wounds of treated animals (Fig. 9b). However, the simvastatin-mediated inhibition of HMGR activity in wound tissue did not translate into any change with respect to VEGF mRNA levels at the wound site (Fig. 9c). By contrast, VEGF protein expression in wound tissue was severely impaired upon simvastatin treatment of mice (Fig. 9d). In good agreement with our in vitro data, the observed loss of wound VEGF protein was paralleled by a strong reduction of 4E-BP1 phosphorylation in wound tissue in connection to the simvastatin-mediated inhibition of HMGR activity in wounds (Fig. 9e). Immunohistochemistry clearly confirmed keratinocytes as the prime cellular source of reduced VEGF levels at wound sites of mock- and simvastatin-treated mice (Fig. 9f).
Additionally, we recognized that reduced levels of wound VEGF might be partially connected to reduced numbers of wound margin keratinocytes upon simvastatin treatment of mice. We discovered about 40% reduction of wound keratinocytes located within the hyperproliferative epithelia at the margins of the wound (Fig. 10). Interestingly, after careful consideration including a series of wound sections from individual mice, we identified a deceleration of wound keratinocyte proliferative potential in simvastatin-treated mice. We used the antibody Ki67 (Fig. 10), which detects a nuclear antigen that is present only in proliferating cells (determining S, G2, and M phase) and thus represents a reliable tool to evaluate the growth fraction of cell populations (39). Here it was remarkable that inhibition of HMGR activity did not impair the Ki67-positive growth fraction of the wound margin epithelia, but simvastatin appeared to interfere with a mechanism temporally down-stream of keratinocyte mitosis, as the Ki67-negative cell fraction was markedly reduced upon simvastatin treatment of animals (Fig. 10, a and b).
FIGURE 10.
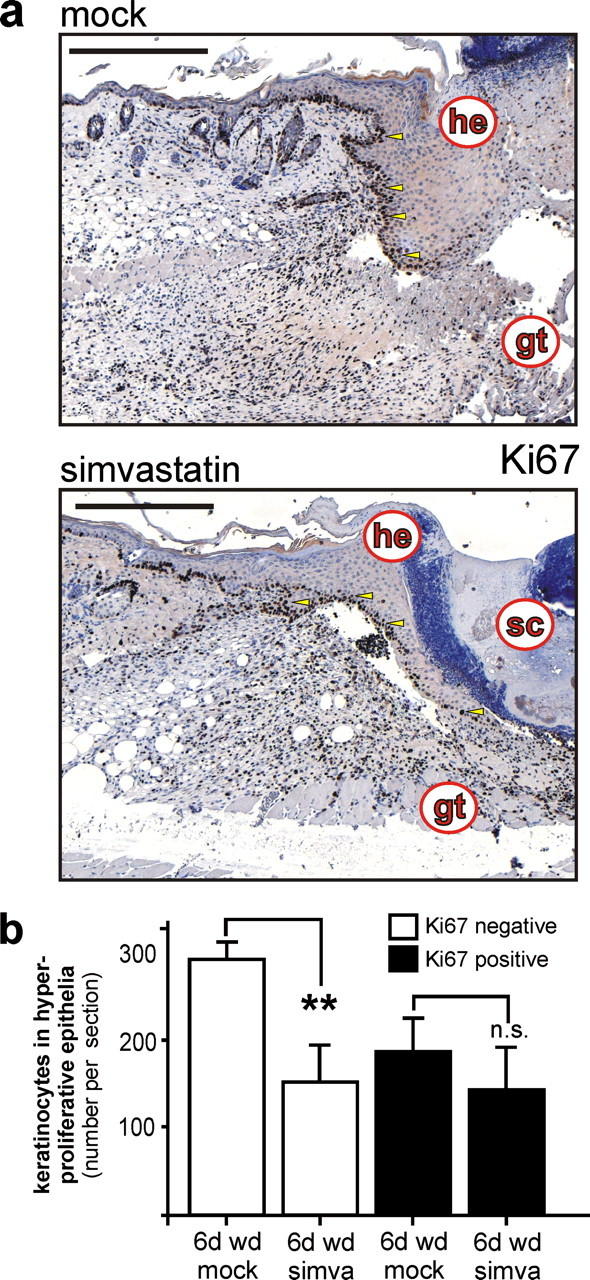
Proliferation of wound keratinocytes in statin-treated mice. a, analysis of keratinocyte proliferation using a rat monoclonal antibody directed against the proliferative antigen marker Ki67 in 6-day wound tissue of untreated (mock) and simvastatin-treated (simva) mice as indicated. b, quantitative analysis of total and Ki67-expressing wound keratinocyte cell numbers from stained wounds. **, p < 0.01; n.s., not significant (unpaired Student's t test) as indicated by brackets. Bars indicate the mean ± S.D. obtained from wounds (n = 6) isolated from three individual animals (n = 3). Sections were stained with the avidin-biotin-peroxidase complex system using 3,3-diaminobenzidine-tetrahydrochloride as a chromogenic substrate, and nuclei were counterstained with hematoxylin. gt, granulation tissue; he, hyperproliferative epithelium; sc, scab. Bars, 100 μm.
