

Abstract
The 70-kDa heat shock proteins (Hsp70s) are highly conserved ATP-dependent molecular chaperones composed of an N-terminal nucleotide binding domain (NBD) and a C-terminal protein substrate binding domain (SBD) in a bilobate structure. Interdomain communication and nucleotide-dependent structural motions are critical for Hsp70 chaperone functions. Our understanding of these functions remains elusive due to insufficient structural information on intact Hsp70s that represent the different states of the chaperone cycle. We report here the crystal structures of DnaK from Geobacillus kaustophilus HTA426 bound with ADP-Mg2+-Pi at 2.37Å and the 70-kDa heat shock cognate protein from Rattus norvegicus bound with ADP-Pi at 3.5Å. The NBD and SBD in these structures are significantly separated from each other, and they might depict the ADP-bound conformation. Moreover, a Trp reporter was introduced at the potential interface region between NBD and the interdomain linker of GkDnaK to probe environmental changes. Results from fluorescence measurements support the notion that substrate binding enhances the domain-disjoining behavior of Hsp70 chaperones.
The 70-kDa heat shock proteins (Hsp70s)2 are molecular chaperones that are highly conserved across many phyla. Hsp70s assist in many crucial cellular functions, including the correct folding of nascent or stress-denatured polypeptides (1-3), protein translocation across membranes (4), and assembly/disassembly of protein complexes (5). Together with J-domain ATPase-activating proteins or nucleotide exchange factors, Hsp70s bind and release their substrates in ATP-driven cycles (6, 7). Previous studies have clearly demonstrated that Hsp70s are composed of two major functional domains. The N-terminal nucleotide binding domain (NBD) contains an adenine nucleotide binding site and possesses intrinsic weak ATPase activity. The C-terminal substrate binding domain (SBD) consists of a substrate-binding pocket covered with a helical subdomain that is responsible for client protein recognition (8, 9). The binding and hydrolysis of ATP in the NBD trigger conformational changes within Hsp70 and alter the functional properties of SBD (10, 11). The ATP-bound state (T-state) has lower substrate affinity than that of the ADP-bound state (R-state), resulting in faster binding and release of client proteins (12, 13). Conversely, SBD binds substrates and interacts with J-domain cochaperone family proteins, resulting in allosteric stimulation of ATP hydrolysis in the NBD (7).
The mechanism for the ATP-dependent intramolecular allosteric regulation between the NBD and SBD of Hsp70s has been the subject of protracted studies. Structural features for interdomain communication and changes upon nucleotide binding have long been sought by chaperone researchers. Previously, Jiang et al. (14) have determined the crystal structure of a C-terminal truncated bovine Hsc70 (bHsc70ΔC, residues 1-554) at a nucleotide-free state and proposed an interacting mode between the two main domains. However, a recent NMR study has concluded that the two main domains are tightly docked under ATP state but disengaged under nucleotide-free, ADP, and substrate binding states (15). In addition, small-angle x-ray scattering study has shown that the two domains are significantly extended in the ADP-bound state, whereas the ATP-bound state is more compact (16). Moreover, data collected from tryptophan fluorescence, tryptic digestion, and infrared spectroscopic studies have consistently shown that the ADP-bound and nucleotide-free states have very similar structures (17, 18). In contrary to these observations, the two domains in the structure shown by Jiang et al. (14) are tightly engaged despite the fact that it is in a nucleotide-free state. Therefore, the structure reported by Jiang et al. (14) might not be physiologically relevant. A more recent crystal structure of ATP-bound yeast Hsp110 (Sse1; residues 2-659), a nucleotide exchange factor and a structural homologue of Hsp70s, demonstrates an ATP-caused intimate domain-conjugating conformation for this superfamily (19). However, structural evidence for domain-disjoining conformation of Hsp70 molecules is still lacking.
In this report we describe the crystal structures of Hsp70 chaperone family proteins in the eubacterial Geobacillus kaustophilus HTA426 DnaK (GkDnaK) and mammalian Rattus norvegicus 70-kDa heat shock cognate protein (rHsc70), in the ADP-Pi state. The GkDnaK is a structural and functional homologue of the well characterized Escherichia coli DnaK (EcoDnaK) (20). Both GkDnaK and rHsc70 constructs contain the NBD and SBD, but the 10-kDa C-terminal helical region has been omitted to avoid aggregation (9). Strikingly, the structures show that the two domains are in fully disengaged conformation, and they could be the first bona fide structures available for Hsp70 molecules in the ADP bound and domain-disjoining state.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The genes encoding wild-type rHsc70ΔC, GkDnaKΔC, and GkDnaJ were cloned into pET-22b, pET-28a, and pET-21b, respectively, with a hexahistidine tag at the C terminus. The GkDnaKΔC-F185W and GkDnaKΔC-F254W mutants were generated according to the Stratagene QuikChange mutagenesis protocol (Stratagene) using the pET28a-GkDnaKΔC plasmid as template. All the recombinant proteins were expressed in E. coli BL21(DE3). The His-tagged proteins were purified with a Ni2+ chelating column (HiTrap, GE Healthcare) followed by size-exclusion chromatography (HiLoad 16/60 Superdex™ 200 preparation grade column; GE Healthcare). The storage buffer for rHsc70ΔC and GkDnaKΔC for crystallization experiments was 20 mm Bicine-NaOH, pH 9.0, 100 mm NaCl. The mutant proteins appeared to be correctly folded based on measurements of secondary structure by circular dichroism spectroscopy (data not shown).
Crystallization and Structure Determination of GkDnaKΔC and rHsc70ΔC—The purified proteins were concentrated with Amicon Ultra® (Millipore) concentrators to 6.5 and 5 mg/ml for GkDnaKΔC and rHsc70ΔC, respectively, then incubated with 1 mm each of ATP and MgCl2 at room temperature for 2 h. Using the hanging drop vapor diffusion technique (1 μl of protein solution + 1 μl of reservoir solution), hexagonal crystals of GkDnaKΔC grew within 1 week in 200 mm ammonium citrate, pH 7.5, 20% (v/v) isopropanol, 15% (w/v) PEG3350 at 298 K. The GkDnaKΔC crystals diffracted to 2.37 Å and belong to space group P212121 with cell dimensions a = 52.75 Å, b = 71.46 Å, and c = 183.86 Å with one GkDnaKΔC molecule per asymmetric unit. For crystallization of rHsc70ΔC, the rHsc70ΔC protein solution (1 μl) was mixed with an equal volume of reservoir solution containing 100 mm CHES buffer, pH 9.5, 23% (w/v) PEG2000, 2% (w/v) PEG8000, and 10 mm threonine at 298 K. The slice shape rHsc70ΔC crystals grew within 3 days and diffracted to 3.5 Å and belong to space group C2 with cell dimensions a = 179.85 Å, b = 94.79 Å, c = 78.23 Å, and β = 93.24°. Both GkDnaKΔC and rHsc70ΔC crystals were flashfrozen in liquid nitrogen followed by cryo-data collection on an ADSC Quantum-315 CCD detector using a synchrotron radiation x-ray source at Beamline BL13B1 of the National Synchrotron Radiation Research Center in Taiwan. Data integration and scaling were carried out with the HKL2000 package (21).
The initial phases of GkDnaKΔC and rHsc70ΔC were solved using the molecular replacement software MOLREP from the CCP4 suite (22) using a truncated EcoDnaK SBD (1DKZ) and a modified bHsc70 NBD (3HSC) as search models. After initial model refinements by CNS (23), the program Xtalview (24) was used to adjust the protein model and verify the presence of the nucleotide by analyzing the Fo - Fc and 2 Fo - Fc electron density maps. The final model for the GkDnaKΔC consists of 504 amino acid residues from Met-1 to Ala-504 with 166 water molecules, an ADP molecule, a magnesium ion, and an inorganic phosphate. The current model has an R-factor of 22.7% for all reflections between 29.58 and 2.37 Å resolution and an Rfree of 27.0% using 5.0% randomly distributed reflections. The final model of the rHsc70ΔC comprises of 381 amino acid residues from Ser-2 to Gly-382 with 94 water molecules, an ADP molecule, and an inorganic phosphate. The R-factor of this model for all reflections between 29.01 and 3.5 Å resolution was refined to 25.7%, and an Rfree value of 27.9% was obtained using 5.0% randomly distributed reflections. The Ramachandran plots for both structures did not violate accepted backbone torsion angles. The PyMol (25) program was used to generate the figures. Statistical data are shown in Table 1.
TABLE 1.
Data collection and refinement statistics for GkDnaKΔC-ADP-Mg2+-Pi and rHsc70ΔC-ADP-Pi structures
| GkDnaKΔC-ADP-Mg2+-Pi | rHsc70ΔC-ADP-Pi | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | C2 |
| Cell dimensions | ||
| a (Å) | 52.75 | 179.85 |
| b (Å) | 71.46 | 94.79 |
| c (Å) | 183.86 | 78.23 |
| β (°) | 93.24 | |
| Wavelength | 1.0000 | 1.0229 |
| Resolution (Å) | 2.37 | 3.50 |
| Rmergea (%) | 6.5 (49.3)b | 13.6 (26.1) |
| I/σ(I) | 14.9 (2.0) | 10.4 (5.0) |
| Completeness (%) | 94.8 (93.9) | 98.4 (97.1) |
| Redundancy |
3.1 (3.1) |
3.3 (3.2) |
| Refinement | ||
| Resolution (Å) | 29.58-2.37 | 29.01-3.50 |
| No. reflections | 25,994 | 15,130 |
| Rwork/Rfree (%) | 22.7/27.0 | 25.7/27.9 |
| No. atoms | ||
| Protein | 3,721 | 5,904 |
| Ligand/ion | 32/1 | 64 |
| Water | 166 | 94 |
| B-factors | ||
| Protein | 43.4 | 71.6 |
| Ligand/ion | 27.5/56.4 | 71.1 |
| Water | 51.8 | 62.6 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.014 | 0.020 |
| Bond angles (°) | 2.4 | 2.6 |
Rmerge = ∑hkl∑i|Ihkl,i - 〈Ihkl〉|/∑hkl〈Ihkl〉.
Numbers in parentheses refer to the highest resolution shell.
Steady-state Fluorescence Measurements—Intrinsic fluorescence spectra of the GkDnaKΔC-F185W mutant were recorded at 298 K on a Varian Cary-Eclipse fluorescence spectrophotometer (Varian Iberica) with an excitation wavelength of 295 nm and an emission wavelength from 310-450 nm. Excitation and emission slit widths were set at 2.5 and 10 nm, respectively. A quartz cuvette with a light path length of 10 mm was used. The experiments were carried out in 40 mm Tris-HCl, pH 7.4, 50 mm KCl, 5 mm MgCl2, and 5 μm GkDnaKΔC in the absence or presence of 0.5 mm ADP or ATP before and after adding 0.5 mm heptapeptide FYQLALT. The incubation time after nucleotide and substrate addition was 20 min for each sample at 298 K. For the GkDnaJ participating assay, ATP was first bound to equilibrium with GkDnaKΔC; afterward, GkDnaJ (1 μm final concentration) was added and incubated at 298 K for 20 min to determine the GkDnaJ-mediated environmental variation of the potential linker-NBD conjugating pocket. All spectra measurements were corrected for background emission and scattering.