Regulation of HMGR Expression and Activity in Diabetes-disturbed Wound Healing—Finally, we complemented our findings on the role of HMGR using a mouse model of diabetes-impaired skin repair. We did so, as we had especially identified insulin as a regulator of HMGR activity and function in cultured keratinocytes. Obese/obese (ob/ob) mice represent a well-established model of diabetes-impaired wound healing (22). These animals were characterized by severe diabetes-obesity syndromes (40) as a consequence of the functional loss of leptin protein encoded by the obese gene (41). As shown in Fig. 11a, diabetes-impaired wound tissue of ob/ob mice did not exhibit the biphasic regulation of HMGR mRNA expression observed for wild-type animals (Fig. 1a). More importantly, the severe loss of HMGR expression and activity compared with wild-type mice is confined primarily to the late phase of repair (Fig. 11, a and b), which is characterized by a nearly complete loss of wound keratinocyte proliferation and formation of a neo-epidermis in diseased animals (Fig. 11c).
FIGURE 11.

Impaired regulation of HMGR in diabetes-disturbed skin repair. a, RNase protection assay for HMGR mRNA expression in skin wounds of diabetic ob/ob mice. The time after injury is indicated at the top of each lane. ctrl skin refers to back skin biopsies of non-wounded mice. 1000 cpm of the hybridization Probe were used as a size marker. Hybridization against GAPDH was used as a loading control. A quantification of HMGR mRNA (PhosphorImager PSL counts per 15 μg of total wound RNA) is shown in the right panel. *, p < 0.05 (ANOVA, Dunnett's method) compared with control skin. #, p < 0.05 (unpaired Student's t test) compared with wild-type (C57) mice. Bars indicate the mean ± S.D. obtained from wounds (n = 48) isolated from animals (n = 12) from three independent animal experiments. b, HMGR activity assays of wound tissue from wild type (C57) and diabetic ob/ob mice (ob/ob) as assessed using 3-methyl [3-14C]glutaryl coenzyme A as substrate. * and §, p < 0.05 (ANOVA, Dunnett's method) compared with the respective control. #, p < 0.05 (unpaired Student's t test) as indicated by brackets. Bars indicate the mean ± S.D. obtained from wounds (n = 24) isolated from animals (n = 12) from three independent animal experiments. c, hematoxylin-counterstained representative sections from 13-day wound tissue of wild-type and diabetic ob/ob mice as indicated. Epithelia are highlighted with the yellow line. gt, granulation tissue; ne, neo-epithelium; sc, scab; wm, wound margin. Bars, 50 μm.
DISCUSSION
Skin tissue functions to protect the organism from harmful environmental conditions. In particular, the epidermal compartment of skin tissue is known to be essential in the prevention of excessive water loss from the body via production of cholesterol, fatty acids and ceramides (2). Here it was important to recognize that skin tissue is a prime source of de novo cholesterol biosynthesis in the body and that this biosynthetic activity had to be attributed to epidermal keratinocytes expressing an active HMGR (3, 4, 8, 9, 42). HMGR is the rate-limiting enzyme in the center of cholesterol biosynthesis and catalyzes the conversion of HMG-CoA into l-mevalonic acid (7). Thus, it was not surprising that a constitutive HMGR expression and activity typifies an important feature of non-wounded skin in mammals (Ref. 8 and this study). The particularly strong signals of HMGR expression and activity observed in the dermal compartment following epidermal-dermal separation in our experiments were consistent with published data that described the pilosebaceous epithelium, remaining embedded in the dermis layer during separation, as the epithelial source of cholesterol biosynthetic activity (3). However, the constitutive HMGR activity in normal skin tissue has been shown to undergo dynamic and adaptive changes upon perturbations of skin integrity. Experimental disruption of the epidermal permeability barrier in hairless mice markedly induced the expression and activity of HMGR (8, 42). Injury-elevated HMGR activity essentially contributed to the restoration of the epidermal lipid barrier, as inhibition of HMGR enzymatic activity by lovastatin abolished repair (9).
Consistent with the epidermal capability to adapt HMGR availability to skin perturbations, we could identify the tissue regeneration process during wound healing as an additional scenario that requested a balanced HMGR induction. Interestingly, we observed a biphasic regulation of HMGR expression and activity upon skin injury. Such a biphasic regulation of gene expression appears to be infrequent but not entirely exceptional during skin repair, as also the matrix protease stromelysin-2 shows a biphasic regulation in wound healing (43). Here it is tempting to argue that the observed maxima of HMGR expression and activity at days 3 and 13 after wounding presumably cover essential functions of the enzyme for this particular phase of repair. Moreover, as wound keratinocytes appeared to be a cellular source contributing to wound HMGR expression, it was reasonable to assume a potential role of HMGR in the control of wound keratinocyte functions. One particular pivotal function of wound keratinocytes in skin repair is to trigger wound angiogenic responses by release of VEGF, for which the cells serve as the favored source in wound tissue (20, 21). At days 3 and 5 post-wounding, obviously, we recognized HMGR expression, activity, and localization (this study) in perfect temporal and spatial match to VEGF expression in wound keratinocytes (21, 28). In keratinocytes, VEGF expression is induced by a series of wound-derived mediators including growth factors and inflammatory cytokines (21). When we characterized diverse wound-derived mediators (44) to co-induce HMGR and VEGF as a functional prerequisite to drive HMGR and VEGF expression cross-talk, it came as a surprise to us that only EGF, TGF-α, and insulin fulfilled this condition. Our data provide strong evidence that regulation of HMGR activity was completely independent from inflammatory cytokines. This finding is quite remarkable, as one has to recognize that HMGR regulation is most likely uncoupled from inflammatory responses at the wound site with accordant consequences and that, by contrast, keratinocyte HMGR regulation appeared to be particularly sensitive to mitogenic stimuli, which serve as important signals in early repair (10, 11). These findings strongly implicate a series of consequences with respect to keratinocyte VEGF production and mitogenic potential.
Importantly, insulin induced HMGR activity in cultured keratinocytes. Our findings confirmed a previous report on HMGR expression in keratinocytes (45) and extend the knowledge beyond expression with respect to HMGR activity (this study). Among all mediators tested, insulin was unique in relation to its absolute dependence on co-induced HMGR activity to drive a robust VEGF expression in keratinocytes. This notion is further supported by findings that report a function of insulin in the expressional control of VEGF in diverse cell types (46, 47). The severe insulin resistance of resident wound cells observed in chronic wounds of diabetic mice (24) is paralleled by markedly reduced levels of VEGF protein (21, 48, 49) and strongly suggests insulin, beside its well-known glucose-controlling endocrine functions, to participate in an as yet unappreciated contribution to wound-derived VEGF production in skin repair. Here, it is noteworthy that insulin appeared to mediate its effects on keratinocyte expression in an indirect manner, as inhibition of insulin-induced HMGR activity by simvastatin completely abrogated VEGF production. Moreover, a role for HMGR activity with respect to VEGF expression has to be appreciated also during wound healing in vivo, as simvastatin treatment of wounded mice provoked a marked reduction of VEGF protein in wound keratinocytes. Insulin mediated its effects via induction of SREBP-2, which is known to be critical for insulin-dependent gene expression (37). SREBP-2 represents the uniquely expressed SREBP isoform in keratinocytes, pivotally regulates HMGR expression in the cells (Ref. 38 and this study) and essentially communicates the insulin signal toward VEGF mRNA translation via HMGR induction in the long run (see also Fig. 12).