RESULTS
Crystal Structure of GkDnaKΔC Bound with ADP-Mg2+-Pi—The overall structure of GkDnaKΔC (residues 1-504) bound with ADP, magnesium ion, and Pi is shown in Fig. 1A. The NBD and SBD are well separated in a stretched-out bilobate shape. The C-terminal region of the NBD (residues 1-353) is connected to the N-terminal region of the SBD (residues 365-504) by an extended and highly conserved hydrophobic linker (residues 354-364). Unlike the nucleotide-free structure of bHsc70ΔC (1YUW) and the ATP-bound structure of yeast Sse1 (Protein Data Bank code 2QXL), there is no direct interaction between the NBD and SBD in the GkDnaKΔC structure. The extended linker is the only connection between these two topologically separated domains. The overall shape of GkDnaKΔCis elongated with a maximum distance (dmax) of 130 Å.
FIGURE 1.

Overall structure of GkDnaKΔC in the ADP-Mg2+-Pi state and intermolecular interactions between symmetry-related GkDnaKΔC molecules. A, ribbon diagram of the GkDnaKΔC-ADP-Mg2+-Pi complex structure and its orthogonal view reveal that the NBD (residues 1-353, blue) and the SBD (residues 365-504, cyan) are connected by an extended linker (residues 354-364, red). The bound nucleotide (ADP, magnesium ion, and Pi) is represented as spheres. B, the extended hydrophobic linker between the NBD and the SBD of GkDnaKΔC (blue) occupies the substrate binding pocket of a neighboring crystallographic symmetry-related GkDnaKΔC molecule (gold).
Interestingly, the hydrophobic linker lies in the substrate binding pocket of a neighboring crystallographic symmetry-related GkDnaKΔC molecule (Fig. 1B). The interactions between the substrate binding pocket and the linker are mainly hydrophobic, but several H-bonds on the linker backbone can be detected (Fig. 2A). These binding forces are similar to those of other Hsp70-peptide complex crystal structures (8, 26). The fact that the linker is bound like a substrate in the substrate binding pocket is not surprising since the linker is very hydrophobic in nature and potentially mimics the preferred substrate peptides for DnaK chaperones. Therefore, the domain-dissociated conformation in our GkDnaKΔC structure may represent a substrate peptide-bound conformation of Hsp70s. This is consistent with NMR study (15) which shows the binding of substrate peptide disrupts the packing between the two domains.
FIGURE 2.

Peptide binding pocket and adenosine nucleotide binding site of GkDnaKΔC. A, stereo view of the environment surrounding the hydrophobic linker which binds in the substrate binding pocket of a symmetry-related SBD from an adjacent molecule. Direct hydrogen bonds between the two molecules are shown as dashed lines. Models of GkDnaKΔC and its symmetry-related molecule are colored blue and gold, respectively. Interacting residues are represented in stick form. B, stereo view of the adenosine nucleotide binding site in the NBD of GkDnaKΔC. The 2 Fo - Fc electron density map for the bound ADP, magnesium ion, and Pi was contoured at the 1.0 σ level and colored in yellow. Hydrogen bonds and salt bridges between the bound nucleotide and protein are represented as dashed lines. C, schematic representation of the interactions involved in binding the hydrolyzed ATP. Hydrogen bonds and salt bridges between the bound nucleotide and protein are represented as dashed lines with lengths shown in angstroms. Three residues (Arg-314, Ile-315, and Lys-240) that interact hydrophobically with the adenine base of the hydrolyzed ATP are boxed.
The Nucleotide Binding Site within GkDnaKΔC—Although the crystals of GkDnaKΔC were grown in the presence of 1 mm ATP and MgCl2, the well defined electron density corresponding to the adenosine nucleotide in the nucleotide binding site reveals that the ATP-Mg2+ has been hydrolyzed to ADP-Mg2+-Pi (Fig. 2B). This observation is consistent with the features of the Hsp70 NBD-nucleotide complex structures in other species (27, 28). A schematic representation of the adenosine nucleotide binding site is shown in Fig. 2C. The main binding forces of the adenine base are hydrophobic through the hydrophobic regions of Lys-240, Arg-314, and Ile-315. The ribose portion is anchored by three H-bonds contributed by Glu-236 and Lys-239. Five additional H-bonds from Thr-12, Asn-13, Gly-170, and Gly-311 stabilize the α- and β-phosphates of the ADP. Furthermore, the Pi group forms a salt bridge with Lys-68, hydrogen bonds to Thr-11, Glu-145, and Thr-172, and bridges with the β-phosphate of ADP via a magnesium ion.
Structural Comparison of GkDnaKΔC NBD with the Nucleotide-free State of EcoDnaK—The only available NBD structure of prokaryotic DnaK in the Protein Data Bank is that of EcoDnaK (Protein Data Bank code 1DKG). Therein, the NBD appears in the nucleotide-free state and is complexed with a nucleotide exchange factor, GrpE (29). The NBD of EcoDnaK has a 58% sequence identity with GkDnaK. As shown in Fig. 3A, the nucleotide binding cavity of GkDnaK NBD in the ADP-Mg2+-Pi-bound form is 7.1 degrees narrower than that of EcoDnaK NBD in the nucleotide-free form. Such “open” and “closed” conformational differences of the NBDs between nucleotide-free and nucleotide-bound states are also observed in the structure of eukaryotic bHsc70 (14). In addition, sequence alignment (data not shown) shows an extensive deletion in GkDnaK that corresponds to amino acid residues 79-102 of EcoDnaK, resulting in a shortened α-helix and a β-strand in the NBD subdomain IB (Fig. 3A). Other related short deletions (corresponding to residues 44, 63, 184, and 208-211 of EcoDnaK) are located in the loop regions exposed to the surface, and none of these differences causes changes in the topology.
FIGURE 3.

Comparison of ADP-Mg2+-Pi bound and nucleotide-free Hsp70 structures. A, structural superposition between the NBD of GkDnaKΔCinthe ADP-Mg2+-Pi state (blue) and that of EcoDnaK in nucleotide-free state (yellow). The most structurally disparate region between GkDnaKΔC and EcoDnaK is colored in red. The bound nucleotide in the GkDnaKΔC NBD is shown in the stick form. The arrows indicate the possible closing behavior of the pocket upon nucleotide binding. B, diagram representation of the large-scale spatial difference for domain arrangement between GkDnaKΔC (blue) and bHsc70ΔC (orange). The NBDs of two structures are superimposed. Helixes A in both structures are colored red. C, stereo view of a structural superimposition of the ADP-Mg2+-Pi state GkDnaKΔC (blue) with the nucleotide-free state bHsc70ΔC (NBD, orange; SBD, yellow). Residues in the NBD of bHsc70ΔC that participate in domain-domain interactions are highly conserved in the GkDnaKΔC NBD. Equivalent residues in the bHsc70ΔC SBD are also highly conserved in the GkDnaKΔC SBD (bHsc70:GkDnaK, Arg-416:Arg-383, Asn-417:Asn-384, Glu-513:Glu-479, Ile-515:Ile-481, Val-519:Ile-485, Ala-522:Ala-488). D, diagram representation of the large-scale spatial difference for domain arrangement between GkDnaKΔC (blue) and yeast Sse1 (magenta). The NBDs of these two structures are superimposed. E, structural superimposition of the NBD and interdomain linker from the GkDnaKΔC (blue) and the yeast Sse1 (magenta). The interdomain linkers from both structure are colored yellow. Residue Phe-185 of GkDnaKΔC is represented as a spheres and colored red.
Large Domain Movement in GkDnaKΔC upon Nucleotide Binding and Hydrolysis—To study the conformational variation of Hsp70 under different states of the chaperon cycle, we compared our GkDnaKΔC structure with that of Jiang et al. (14), bHsc70ΔC. Although bHsc70ΔC is in a nucleotide-free state, the structure shows a domain-conjugating conformation. The superimposition of the NBD portions of GkDnaKΔC and bHsc70ΔC reveals a dramatic conformational rearrangement for SBDs (Fig. 3B). In the bHsc70ΔC structure, helix A of the SBD locates in the groove between subdomains IA and IIA of NBD and serves as a communicating bridge between the NBD and SBD. In our GkDnaKΔC structure, however, helix A of SBD faces away from and does not contact with the NBD. The SBD of GkDnaKΔC in the ADP-Mg2+-Pi bound state is only connected to the NBD by an extended hydrophobic linker and lacks any direct interdomain interactions. Structural comparison nevertheless shows that the bHsc70 residues responsible for the NBD-SBD interactions are highly conserved in GkDnaK (Fig. 3C). Conceivably, a similar interdomain communicating mode might also happen in GkDnaK. Moreover, whereas the dimension of our GkDnaKΔC structure (dmax = 130 Å) is in good agreement with the result of a small angle x-ray scattering study for ADP-bound bHsc70ΔC (dmax = 125 Å) (16), the dimension of the nucleotide-free form of bHsc70ΔC(dmax = 105 Å) (14) is significantly smaller than the value previously reported.
Recently, the crystal structure of yeast Hsp110 (Sse1) at ATP-bound state was determined (19). Hsp110 proteins constitute a heterogeneous family of abundant molecular chaperones related to the Hsp70 proteins and act as an efficient nucleotide exchange factor. In the Sse1 structure, the NBD and SBD associate intimately. The bound ATP remains unhydrolyzed despite the fact that ATP is generally not stably maintained in Hsp70s (27, 28). This particular phenomenon may originate from the weak intrinsic ATPase activity and functional non-necessity of ATP hydrolysis in Sse1 (30). We also compared our structure with that of the ATP-bound Sse1 (Fig. 3D). The Sse1-ATP demonstrates a significant positional difference in the interdomain linker as well as SBD to those of GkDnaKΔC. The interdomain linker of Sse1 fits into the groove between subdomains IA and IIA of NBD and forms an interdomain β-sheet. The SBD of Sse1 tightly conjugates with NBD on subdomains IA and IIA. With the intimate domain-associating conformation and the bound non-hydrolyzed ATP molecule in NBD, the Sse1-ATP structure may mimic an ATP state conformation for Hsp70. In comparison, with the observed bound substrate in SBD and the hydrolyzed ADP-Mg2+-Pi in NBD, the GkDnaKΔC structure incurs a conformational state that represents a distinctive stage in the Hsp70 functional cycle.
Examination of the Nucleotide and Substrate-induced Positional Variation of the Interdomain Linker—Previous studies have shown that binding of substrate peptides at SBD stimulates ATP hydrolysis in the NBD of Hsp70s (7). The structural comparison of substrate-bound GkDnaKΔC-ADP-Mg2+-Pi with the yeast Sse1-ATP further demonstrates a significant positional variation of the interdomain hydrophobic linker (Fig. 3E). It is likely that the substrate-induced interdomain linker relocation may play an important role in the allosteric regulation between the two main domains. To test this hypothesis, steady-state fluorescence measurements were performed. Because GkDnaKΔC does not contain any endogenous tryptophan in its amino acid sequence, a tryptophan residue (F185W) was introduced onto the interface region to serve as a fluorescence probe. The equivalent position of F185W in yeast Sse1-ATP structure is located between the NBD and the interdomain hydrophobic linker (19). However, the corresponding position of F185W in bovine Hsc70 is located at the interface between NBD and SBD (14). A heptapeptide FYQLALT that exhibits high affinity for Hsp70 (31) was utilized as substrate in this study. The fluorescence emission spectra were monitored before and after GkDnaKΔC-F185W were incubated with FYQLALT in the presence and absence of nucleotides. As shown in Fig. 4A, in the presence of ATP and before adding the peptide FYQLALT, the intrinsic fluorescence intensity was lower than that of the ADP-bound or nucleotide-free states. Strikingly, a unanimous change of fluorescent spectra for each nucleotide states was observed upon adding the substrate FYQLALT. The emission fluorescent intensity of the tryptophan residue demonstrated an environment similar to that of the ADP- or nucleotide-free state. This phenomenon is in agreement with the results of NMR study, indicating that substrate binding uncoupled the two main domains of EcoDnaK and precipitated an ADP state-like overall conformation (15). In addition, fluorescence was measured with a GkDnaKΔC-F254W mutant wherein the Trp-reporter residue was located away from the NBD-SBD or NBD-linker interface. In this case, no substantial change was observed in intrinsic fluorescence after incubating with ATP or ADP (Fig. 4B).