FIGURE 12.

Regulatory pathways controlling insulin-induced VEGF protein expression in keratinocytes. (↓), stimulation; (⊥), inhibition, (...), inhibited process; (—), ongoing process; P, phosphate.
Keratinocyte proliferation, starting at the wound edge and from hair follicle remnants at the wound site, characterizes a central movement toward wound closure. Besides the effects of HMGR on VEGF expression, we additionally recognized both potent epidermal growth factors EGF and TGF-α to serve as inducers of HMGR expression and activity in keratinocytes. By contrast, EGF-induced HMGR activity did not contribute to VEGF expression and highlighted the individual properties of EGF and insulin in relation to keratinocyte stimulation. It is important to note that keratinocyte proliferation was partially dependent on HMGR activity in vitro and in vivo, as proliferative conditions in cultured and also wound margin keratinocytes were impaired upon administration of simvastatin, respectively, and as a reduced activity of HMGR was associated with the loss of neo-epithelial structures in chronic, diabetes-impaired wounds. A series of reports have shown the anti-proliferative capacity of statins in a variety of primary and transformed cell types (16-19). These effects are now evidenced to be caused by inhibition of isoprenylation and thus functional impairment of small GTPases implicated in cell cycle control (14, 15).
An additional line of reasoning might explain the observed thinning of wound edges in simvastatin-treated wild-type mice because a block on cholesterol biosynthesis could be discussed to cause a defective assembly of stratum corneum lipids (1-4). The resulting abnormal stratum corneum formation might subsequently lead to a premature sloughing and loss of stratum corneum keratinocytes, which creates a new equilibrium of the epidermal transit time, resulting in a thinner overall epidermis. However, as HMGR activity was highest during very late stages of normal repair, it is here reasonable to argue for HMGR to also participate in the re-adjustment of a functional permeability barrier in the newly formed neo-epidermis at the very end of acute wound healing processes. In summary, our data show an induction of HMGR expression and activity in acute skin wounds and evidence a role of this enzyme in the control of keratinocyte angiogenic and proliferative potential.
Acknowledgments
We thank Dr. Elke Müller for the assessment of wound VEGF protein.
This work was supported in part by the Deutsche Forschungsgemeinschaft (SFB 553, Grant FR 1540/1-2, GK 1172, Excellence Cluster Cardiopulmonary System) and by Eicosanox, EC FP6 funding (LSHM-CT-2005-005033). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; SREBP-2, sterol regulatory element-binding protein; HMGR, HMG-CoA reductase; VEGF, vascular endothelial growth factor; 4E-BP1, eukaryotic initiation factor 4 E-binding protein 1; EGF, epidermal growth factor; TGF, transforming growth factor; PBS, phosphate-buffered saline; TNF, tumor necrosis factor; IL, interleukin; DTT, dithiothreitol; ELISA, enzyme-linked immunosorbent assay; ANOVA, analysis of variance; PI3K, phosphatidylinositol 3-kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