FIGURE 4.
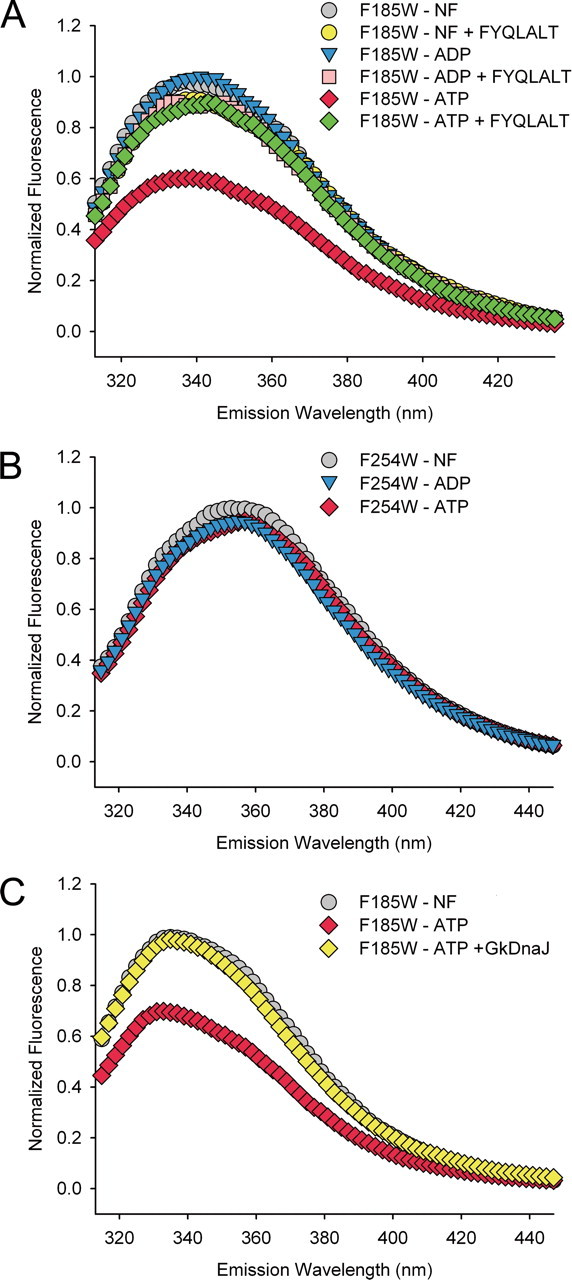
Examination of the nucleotide- and substrate-induced conformational changes in the GkDnaKΔC. A, the normalized fluorescence emission spectra of GkDnaKΔC-F185W in the nucleotide-free state (NF, gray), ADP-bound state (blue), ATP-bound state (red), nucleotide-free state with substrate FYQLALT (yellow), ADP-bound state with substrate FYQLALT (pink), and ATP-bound state with substrate FYQLALT (green). B, the normalized fluorescence emission spectra of GkDnaKΔC-F254W mutant in different nucleotide binding states. C, the GkDnaJ-induced effect (yellow) on the reversal of the ATP-induced fluorescence quenching for the GkDnaKΔC-F185W mutant.
DnaJ-induced Environmental Variation—During the functional cycle of DnaK, the J-domain cochaperone (DnaJ) can stimulate the ATP hydrolysis of DnaK and trigger the release of inorganic phosphate (2). We next investigated if DnaJ may also cause the dissociation of the SBD from the NBD of GkDnaK, which reflects the transition from the ATP- to ADP-bound state. A steady-state fluorescence measurement assay was performed to examine this putative conformational switch. The GkDnaKΔC-F185W was first allowed to bind ATP. Subsequently, purified GkDnaJ was added to the reaction for immediate analysis. As shown in Fig. 4C, the ATP-induced decrease of fluorescence intensities was fully recovered, implying that GkDnaJ binding can elicit an ADP-like environment and induce the disengagement of the SBD from the NBD of GkDnaKΔC.
Crystal Structure of rHsc70ΔC at ADP-Pi-bound State—To determine whether the dissociation of the NBD from the SBD was a general characteristic for Hsp70s, we also studied the crystal structure of the 70-kDa heat shock cognate protein from R. norvegicus (rHsc70ΔC). The rHsc70ΔC construct used in this analysis spans residues 1-543, which corresponds to the same region of GkDnaKΔC and shares a 51% sequence identity. Surprisingly, after structure determination, we could only solve the NBD portion (residues 2-382, Fig. 5A) of the structure. The electron densities for residues 383-543 of rHsc70ΔC, which encompass the SBD and the hydrophobic linker, were too weak to be integrated into the structure. To verify if the crystallized protein still remained intact (residues 1-543), crystals were dissolved and subjected to SDS-PAGE analysis. As shown in Fig. 5B, the rHsc70ΔC proteins in the crystal were not degraded (Fig. 5B). Furthermore, the crystal packing of rHsc70ΔC shows a large void space with no molecular contact between the two layers of the NBD regions (Fig. 5C). Although we have difficulty locating the SBD structure in the gap region, this empty space should be occupied by the SBD to maintain the crystal lattice. Conceivably, SBD was properly folded in the crystals of the rHsc70ΔC. Inability to resolve the SBD structure might imply the linkers between the NBD and the SBD were highly flexible, resulting in coexistence of different conformations. Nevertheless, a structure similar to that of bHsc70ΔC (14) was not likely to be the predominant conformation of rHsc70ΔC in crystalline state, since such a structure cannot be fitted into this crystal packing (Fig. 5C). Hence, it is reasonable to assume that NBD and SBD were not docked together or associated with each other in rHsc70ΔC. Nevertheless, the electron density map corresponding to the bound nucleotide in the NBD revealed that the ATP had also been hydrolyzed to ADP-Pi (Fig. 5D). The hydrolyzed ATP is bound in a manner similar to that of the GkDnaKΔC structure, except that the interaction contributed by the magnesium ion is substituted by Asp-199.
FIGURE 5.

Overall structure, adenosine nucleotide binding site, and the highly flexible SBD of rHsc70ΔC in the ADP-Pi state. A, ribbon drawing of the rHsc70ΔC-ADP-Pi complex. The NBD (residues 2-382) is colored green, and the bound nucleotide (ADP and Pi) are represented as spheres. The SBD (residues 383-543) and hydrophobic linker region are unassignable here. B, the crystals of rHsc70ΔC were analyzed by SDS-PAGE and visualized by dye-staining. Lane 1, the rHsc70ΔC solution used in the crystallization reaction. Lane 2, the rHsc70ΔC crystals. C, the superimposition of ADP-Mg2+-Pi-bound form of GkDnaKΔC (blue) and nucleotide-free form of bHsc70ΔC (orange) on the crystal packing of the ADP-Pi-bound form of rHsc70ΔC (gray). The plausible space for the SBD of rHsc70ΔCis encircled by magenta dashed lines. D, stereo view of the environment surrounding the adenosine nucleotide binding site in the NBD of rHsc70ΔC. The 2 Fo - Fc electron density map for the bound ADP-Pi was contoured at 1.0 σ (gray) and 3.5 σ (yellow) levels. The stick models indicate residues interacting with ADP or Pi. Dashed lines indicate hydrogen bonds and salt bridges between the bound nucleotide and protein. E, the superimposition of bound adenosine nucleotides among GkDnaKΔC-ADP-Mg2+-Pi, rHsc70ΔC-ADP-Pi, and bovine NBD (K71M)-ATP-Mg2+-K+ precatalytic state structures. All atoms are represented by sticks except Mg2+, which is represented by spheres. Coloring has the following pattern: carbon (gray), nitrogen (cyan), oxygen (red), phosphorous and Mg2+ of GkDnaKΔC (blue), phosphorous and Mg2+ of bovine NBD (K71M) (green), phosphorous of rHsc70ΔC (yellow), and Asp-199 of rHsc70ΔC (light yellow) and its corresponding residues in GkDnaKΔC (light blue) and bovine NBD (K71M) (light green). The β- and γ-phosphate of pre- of post-hydrolyzed ATP are linked by dashed lines with lengths shown in angstroms.
Comparison of Bound Nucleotide in Various States—The conformational comparison of bound adenosine nucleotides among structures of GkDnaKΔC-ADP-Mg2+-Pi, rHsc70ΔC-ADP-Pi, and bovine NBD(K71M)-ATP-Mg2+-K+ in the precatalytic state (32) clearly displays significant differences in the distance between the β- and γ-phosphate (Fig. 5E). In the K71M mutant of bovine NBD, the unhydrolyzed ATP shows a common distance of 2.95 Å between β- and γ-phosphate as well as that between α- and β-phosphate. For GkDnaKΔC-ADP-Mg2+-Pi, the measured distance between the β- and γ-phosphate is 4.54 Å, which is well conserved among other Hsp70 NBD structures (27, 28). On the other hand, for rHsc70ΔC, the absence of magnesium ion causes a shortening of this distance to 3.78 Å. The biological significance of this unique observation, however, needs to be elucidated.
DISCUSSION
Herein, we report the crystal structures of the C-terminal truncated GkDnaK and rHsc70. The structure of GkDnaKΔC clearly shows that, for a single molecule in the ADP-Mg2+-Pi state, there is no contact between the NBD and the SBD. Furthermore, the linker connecting the NBD and the SBD is fully extended and locates in the substrate binding pocket of a neighboring GkDnaKΔC. The linker somehow behaves like a peptide substrate. This is not entirely surprising, since the linker contains a sequence very similar to that of peptides with high affinity for DnaK (33). Therefore, the stable domain-dissociation structure of GkDnaKΔC reported here might be one of the domain-disengaged conformations selected by crystallization. Nevertheless, this structure does suggest that GkDnaK may be polymerized in the ADP-bound state. Indeed, it was previously shown that many Hsp70 species have self-associated properties in solution (34-41), and the oligomeric conformations can be dissociated into monomers in the presence of ATP (42). On the other hand, only the NBD can be resolved in rHsc70ΔC despite the fact that SBD is present in the protein crystal. In this case, the sequence of the linker differs from that of the high affinity heptapeptide for Hsc70 (31). The linker, therefore, is less likely to be stabilized by an adjacent rHsc70ΔC. If so, the linker should be flexible, and the location of the SBD relative to that of NBD in rHsc70ΔC may be variable. However, it is unlikely that the NBD and the SBD in rHsc70ΔC will adopt the conformation reported by Jiang et al. (14), since large empty space will be generated in the protein crystals. In any event, it is reasonable to assume that the NBD and the SBD of rHsc70DC with ADP bound may appropriate conformations with few contact points, albeit there may be several such states.
Our structure with well separated NBD and SBD represents only an ADP state of the molecule. Conceivably, this structure should differ significantly from that of GkDnaKΔC in the ATP-bound state. Indeed, our results from fluorescent measurements imply that binding of peptide substrate to GkDnaKΔCin the ATP state triggers conformational changes bringing about an enhancement in ATP hydrolysis and disengaging the SBD from the NBD. This conclusion is in good agreement with the results from NMR study of EcoDnaK (15). Therein, binding of substrate reverses domain packing induced by ATP, and SBD and NBD behave almost like independent domains. More importantly, the crystal structure of Sse1 (a Hsp70 structural homologue in yeast) clearly demonstrates that the SBD and the NBD may communicate intimately in the ATP-bound conformation (19). However, it remains to be determined if DnaK or Hsp70 may adopt a similar conformation in the ATP state.