I. Goren, unpublished observation.
References
- 1.Grubauer, G., Feingold, K. R., Harris, R. M., and Elias, P. M. (1989) J. Lipid Res. 30 89-96 [PubMed] [Google Scholar]
- 2.Lampe, M. A., Burlingame, A. L., Whitney, J., Williams, M. L., Brown, B. E., Roitman, E., and Elias, P. M. (1983) J. Lipid Res. 24 120-130 [PubMed] [Google Scholar]
- 3.Feingold, K. R., Brown, B. E., Lear, S. R., Moser, A. H., and Elias, P. M. (1983) J. Investig. Dermatol. 81 365-369 [DOI] [PubMed] [Google Scholar]
- 4.Feingold, K. R., Wiley, M. H., Moser, A. H., Lau, D. T., Lear, S. R., and Siperstein, M. D. (1982) J. Lab. Clin. Med. 100 405-410 [PubMed] [Google Scholar]
- 5.Menon, G. K., Feingold, K. R., Moser, A. H., Brown, B. E., and Elias, P. M. (1985) J. Lipid Res. 26 418-427 [PubMed] [Google Scholar]
- 6.Grubauer, G., Feingold, K. R., and Elias, P. M. (1987) J. Lipid Res. 28 746-752 [PubMed] [Google Scholar]
- 7.Rodwell, V. W., Nordstrom, J. L., and Mitschelen, J. H. (1976) Adv. Lipid Res. 14 1-74 [DOI] [PubMed] [Google Scholar]
- 8.Proksch, E., Elias, P. M., and Feingold, K. R. (1990) J. Clin. Investig. 85 874-882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feingold, K. R., Mao-Qiang, M., Menon, G. K., Cho, S. S., Brown, B. E., and Elias, P. M. (1990) J. Clin. Investig. 86 1738-1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin, P. (1997) Science 276 75-81 [DOI] [PubMed] [Google Scholar]
- 11.Singer, A. J., and Clark, R. A. F. (1999) N. Engl. J. Med. 341 738-746 [DOI] [PubMed] [Google Scholar]
- 12.Istvan, E. S., and Deisenhofer, J. (2001) Science 292 1160-1164 [DOI] [PubMed] [Google Scholar]
- 13.Goldstein, J. L., and Brown, M. S. (1990) Nature 343 425-430 [DOI] [PubMed] [Google Scholar]
- 14.Liao, J. K. (2002) J. Clin. Investig. 110 285-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker, K., and Olson, M. F. (2005) Curr. Opin. Genet. Dev. 15 62-68 [DOI] [PubMed] [Google Scholar]
- 16.Kotamraju, S., Williams, C. L., and Kalyanaraman, B. (2007) Cancer Res. 67 7386-7394 [DOI] [PubMed] [Google Scholar]
- 17.Vrtovsnik, F., Couette, S., Prie, D., Lallemand, D., and Friedlander, G. (1997) Kidney Int. 52 1016-1027 [DOI] [PubMed] [Google Scholar]
- 18.Carlin, C. M., Peacock, A. J., and Welsh, D. J. (2007) Am. J. Respir. Cell Mol. Biol. 37 447-456 [DOI] [PubMed] [Google Scholar]
- 19.Denoyelle, C., Vasse, M., Körner, M., Mishal, Z., Ganne, F., Vannier, J. P., Soria, J., and Soria, C. (2001) Carcinogenesis 22 1139-1148 [DOI] [PubMed] [Google Scholar]
- 20.Brown, L. F., Yeo, K. T., Berse, B., Yeo, T. K., Senger, D. R., Dvorak, H. F., and van der Water, L. (1992) J. Exp. Med. 176 1375-1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank, S., Hübner, G., Breier, G., Longaker, M. T., Greenhalgh, D. G., and Werner, S. (1995) J. Biol. Chem. 270 12607-12613 [DOI] [PubMed] [Google Scholar]
- 22.Frank, S., Stallmeyer, B., Kämpfer, H., Kolb, N., and Pfeilschifter, J. (2000) J. Clin. Investig. 106 501-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stallmeyer, B., Pfeilschifter, J., and Frank, S. (2001) Diabetologia 44 471-479 [DOI] [PubMed] [Google Scholar]
- 24.Goren, I., Müller, E., Pfeilschifter, J., and Frank, S. (2006) Am. J. Pathol. 168 765-777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung, B. P., Sattar, N., Crilly, A., Prach, M., McCarey, D. W., Payne, H., Madhock, R., Campbell, C., Gracie, J. A., Liew, F. Y., and McInnes, I. B. (2003) J. Immunol. 170 1524-1530 [DOI] [PubMed] [Google Scholar]