Because the domain-communicating interfaces are highly conserved across all species of the Hsp70 chaperone family (19), this nucleotide-dependent domain movement should be universal. Results from our laboratory and others permit us to draw a picture of the domain rearrangement behavior for Hsp70 at different stages of the chaperone cycle (Fig. 6A). With the assistance of J-domain cochaperone, the binding of substrate and hydrolysis of ATP could lead Hsp70 to an open conformation. Then, the nucleotide exchange factor causes ADP-ATP replacement and the releases of bound-substrate resulting in a close conformation for Hsp70. On the basis of thermodynamic consideration (43), an “entropic pulling” mechanism has recently been proposed for Hsp70 chaperone activity. According to this model, Hsp70s use the entropy loss due to excluded-volume effects to perform their disparate cellular function. The excluded-volume effect is proportional to the space occupied by the Hsp70 molecule, and chaperones that occupy larger space have higher functional activity. The structure of GkDnaKΔC described here clearly illustrates how Hsp70s may enlarge the excluded-volume effect by extending the two-lobed conformation (Fig. 6B). Here, the distance from the bound peptide to the bottom of the NBD is about 115 Å in the domain-separated GkDnaKΔC structure. This value represents the radius of excluded volume for substrate-bound GkDnaKΔC and corresponds to ∼31 residues in a fully extended polypeptide chain (44). It is in agreement with the result obtained in the entropic pulling study, demonstrating that Hsp70 has the capacity to generate a strong pulling force for a length of ∼30 imported residues (43). This number also matches with the reported average distance (25-35 residues) between each typical Hsp70-binding site on the translocating protein (45). When the SBD of Hsp70 anchors onto the substrate, the flexible inter-domain linker could let the disengaged NBD act as a plummet with high degree of freedom. This particular behavior may assist Hsp70 chaperones to handle shorter exposed substrate peptide in various situations. For instance, it could disassociate compact protein aggregates by plucking the slight exposed hydrophobic segments. Subsequently, the substrate induces domain-disengaged conformation, and the dispersive movement of NBD could generate an excluded-volume effect despite the fact that the SBD is fixed on the substrate. Hence, the characteristic domain-disjoining behavior of Hsp70s could be linked to their crucial cellular functions through the entropy pulling model.
FIGURE 6.

Implication of Hsp70 chaperone cycle and benefits for domain-disjoining conformation of Hsp70s. A, schematic representation for the domain rearrangement of Hsp70. The NBD, SBD, and linker of Hsp70 are colored blue, cyan, and gray, respectively. The cochaperones J-domain ATPase activating protein and nucleotide exchange factor are symbolized by letters J and NEF, respectively. The substrate of Hsp70 is represented as red ribbons, and ATP and ADP are as magenta ellipsoids. B, schematic diagram depicting how luminal Hsp70s enlarge their excluded-volume effect by extending the two-lobed conformation (left, before; right, after) while conducting the post-translational translocation of precursor proteins across membranes. The two-lobed structures are Hsp70 molecules in which the NBDs are colored blue, the SBDs are colored cyan, and the interdomain linkers are colored gray. The brown structures represent the channel complexes that span two-layer membranes, and ribbons represent precursor proteins being imported into the lumen. The possible distributing area of the substrate-bound Hsp70 are represented by paler versions of Hsp70 molecules and circled by red dashed lines.
In summary, we report here the structures of the 60-kDa fragment of GkDnaK and rHsc70. In both structures, the bound nucleotides are ADP-Pi, and the nucleotide binding domain and peptide-substrate binding domain do not contact with each other except for the connecting linker. The mechanism for domain disengagement and the role of the 10-kDa fragment at the C terminus remain to be elucidated.
Acknowledgments
We are grateful for access to synchrotron radiation beamline 13B1 at the National Synchrotron Radiation Research Center in Taiwan.
The atomic coordinates and structure factors (code 2V7Y and 2V7Z) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by Taiwan Academia Sinica and National Science Council Research Grants NSC95-2311-B-001-064 (to C.-D. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Hsp70, 70-kDa heat shock protein; NBD, nucleotide binding domain; SBD, substrate binding domain; GkDnaK, G. kaustophilus HTA426 DnaK; rHsc70, R. norvegicus 70-kDa heat shock cognate protein; Bicine, N,N-bis(2-hydroxyethyl)glycine; CHES, 2-(cyclohexylamino)ethanesulfonic acid.