- 26.McKay, A., Leung, B. P., McInnes, I. B., Thomson, N. C., and Liew, F. Y. (2004) J. Immunol. 172 2903-2908 [DOI] [PubMed] [Google Scholar]
- 27.Stallmeyer, B., Kämpfer, H., Kolb, N., Pfeilschifter, J., and Frank, S. (1999) J. Investig. Dermatol. 113 1090-1098 [DOI] [PubMed] [Google Scholar]
- 28.Frank, S., Stallmeyer, B., Kämpfer, H., Kolb, N., and Pfeilschifter, J. (2002) FASEB J. 13 2002-2014 [PubMed] [Google Scholar]
- 29.Chomczynski, P., and Sacchi, N. (1987) Anal. Biochem. 162 156-159 [DOI] [PubMed] [Google Scholar]
- 30.Weindel, K., Marme, D., and Weich, H. A. (1992) Biochem. Biophys. Res. Commun. 183 1167-1174 [DOI] [PubMed] [Google Scholar]
- 31.Boukamp, P., Petrussevska, R. T., Breitkreutz, D., Hornung, J., Markham, A., and Fusenig, N. E. (1988) J. Cell Biol. 106 761-771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kämpfer, H., Kalina, U., Mühl, H., Pfeilschifter, J., and Frank, S. (1999) J. Investig. Dermatol. 113 369-374 [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi, C. M., Emanuelli, B., and Kahn, C. R. (2006) Nat. Rev. Mol. Cell. Biol. 7 85-96 [DOI] [PubMed] [Google Scholar]
- 34.Pastore, S., Mascia, F., Mariani, V., and Girolomoni, G. (2007) J. Investig. Dermatol., in press [DOI] [PubMed]
- 35.Segrelles, C., Ruiz, S., Santos, M., Martinez-Palacio, J., Lara, M. F., and Paramio, J. M. (2004) Carcinogenesis 25 1137-1147 [DOI] [PubMed] [Google Scholar]
- 36.Gingras, A. C., Raught, B., and Sonenberg, N. (2001) Genes Dev. 15 807-826 [DOI] [PubMed] [Google Scholar]
- 37.Bengoechea-Alonso, M. T., and Ericsson, J. (2007) Curr. Opin. Cell Biol. 19 215-222 [DOI] [PubMed] [Google Scholar]
- 38.Harris, I. R., Farrell, A. M., Holleran, W. M., Jackson, S., Grunfeld, C., Elias, P. M., and Feingold, K. M. (1998) J. Lipid Res. 39 412-422 [PubMed] [Google Scholar]
- 39.Gerdes, J., Lemke, H., Baisch, H., Wacker, H. H., Schwab, U., and Stein, H. (1984) J. Immunol. 133 1710-1715 [PubMed] [Google Scholar]
- 40.Coleman, D. L. (1978) Diabetologia 14 141-148 [DOI] [PubMed] [Google Scholar]
- 41.Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994) Nature 372 425-432 [DOI] [PubMed] [Google Scholar]
- 42.Harris, I. R., Farrell, A. M., Grunfeld, C., Holleran, W. M., Elias, P. M., and Feingold, K. R. (1997) J. Investig. Dermatol. 109 783-787 [DOI] [PubMed] [Google Scholar]
- 43.Madlener, M., Mauch, C., Conca, W., Brauchle, M., Parks, W. C., and Werner, S. (1996) Biochem. J. 320 659-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Werner, S., and Grose, R. (2003) Physiol. Rev. 83 835-870 [DOI] [PubMed] [Google Scholar]
- 45.Harris, I. R., Höppner, H., Siefken, W., Farrell, A. M., and Wittern, K. P. (2000) J. Investig. Dermatol. 114 83-87 [DOI] [PubMed] [Google Scholar]
- 46.Treins, C., Giorgetti-Peraldi, S., Murdaca, J., Semenza, G. L., and Van Obberghen, E. (2002) J. Biol. Chem. 277 27975-27981 [DOI] [PubMed] [Google Scholar]
- 47.Jiang, Z. Y., He, Z., King, B. L., Kuroki, T., Opland, D. M., Suzuma, K., Suzuma, I., Ueki, K., Kulkarni, R. N., Kahn, C. R., and King, G. L. (2003) J. Biol. Chem. 278 31964-31971 [DOI] [PubMed] [Google Scholar]
- 48.Lauer, G., Sollberg, S., Cole, M., Flamme, I., Sturzebecher, J., Mann, K., Krieg, T., and Eming, S. A. (2000) J. Investig. Dermatol. 115 12-18 [DOI] [PubMed] [Google Scholar]
- 49.Kämpfer, H., Pfeilschifter, J., and Frank, S. (2001) Lab. Investig. 81 361-373 [DOI] [PubMed] [Google Scholar]