References
- 1.Young, J. C., Agashe, V. R., Siegers, K., and Hartl, F. U. (2004) Nat. Rev. Mol. Cell Biol. 5 781-791 [DOI] [PubMed] [Google Scholar]
- 2.Bukau, B., and Horwich, A. L. (1998) Cell 92 351-366 [DOI] [PubMed] [Google Scholar]
- 3.Hartl, F. U. (1996) Nature 381 571-579 [DOI] [PubMed] [Google Scholar]
- 4.Neupert, W., and Brunner, M. (2002) Nat. Rev. Mol. Cell Biol. 3 555-565 [DOI] [PubMed] [Google Scholar]
- 5.Wegele, H., Muller, L., and Buchner, J. (2004) Rev. Physiol. Biochem. Pharmacol. 151 1-44 [DOI] [PubMed] [Google Scholar]
- 6.Liberek, K., Marszalek, J., Ang, D., Georgopoulos, C., and Zylicz, M. (1991) Proc. Natl. Acad. Sci. U. S. A. 88 2874-2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laufen, T., Mayer, M. P., Beisel, C., Klostermeier, D., Mogk, A., Reinstein, J., and Bukau, B. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 5452-5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu, X., Zhao, X., Burkholder, W. F., Gragerov, A., Ogata, C. M., Gottesman, M. E., and Hendrickson, W. A. (1996) Science 272 1606-1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou, C. C., Forouhar, F., Yeh, Y. H., Shr, H. L., Wang, C., and Hsiao, C. D. (2003) J. Biol. Chem. 278 30311-30316 [DOI] [PubMed] [Google Scholar]
- 10.Liberek, K., Skowyra, D., Zylicz, M., Johnson, C., and Georgopoulos, C. (1991) J. Biol. Chem. 266 14491-14496 [PubMed] [Google Scholar]
- 11.Mayer, M. P., Schroder, H., Rudiger, S., Paal, K., Laufen, T., and Bukau, B. (2000) Nat. Struct. Biol. 7 586-593 [DOI] [PubMed] [Google Scholar]
- 12.Palleros, D. R., Reid, K. L., Shi, L., Welch, W. J., and Fink, A. L. (1993) Nature 365 664-666 [DOI] [PubMed] [Google Scholar]
- 13.Schmid, D., Baici, A., Gehring, H., and Christen, P. (1994) Science 263 971-973 [DOI] [PubMed] [Google Scholar]
- 14.Jiang, J., Prasad, K., Lafer, E. M., and Sousa, R. (2005) Mol. Cell 20 513-524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swain, J. F., Dinler, G., Sivendran, R., Montgomery, D. L., Stotz, M., and Gierasch, L. M. (2007) Mol. Cell 26 27-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilbanks, S. M., Chen, L., Tsuruta, H., Hodgson, K. O., and McKay, D. B. (1995) Biochemistry 34 12095-12106 [DOI] [PubMed] [Google Scholar]
- 17.Buchberger, A., Theyssen, H., Schroder, H., McCarty, J. S., Virgallita, G., Milkereit, P., Reinstein, J., and Bukau, B. (1995) J. Biol. Chem. 270 16903-16910 [DOI] [PubMed] [Google Scholar]
- 18.Moro, F., Fernandez-Saiz, V., and Muga, A. (2006) Protein Sci. 15 223-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, Q., and Hendrickson, W. A. (2007) Cell 131 106-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richarme, G., and Kohiyama, M. (1993) J. Biol. Chem. 268 24074-24077 [PubMed] [Google Scholar]
- 21.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 22.Collaborative Computational Project, N. (1994) Acta Crystallogr. Sect. D 50 760-76315299374 [Google Scholar]
- 23.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D 54 905-921 [DOI] [PubMed] [Google Scholar]
- 24.McRee, D. E. (1999) J. Struct. Biol. 125 156-165 [DOI] [PubMed] [Google Scholar]
- 25.DeLano, W. L. (2002) The PyMOL User's Manual, Delano Scientific, San Carlos, CA
- 26.Cupp-Vickery, J. R., Peterson, J. C., Ta, D. T., and Vickery, L. E. (2004) J. Mol. Biol. 342 1265-1278 [DOI] [PubMed] [Google Scholar]
- 27.Flaherty, K. M., DeLuca-Flaherty, C., and McKay, D. B. (1990) Nature 346 623-628 [DOI] [PubMed] [Google Scholar]
- 28.Sriram, M., Osipiuk, J., Freeman, B., Morimoto, R., and Joachimiak, A. (1997) Structure 5 403-414 [DOI] [PubMed] [Google Scholar]
- 29.Harrison, C. J., Hayer-Hartl, M., Di Liberto, M., Hartl, F., and Kuriyan, J. (1997) Science 276 431-435 [DOI] [PubMed] [Google Scholar]
- 30.Raviol, H., Sadlish, H., Rodriguez, F., Mayer, M. P., and Bukau, B. (2006) EMBO J. 25 2510-2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takenaka, I. M., Leung, S. M., McAndrew, S. J., Brown, J. P., and Hightower, L. E. (1995) J. Biol. Chem. 270 19839-19844 [DOI] [PubMed] [Google Scholar]
- 32.O'Brien, M. C., Flaherty, K. M., and McKay, D. B. (1996) J. Biol. Chem. 271 15874-15878 [DOI] [PubMed] [Google Scholar]
- 33.Rudiger, S., Germeroth, L., Schneider-Mergener, J., and Bukau, B. (1997) EMBO J. 16 1501-1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freiden, P. J., Gaut, J. R., and Hendershot, L. M. (1992) EMBO J. 11 63-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palleros, D. R., Reid, K. L., Shi, L., and Fink, A. L. (1993) FEBS Lett. 336 124-128 [DOI] [PubMed] [Google Scholar]
- 36.Carlino, A., Toledo, H., Skaleris, D., DeLisio, R., Weissbach, H., and Brot, N. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 2081-2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blond-Elguindi, S., Fourie, A. M., Sambrook, J. F., and Gething, M. J. (1993) J. Biol. Chem. 268 12730-12735 [PubMed] [Google Scholar]
- 38.Brown, C. R., Martin, R. L., Hansen, W. J., Beckmann, R. P., and Welch, W. J. (1993) J. Cell Biol. 120 1101-1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson, J. V., Haskell, D. W., and Guy, C. L. (1994) Plant Physiol. 104 1371-1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schonfeld, H. J., Schmidt, D., Schroder, H., and Bukau, B. (1995) J. Biol. Chem. 270 2183-2189 [DOI] [PubMed] [Google Scholar]
- 41.Bhattacharyya, T., Karnezis, A. N., Murphy, S. P., Hoang, T., Freeman, B. C., Phillips, B., and Morimoto, R. I. (1995) J. Biol. Chem. 270 1705-1710 [DOI] [PubMed] [Google Scholar]
- 42.Benaroudj, N., Triniolles, F., and Ladjimi, M. M. (1996) J. Biol. Chem. 271 18471-18476 [DOI] [PubMed] [Google Scholar]
- 43.De Los Rios, P., Ben-Zvi, A., Slutsky, O., Azem, A., and Goloubinoff, P. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 6166-6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Creighton, T. E. (1993) Proteins: Structures and Molecular Properties, 2nd Ed., p. 5, W. H. Freeman, New York
- 45.Rudiger, S., Buchberger, A., and Bukau, B. (1997) Nat. Struct. Biol. 4 342-349 [DOI] [PubMed] [Google Scholar]